THE LIVING WORLD
Unit Three. The Continuity of Life
8.7. Cancer and Control of the Cell Cycle
Cancer can be caused by chemicals like those in cigarette smoke, by environmental factors such as UV rays that damage DNA, or in some instances by viruses that circumvent the cell’s normal growth and division controls. Whatever the immediate cause, however, all cancers are characterized by unrestrained cell growth and division. The cell cycle never stops in a cancerous line of cells.
Cancer results from damaged genes failing to control cell division. Researchers have identified several of these genes. One particular gene seems to be a key regulator of the cell cycle. Officially dubbed p53 (researchers italicize the gene symbol to differentiate it from the protein), this gene plays a key role in the G1 checkpoint of cell division. Figure 8.13 illustrates how the product of this gene, the p53 protein, monitors the integrity of DNA, checking that it has been successfully replicated and is undamaged. If the p53 protein detects damaged DNA, as it does in the upper panel, it halts cell division and stimulates the activity of special enzymes to repair the damage. Once the DNA has been repaired, p53 allows cell division to continue, indicated by the upper path of arrows. In cases where the DNA cannot be repaired, p53 then directs the cell to kill itself, activating an apoptosis (cell suicide) program, indicated by the lower path of arrows.
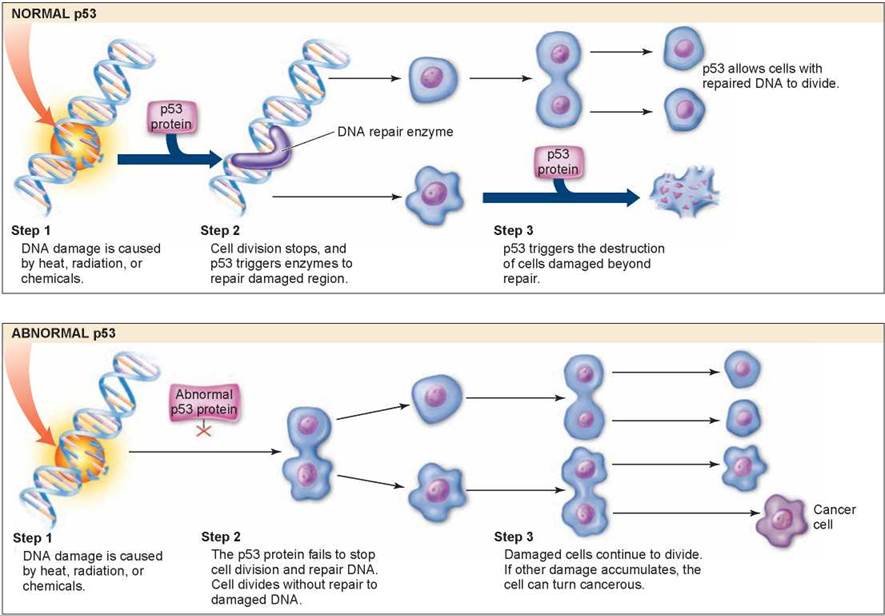
Figure 8.13. Cell division and p53 protein.
Normal p53 protein monitors DNA, destroying cells with irreparable damage to their DNA. Abnormal p53 protein fails to stop cell division and repair DNA. As damaged cells proliferate, cancer develops.
By halting division in damaged cells, p53 prevents the formation of tumors (even though its activities are not limited to cancer prevention). Scientists have found that p53 is itself damaged beyond use in most human cancers they have examined. It is precisely because p53 is nonfunctional that these cancer cells are able to repeatedly undergo cell division without being halted at the Gj checkpoint. The lower panel of figure 8.13 shows what happens when p53 doesn’t function properly. The abnormal p53 does not stop cell division and the damaged strand is replicated, which results in damaged cells. As more and more damage occurs to these cells, they become cancerous. To test this, scientists administered healthy p53 protein to rapidly dividing cancer cells in a petri dish: The cells soon ceased dividing and died. Scientists have further reported that cigarette smoke causes mutations in the p53 gene, reinforcing the strong link between smoking and cancer that you will encounter in chapter 24, section 24.6.
In about 50% of cancers, the p53 cancer defense malfunctions because the p53 gene itself has been damaged by chemicals or radiation, so that the protein the gene encodes no longer functions properly. In the other 50%, however, the defects lie in other genes. In many instances, the DNA is damaged at a site suppressing the production of a small molecule called MDM2 that is a potent natural inhibitor of the p53 protein. When the MDM2 gene becomes overactive as a result of mutation, its protein product suppresses p53’s activity.
A very promising approach to cancer prevention involves this second kind of p53 malfunction. Researchers studying the MDM2 protein found a relatively deep and well-defined pocket on its surface that proved to be the site that makes contact with the p53 protein. Perhaps, they hypothesized, a small molecule could be found that would fit into the site, and in doing so prevent p53 from binding. Such a molecule might prevent 50% of cancers! Searching for the key to fit this lock, they identified a family of synthetic chemicals they called “nutlins.” When tumor cells that had functional p53 genes were treated with nutlin, levels of p53 protein in the treated cells went up, and the tumor cells were killed, while normal cells treated in the same way were not killed. Nutlin is one of a host of new cancer therapies under active investigation.
Key Learning Outcome 8.7. Mutations disabling key elements of the G1 checkpoint are associated with many cancers.
Biology and Staying Healthy
Curing Cancer
Half of all Americans will face cancer at some point in their lives. Potential cancer therapies are being developed on many fronts. Some act to prevent the start of cancer within cells. Others act outside cancer cells, preventing tumors from growing and spreading. The figure on the right indicates targeted areas for the development of cancer treatments. The following discussion will examine each of these areas.
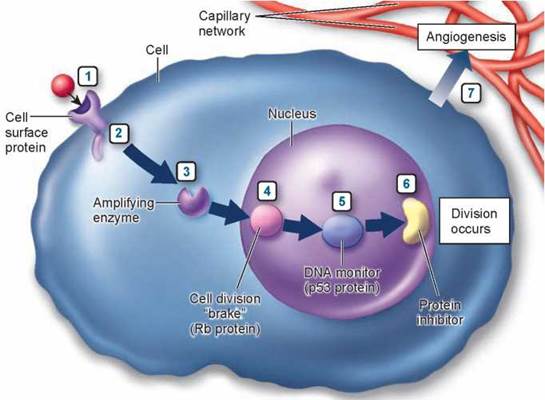
Seven different stages in the cancer process.
(1) On the cell surface, a growth factor's signal to divide is increased. (2) Just inside the cell, a protein relay switch that passes on the divide signal gets stuck in the "ON" position. (3) In the cytoplasm, enzymes that amplify the signal are amplified even more. In the nucleus, (4) a "brake" preventing DNA replication is inoperable, (5) proteins that check for damage in the DNA are inactivated, and (6) other proteins that inhibit the elongation of chromosome tips are destroyed. (7) The new tumor promotes angiogenesis, the formation of new blood vessels that promote growth.
Preventing the Start of Cancer
Many promising cancer therapies act within potential cancer cells, focusing on different stages of the cell's "Shall I divide?" decision-making process.
1. Receiving the Signal to Divide. The first step in the decision process is receiving a "divide" signal, usually a small protein called a growth factor released from a neighboring cell. The growth factor, the red ball at #1 in the figure, is received by a protein receptor on the cell surface. Like banging on a door, its arrival signals that it's time to divide. Mutations that increase the number of receptors on the cell surface amplify the division signal and so lead to cancer. Over 20% of breast cancer tumors prove to overproduce a protein called HER2 associated with the receptor for epidermal growth factor (EGF).
Therapies directed at this stage of the decision process utilize the human immune system to attack cancer cells. Special protein molecules called monoclonal antibodies, created by genetic engineering, are the therapeutic agents. These monoclonal antibodies are designed to seek out and stick to HER2. Like waving a red flag, the presence of the monoclonal antibody calls down attack by the immune system on the HER2 cell. Because breast cancer cells overproduce HER2, they are killed preferentially. The biotechnology research company Genentech's recently approved monoclonal antibody, called herceptin, has given promising results in clinical tests.
Up to 70% of colon, prostate, lung, and head/neck cancers have excess copies of a related receptor, epidermal growth factor 1 (HER1). The monoclonal antibody C225, directed against HER1, has succeeded in shrinking 22% of advanced, previously incurable colon cancers in early clinical trials. Apparently blocking HER1 interferes with the ability of tumor cells to recover from chemotherapy or radiation.
2. Passing the Signal via a Relay Switch. The second step in the decision process is the passage of the signal into the cell's interior, the cytoplasm. This is carried out in normal cells by a protein called Ras that acts as a relay switch, #2 in the figure. When growth factor binds to a receptor like EGF, the adjacent Ras protein acts like it has been "goosed," contorting into a new shape. This new shape is chemically active, and initiates a chain of reactions that passes the "divide" signal inward toward the nucleus. Mutated forms of the Ras protein behave like a relay switch stuck in the "ON" position, continually instructing the cell to divide when it should not. Thirty percent of all cancers have a mutant form of Ras. So far, no effective therapies have been developed targeting this step.
3. Amplifying the Signal. The third step in the decision process is the amplification of the signal within the cytoplasm. Just as a TV signal needs to be amplified in order to be received at a distance, so a "divide" signal must be amplified if it is to reach the nucleus at the interior of the cell, a very long journey at a molecular scale. To get a signal all the way into the nucleus, the cell employs a sort of pony express. The "ponies" in this case are enzymes called tyrosine kinases, #3 in the figure. These enzymes add phosphate groups to proteins, but only at a particular amino acid, tyrosine. No other enzymes in the cell do this, so the tyrosine kinases form an elite core of signal carriers not confused by the myriad of other molecular activities going on around them.
Cells use an ingenious trick to amplify the signal as it moves toward the nucleus. Ras, when "ON,” activates the initial protein kinase. This protein kinase activates other protein kinases that in their turn activate still others. The trick is that once a protein kinase enzyme is activated, it goes to work like a demon, activating hoards of others every second! And each and every one it activates behaves the same way too, activating still more, in a cascade of everwidening effect. At each stage of the relay, the signal is amplified a thousandfold.
Mutations stimulating any of the protein kinases can dangerously increase the already amplified signal and lead to cancer. Some 15 of the cell's 32 internal tyrosine kinases have been implicated in cancer. Five percent of all cancers, for example, have a mutant hyperactive form of the protein kinase Src. The trouble begins when a mutation causes one of the tyrosine kinases to become locked into the "ON” position, sort of like a stuck doorbell that keeps ringing and ringing.
To cure the cancer, you have to find a way to shut the bell off. Each of the signal carriers presents a different problem, as you must quiet it without knocking out all the other signal pathways the cell needs. The cancer therapy drug Gleevec, a monoclonal antibody, has just the right shape to fit into a groove on the surface of the tyrosine kinase called "abl.” Mutations locking abl "ON” are responsible for chronic myelogenous leukemia, a lethal form of white blood cell cancer. Gleevec totally disables abl. In clinical trials, blood counts revert to normal in more than 90% of cases.
4. Releasing the Brake. The fourth step in the decision process is the removal of the "brake” the cell uses to restrain cell division. In healthy cells this brake, a tumor-suppressor protein called Rb, blocks the activity of a protein called E2F, #4 in the figure. When free, E2F enables the cell to copy its DNA. Normal cell division is triggered to begin when Rb is inhibited, unleashing E2F. Mutations that destroy Rb release E2F from its control completely, leading to ceaseless cell division. Forty percent of all cancers have a defective form of Rb.
Therapies directed at this stage of the decision process are only now being attempted. They focus on drugs able to inhibit E2F, which should halt the growth of tumors arising from inactive Rb. Experiments in mice in which the E2F genes have been destroyed provide a model system to study such drugs, which are being actively investigated.
5. Checking That Everything Is Ready. The fifth step in the decision process is the mechanism used by the cell to ensure that its DNA is undamaged and ready to divide. This job is carried out in healthy cells by the tumor- suppressor protein p53, which inspects the integrity of the DNA, #5 in the figure. When it detects damaged or foreign DNA, p53 stops cell division and activates the cell's DNA repair systems. If the damage doesn't get repaired in a reasonable time, p53 pulls the plug, triggering events that kill the cell. In this way, mutations such as those that cause cancer are either repaired or the cells containing them eliminated. If p53 is itself destroyed by mutation, future damage accumulates unrepaired. Among this damage are mutations that lead to cancer. Fifty percent of all cancers have a disabled p53. Fully 70% to 80% of lung cancers have a mutant inactive p53—the chemical benzo[a]pyrene in cigarette smoke is a potent mutagen of p53.
6. Stepping on the Gas. Cell division starts with replication of the DNA. In healthy cells, another tumor suppressor "keeps the gas tank nearly empty” for the DNA replication process by inhibiting production of an enzyme called telomerase. Without this enzyme, a cell's chromosomes lose material from their tips, called telomeres. Every time a chromosome is copied, more tip material is lost. After some 30 divisions, so much is lost that copying is no longer possible. Cells in the tissues of an adult human have typically undergone 25 or more divisions. Cancer can't get very far with only the five remaining cell divisions, so inhibiting telomerase is a very effective natural brake on the cancer process, #6 in the figure. It is thought that almost all cancers involve a mutation that destroys the telomerase inhibitor, releasing this brake and making cancer possible. It should be possible to block cancer by reapplying this inhibition. Cancer therapies that inhibit telomerase are just beginning clinical trials.
Preventing the Spread of Cancer
7. Stopping Tumor Growth. Once a cell begins cancerous growth, it forms an expanding tumor. As the tumor grows ever-larger, it requires an increasing supply of food and nutrients, obtained from the body's blood supply. To facilitate this necessary grocery shopping, tumors leak out substances into the surrounding tissues that encourage the formation of small blood vessels, a process called angiogenesis, #7 in the figure. Chemicals that inhibit this process are called angiogenesis inhibitors. Two such natural angiogenesis inhibitors, angiostatin and endostatin, caused tumors to regress to microscopic size in mice, but initial human trials were disappointing.
Laboratory drugs are more promising. A monoclonal antibody drug called Avastin, targeted against a blood vessel growth-promoting substance called vascular endothelial growth factor (VEGF), destroys the ability of VEGF to carry out its blood-vessel-forming job. Given to hundreds of advanced colon cancer patients in 2003 as part of a large clinical trial, Avastin improved colon cancer patients's chance of survival by 50% over chemotherapy.
Inquiry & Analysis
Why Do Human Cells Age?
Human cells appear to have built-in life spans. As you learned in this chapter, cell biologist Leonard Hayflick reported in 1961 the startling result that skin cells growing in tissue culture, such as those growing in culture flasks in the photo below, will divide only a certain number of times. After about 50 population doublings, cell division stops (a doubling is a round of cell division producing two daughter cells for each dividing cell; for example, going from a population of 30 cells to 60 cells). If a cell sample is taken after 20 doublings and frozen, when thawed it resumes growth for 30 more doublings, and then stops. An explanation of the "Hayflick limit” was suggested in 1978 when researchers first glimpsed an extra length of DNA at the end of chromosomes. Dubbed telomeres, these lengths proved to be composed of the simple DNA sequence TTAGGG, repeated nearly a thousand times. Importantly, telomeres were found to be substantially shorter in the cells of older body tissues. This led to the hypothesis that a run of some 16 TTAGGGs was where the DNA replicating enzyme, called polymerase, first sat down on the DNA (16 TTAGGGs being the size of the enzyme's "footprint”), and because of being its docking spot, the polymerase was unable to copy that bit. Thus a 100-base portion of the telomere was lost by a chromosome during each doubling as DNA replicated. Eventually, after some 50 doubling cycles, each with a round of DNA replication, the telomere would be used up and there would be no place for the DNA replication enzyme to sit. The cell line would then enter senescence, no longer able to proliferate.
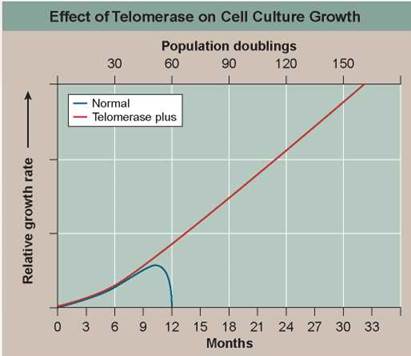
This hypothesis was tested in 1998. Using genetic engineering, researchers transferred into newly established human cell cultures a gene that leads to expression of an enzyme called telomerase that all cells possess but no body cell uses. This enzyme adds TTAGGG sequences back to the end of telomeres, in effect rebuilding the lost portions of the telomere. Laboratory cultures of cell lines with (telomerase plus) and without (normal) this gene were then monitored for many generations. The graph above displays the results.
1. Applying Concepts
a. Variable. In the graph, what is the dependent variable?
b. Comparing Continuous Processes. How do normal skin cells (blue line) differ in their growth history from telomerase plus cells with the telomerase gene (red line)?
2. Interpreting Data
a. After how many doublings do the normal cells cease to divide? Is this consistent with the telomerase hypothesis?
b. After how many doublings do the telomerase plus cells cease to divide in this experiment?
3. Making Inferences After nine population doublings, would the rate of cell division be different between the two cultures? After 15? Why?
4. Drawing Conclusions How does the addition of the telomerase gene affect the senescence of skin cells growing in culture? Does this result confirm the telomerase hypothesis this experiment had set out to test?
5. Further Analysis
a. Cancer cells are thought to possess mutations disabling the cell's ability to keep the telomerase gene shut off. How would you test this hypothesis?
b. Sperm-producing cells continue to divide throughout a male's adult life. How might this be possible? How would you test this idea?

Test Your Understanding
1. Prokaryotes reproduce new cells by
a. copying DNA then undergoing binary fission.
b. splitting in half.
c. undergoing mitosis.
d. copying DNA then undergoing the M phase.
2. The eukaryotic cell cycle is different from prokaryotic cell division in all the following ways except
a. the amount of DNA present in the cells.
b. how the DNA is packaged.
c. in the production of genetically identical daughter cells.
d. the involvement of microtubules.
3. In eukaryotes, the genetic material is found in chromosomes
a. and the more complex the organism, the more pairs of chromosomes it has.
b. and many organisms have only one chromosome.
c. and most eukaryotes have between 10 and 50 pairs of chromosomes.
d. and most eukaryotes have between 2 and 10 pairs of chromosomes.
4. Homologous chromosomes
a. are also referred to as sister chromatids.
b. are genetically identical.
c. carry information about the same traits located in the same places on the chromosomes.
d. are connected to each other at their centromeres.
5. Eukaryotic chromosomes are composed of
a. DNA
b. proteins.
c. histones.
d. All of the above.
6. In mitosis, when the duplicated chromosomes line up in the center of the cell, that stage is called
a. prophase.
b. metaphase.
c. anaphase.
d. telophase.
7. The division of the cytoplasm in the eukaryotic cell cycle is called
a. interphase.
b. mitosis.
c. cytokinesis.
d. binary fission.
8. The cell cycle is controlled by
a. a series of checkpoints.
b. telomerase.
c. the G0 phase.
d. All of the above.
9. When cell division becomes unregulated, and a cluster of cells begins to grow without regard for the normal controls, that is called
a. a mutation.
b. cancer.
c. metastases.
d. oncogenes.
10. The normal function of thep53 gene in the cell is
a. as a tumor-suppressor gene.
b. to monitor the DNA for damage.
c. to trigger the destruction of cells not capable of DNA repair.
d. All of the above.