THE LIVING WORLD
Unit two. The Living Cell
4.6. The Endomembrane System
Surrounding the nucleus within the interior of the eukaryotic cell is a tightly packed mass of membranes. They fill the cell, dividing it into compartments, channeling the transport of molecules through the interior of the cell and providing the surfaces on which enzymes act. The system of internal compartments created by these membranes in eukaryotic cells constitutes the most fundamental distinction between the cells of eukaryotes and prokaryotes.
Endoplasmic Reticulum: The Transportation System
The extensive system of internal membranes is called the endoplasmic reticulum, often abbreviated ER. The ER creates a series of channels and interconnections, and it also isolates some spaces as membrane-enclosed sacs called vesicles. The surface of the ER is the place where the cell makes proteins intended for export (such as enzymes secreted from the cell surface). The surface of those regions of the ER devoted to the synthesis of transported proteins is heavily studded with ribosomes and appears pebbly, like the surface of sandpaper, when seen through an electron microscope. For this reason, these regions are called rough ER. Regions in which ER-bound ribosomes are relatively scarce are correspondingly called smooth ER. The surface of the smooth ER is embedded with enzymes that aid in the manufacture of carbohydrates and lipids.
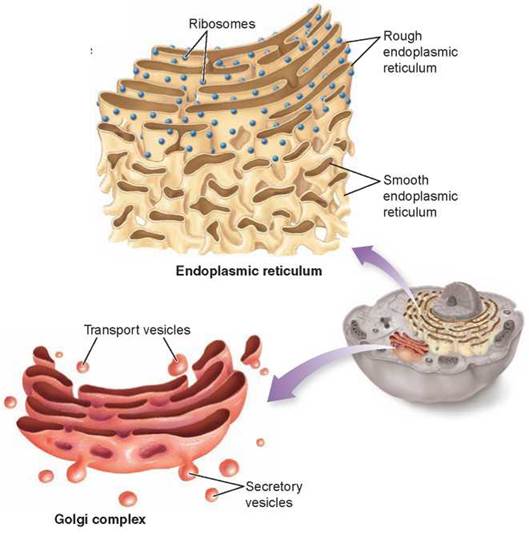
The Golgi Complex: The Delivery System
As new molecules are made on the surface of the ER, they are passed from the ER to flattened stacks of membranes called Golgi bodies. The number of Golgi bodies a cell contains ranges from 1 or a few in protists, to 20 or more in animal cells, and several hundred in certain plant cells. Golgi bodies function in the collection, packaging, and distribution of molecules manufactured in the cell. Scattered throughout the cytoplasm, Golgi bodies are collectively referred to as the Golgi complex.
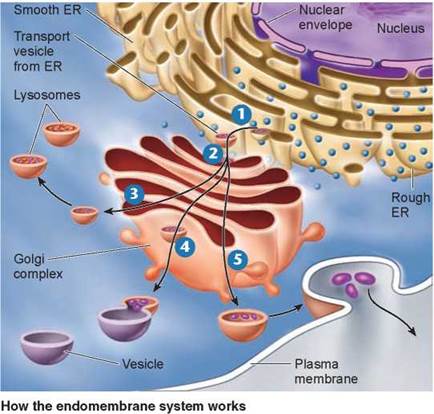
The rough ER, smooth ER, and Golgi work together as a transport system in the cell. Proteins and lipids that are manufactured on the ER membranes are transported throughout the channels of the ER and are packaged into transport vesicles that bud off from the ER 1. The vesicles fuse with the membrane of the Golgi bodies, dumping their contents into the Golgi 2. Within the Golgi bodies the molecules may take one of many paths, indicated by the branching arrows in the figure. Many of these molecules become tagged with carbohydrates. The molecules collect at the ends of the membranous folds of the Golgi bodies. Vesicles that pinch off from the ends carry the molecules to the different compartments of the cell 3 and 4, or to the inner surface of the plasma membrane, where molecules to be secreted are released to the outside 5.
Lysosomes: Recycling Centers
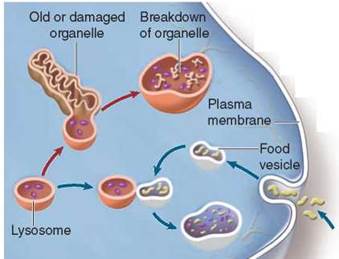
Other organelles called lysosomes arise from the Golgi complex (the light orange vesicle budding at 3 and contain a concentrated mix of the powerful enzymes manufactured in the rough ER that break down macromolecules. Lysosomes are also the recycling centers of the cell, digesting worn-out cell components to make way for newly formed ones while recycling the proteins and other materials of the old parts. Large organelles called mitochondria are replaced in some human tissues every 10 days, with lysosomes digesting the old ones as the new ones are produced. In addition to breaking down organelles and other structures within cells, lysosomes also eliminate particles (including other cells) that the cell has engulfed.
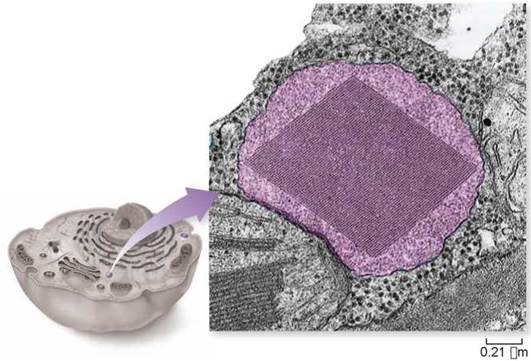
Peroxisomes: Chemical Specialty Shops
The interior of the eukaryotic cell contains a variety of membrane-bounded spherical organelles derived from the ER that carry out particular chemical functions. Almost all eukaryotic cells, for example, contain peroxisomes. Peroxisomes are spherical organelles that may contain a large crystal structure composed of protein (a peroxisome containing a crystal has been colored purple in the micrograph below). Peroxisomes contain two sets of enzymes. One set found in plant seeds converts fats to carbohydrates, and the other set found in all eukaryotes detoxifies various potentially harmful molecules—strong oxidants— that form in cells. They do this by using molecular oxygen to remove hydrogen atoms from specific molecules. These chemical reactions would be very destructive to the cell if not confined to the peroxisomes.
The Endomembrane System and Your Health
Several serious human diseases result when the endomem-brane system does not work as it should. One of the most serious of these involves a breakdown in the ability of the Golgi complex to properly address proteins destined for delivery to the cell’s lysosomes. In a normal cell, the address label is a modified sugar called mannose-6-phosphate, which the Golgi complex attaches to lysosome-bound proteins with a special enzyme. In individuals lacking this enzyme, the proteins that should have gone to lysosomes lack the mannose-6- phosphate address tag, and instead are delivered to the plasma membrane and secreted from the cell. Because almost all the recycling enzymes normally found in lysosomes are missing, the lysosomes swell with undegraded materials. Viewed under a microscope, the lysosomes appear to contain numerous inclusions where the undegraded material has clumped together, giving this condition its name, inclusion-cell disease. The swollen lysosomes ultimately cause serious damage to the cells of a developing human embryo, leading to facial and skeletal abnormalities and mental retardation.
About 40 different human diseases are caused by failure of the endomembrane system to deliver particular enzymes to the lysosomes. Called lysosome storage disorders, these disorders follow the pattern you have seen for inclusion-cell disease. For lack of the necessary recycling enzyme, a particular cell material accumulates undegraded in the lysosomes, eventually leading to cell damage and death. Most of these disorders are fatal in early childhood.
In the lysosome storage disorder called Pompe’s disease, lysosomes lack an enzyme necessary to digest glycogen. Glycogen, which the body uses as a ready source of energy, is stored in muscle and liver cells. When energy is required, lysosomes digest the glycogen, chopping off sugar units that the cell can use to produce energy (we explore how this happens in chapter 7). Pompe’s disease lysosomes, because they lack the necessary enzyme, do not degrade glycogen. Instead, harmful amounts of glycogen accumulate in the muscle and liver cells.
In the lysosome storage disease called Tay-Sachs disease, lysosomes are missing the enzyme necessary to degrade specific glycolipids called gangliosides that are abundant in the plasma membranes of brain cells. In Tay-Sachs individuals, the glycolipids accumulate in brain cells, which swell and eventually burst, releasing oxidative enzymes that kill the brain cells. The affected individual begins to exhibit rapid mental deterioration at about six to eight months of age. Within a year after birth, affected children are blind. Paralysis and death follow within five years.
Key Learning Outcome 4.6. An extensive system of interior membranes organizes the interior of the cell into functional compartments that manufacture and deliver proteins and carry out a variety of specialized chemical processes.