Medical Microbiology
Section 2 The adversaries–host defences
9 The innate defences of the body
Introduction
In the preceding chapters, we have outlined some of the fundamental characteristics of the myriad types of microparasites and macroparasites that may infect the body. We now turn to consider the ways in which the body seeks to defend itself against infection by these organisms.
The body has both ‘innate’ and ‘adaptive’ immune defences
When an organism infects the body, the defence systems already in place may well be adequate to prevent replication and spread of the infectious agent, thereby preventing development of disease. These established mechanisms are referred to as constituting the ‘innate’ immune system. However, should innate immunity be insufficient to parry the invasion by the infectious agent, the so-called ‘adaptive’ immune system then comes into action, although it takes time to reach its maximum efficiency (Fig. 9.1). When it does take effect, it generally eliminates the infective organism, allowing recovery from disease.
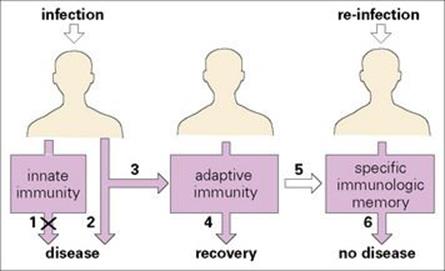
Figure 9.1 Innate and adaptive immunity. An infectious agent first encounters elements of the innate immune system. These may be sufficient (1) to prevent disease but if not, disease may result (2). The adaptive immune system is then activated (3) to produce recovery (4) and a specific immunologic memory (5). Following re-infection with the same agent, no disease results (6) and the individual has acquired immunity to the infectious agent.
The main feature distinguishing the adaptive response from the innate mechanism is that specific memory of infection is imprinted on the adaptive immune system, so that should there be a subsequent infection by the same agent, a particularly effective response comes into play with remarkable speed. It is worth emphasizing, however, that there is close synergy between the two systems, with the adaptive mechanism greatly improving the efficiency of the innate response.
The contrasts between these two systems are set out in Table 9.1. On the one hand, the soluble factors such as lysozyme and complement, together with the phagocytic cells, contribute to the innate system, while on the other the lymphocyte-based mechanisms that produce antibody and T lymphocytes are the main elements of the adaptive immune system. Not only do these lymphocytes provide improved resistance by repeated contact with a given infectious agent, but the memory with which they become endowed shows very considerable specificity to that infection. For instance, infection with measles virus will induce a memory to that microorganism alone and not to another virus such as rubella.
Table 9.1 Comparison of innate and adaptive effector immune systems
|
Innate immune system |
Adaptive immune system |
|
|
Major elements |
||
|
Soluble factors |
Lysozyme, complement, acute phase proteins, e.g. C-reactive protein, interferon |
Antibody |
|
Cells |
Phagocytes |
T lymphocytes |
|
Response to microbial infection |
||
|
First contact |
+ |
+ + |
|
Second contact |
+ |
+ + + + |
|
Non-specific; no memory |
Specific; memory |
|
Innate immunity is sometimes referred to as ‘natural’, and adaptive as ‘acquired’. There is considerable interaction between the two systems. ‘Humoral’ immunity due to soluble factors contrasts with immunity mediated by cells. Primary contact with antigen produces both adaptive and innate responses, but if the same antigen persists or is encountered a second time the specific adaptive response to that antigen is much enhanced.
Defences against entry into the body
A variety of biochemical and physical barriers operate at the body surfaces
Before an infectious agent can penetrate the body, it must overcome biochemical and physical barriers that operate at the body surfaces. One of the most important of these is the skin, which is normally impermeable to the majority of infectious agents. Many bacteria fail to survive for long on the skin because of the direct inhibitory effects of lactic acid and fatty acids present in sweat and sebaceous secretions and the lower pH to which they give rise (Fig. 9.2). However, should there be skin loss, as can occur in burns, for example, infection becomes a major problem.
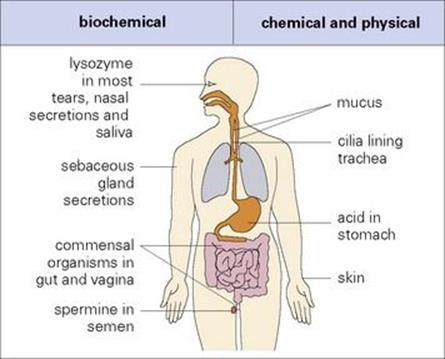
Figure 9.2 Exterior defences. Most of the infectious agents encountered by an individual are prevented from entering the body by a variety of biochemical and physical barriers. The body tolerates a variety of commensal organisms, which compete effectively with many potential pathogens.
The membranes lining the inner surfaces of the body secrete mucus, which acts as a protective barrier, inhibiting the adherence of bacteria to the epithelial cells, thereby preventing them from gaining access to the body. Microbial and other foreign particles trapped within this adhesive mucus may be removed by mechanical means such as ciliary action, coughing and sneezing. The flushing actions of tears, saliva and urine are other mechanical strategies that help to protect the epithelial surfaces. In addition, many of the secreted body fluids contain microbicidal factors, e.g. the acid in gastric juice, spermine and zinc in semen, lactoperoxidase in milk, and lysozyme in tears, nasal secretions and saliva.
The phenomenon of microbial antagonism is associated with the normal bacterial flora of the body. These commensal organisms suppress the growth of many potentially pathogenic bacteria and fungi at superficial sites, first by virtue of their physical advantage of previous occupancy, especially on epithelial surfaces, second by competing for essential nutrients, or third by producing inhibitory substances such as acid or colicins. The latter are a class of bactericidins that bind to the negatively charged surface of susceptible bacteria and form a voltage-dependent channel in the membrane, which kills by destroying the cell’s energy potential.
Defences once the microorganism penetrates the body
Despite the general effectiveness of the various barriers, microorganisms successfully penetrate the body on many occasions. When this occurs, two main defensive strategies come into play, based on:
• the mechanism of phagocytosis, involving engulfment and killing of microorganisms by specialized cells, the ‘professional phagocytes’
• the destructive effect of soluble chemical factors, such as bactericidal enzymes.
Two types of professional phagocyte
Perhaps because of the belief that professionals do a better job than amateurs, the cells that shoulder the main burden of our phagocytic defences have been labelled ‘professional phagocytes’. These consist of two major cell families, as originally defined by Elie Metchnikoff, the Russian zoologist (Box 9.1; Fig. 9.3):
• the large macrophages
• the smaller polymorphonuclear granulocytes, which are generally referred to as polymorphs or neutrophils because their cytoplasmic granules do not stain with haematoxylin and eosin.
![]()
Box 9.1  Lessons in Microbiology
Lessons in Microbiology
Elie Metchnikoff (1845–1916)
This perceptive Russian zoologist can legitimately be regarded as the father of the concept of cellular immunity, in which it is recognized that certain specialized cells mediate the defence against microbial infections. He was intrigued by the motile cells of transparent starfish larvae and made the critical observation that a few hours after introducing a rose thorn into the larvae, the rose thorn became surrounded by the motile cells. He extended his investigations to mammalian leukocytes, showing their ability to engulf microorganisms, a process that he termed ‘phagocytosis’ (literally, eating by cells).
Because he found this process to be even more effective in animals recovering from an infection, he came to the conclusion that phagocytosis provided the main defence against infection. He defined the existence of two types of circulating phagocytes: the polymorphonuclear leukocyte, which he termed a ‘microphage’, and the larger ‘macrophage’.
Although Metchnikoff held the somewhat polarized view that cellular immunity based upon phagocytosis provided the main, if not the only, defence mechanism against infectious microorganisms, we now know that the efficiency of the phagocytic system is enormously enhanced through cooperation with humoral factors, in particular antibody and complement.
![]()

Figure 9.3 Elie Metchnikoff (1845–1916).
(Courtesy of the Wellcome Institute Library, London.)
As a very crude generalization, it may be said that the polymorphs provide the major defence against pyogenic (pus-forming) bacteria, while the macrophages are thought to be at their best in combating organisms capable of living within the cells of the host.
Macrophages are widespread throughout the tissues
Macrophages originate as bone marrow promonocytes, which develop into circulating blood monocytes (Fig. 9.4) and finally become the mature macrophages, which are widespread throughout the tissues and collectively termed the ‘mononuclear phagocyte system’ (Fig. 9.5). These macrophages are present throughout the connective tissue and are associated with the basement membrane of small blood vessels. They are particularly concentrated in the lung (alveolar macrophages), liver (Kupffer cells) and the lining of lymph node medullary sinuses and splenic sinusoids (Fig. 9.6), where they are well placed to filter off foreign material (Fig. 9.7). Other examples are the brain microglia, kidney mesangial cells, synovial A cells and osteoclasts in bone. In general, these are long-lived cells that depend upon mitochondria for their metabolic energy and show elements of rough-surfaced endoplasmic reticulum (Fig. 9.8) related to the formidable array of different secretory proteins that these cells generate.
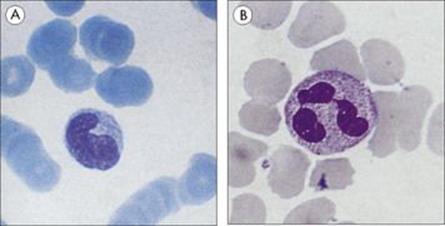
Figure 9.4 Phagocytic cells. (A) Blood monocyte and (B) polymorphonuclear neutrophil, both derived from bone marrow stem cells.
(Courtesy of P.M. Lydyard.)
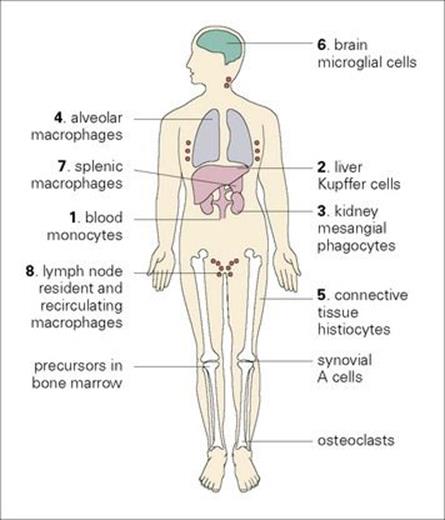
Figure 9.5 The mononuclear phagocyte system. Tissue macrophages are derived from blood monocytes, which are manufactured in the bone marrow. (The numbers relate to those in Fig. 9.6.)
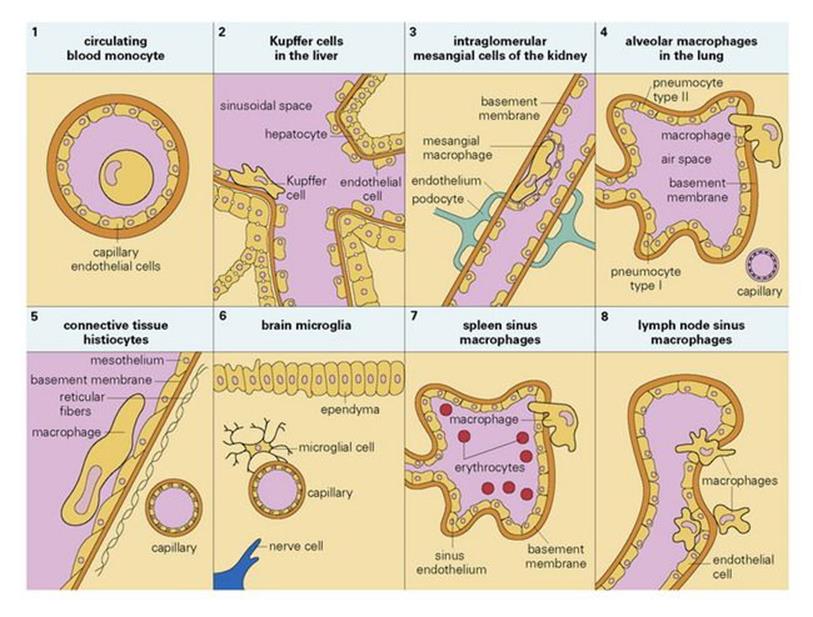
Figure 9.6 Tissue location of mononuclear phagocytes.
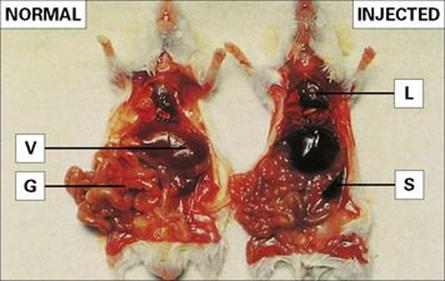
Figure 9.7 Localization of intravenously injected particles in the mononuclear phagocyte system. (Right) A mouse was injected with fine carbon particles and killed 5 min later. Carbon accumulates in organs rich in mononuclear phagocytes: lungs (L), liver (V), spleen (S) and areas of the gut wall (G). (Left) Normal organ colour shown in a control mouse.
(Courtesy of P.M. Lydyard.)
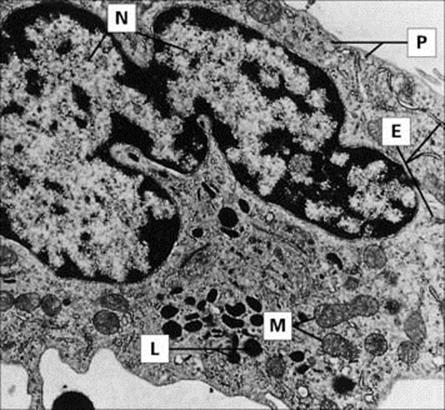
Figure 9.8 Monocyte (× 8000), with ‘horseshoe’ nucleus (N). Phagocytic and pinocytic vesicles (P), lysosomal granules (L), mitochondria (M) and isolated profiles of rough-surfaced endoplasmic reticulum (E) are evident.
(Courtesy of B. Nichols; © Rockefeller University Press.)
Polymorphs possess a variety of enzyme-containing granules
The polymorph is the dominant white cell in the bloodstream and, like the macrophage, shares a common haemopoietic stem cell precursor with the other formed elements of the blood. It has no mitochondria, but uses its abundant cytoplasmic glycogen stores for its energy requirements; therefore, glycolysis enables these cells to function under anaerobic conditions, such as those in an inflammatory focus. The polymorph is a non-dividing, short-lived cell, with a segmented nucleus; the cytoplasm is characterized by an array of granules, which are illustrated in Figure 9.9.
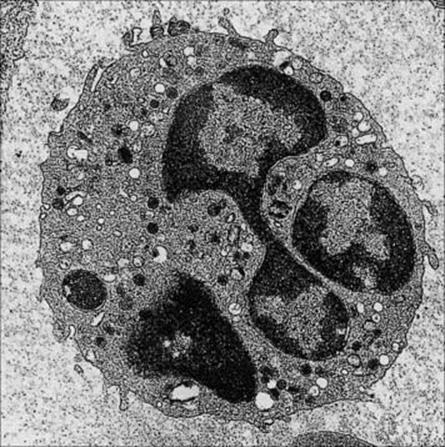
Figure 9.9 Neutrophil. The multi-lobed nucleus and primary azurophilic, secondary specific and tertiary lysosomal granules are well displayed. In some granules there is an overlap in the contents between azurophilic and secondary granules. Typical conventional lysosomes with acid hydrolase are also seen.
(Courtesy of D. McLaren.)
|
Azurophil granules |
Specific granules |
|
0.5 μm |
0.2 μm |
|
1500/cell |
3000/cell |
|
Lysozyme |
Lysozyme |
|
Myeloperoxidase |
Cytochrome b558 |
|
Elastase |
OH phosphatase |
|
Cathepsin G |
Lactoferrin |
|
H+ hydrolases |
|
|
Defensins |
Vitamin B12 binding protein |
|
BPI (bactericidal permeability increasing protein) |
Phagocytosis and killing
Phagocytes recognize pathogen-associated molecular patterns (PAMPs)
The first event in the uptake and digestion of a microorganism by the professional phagocyte involves the attachment of the microbe to the surface of the cell through the recognition of repeating pathogen-associated molecular patterns (PAMPs) on the microbe by pattern recognition receptors (PRRs) on the phagocyte surface (Fig. 9.10). A major subset of these PRRs belongs to the class of so-called ‘Toll-like receptors’ (TLRs) because of their similarity to the Toll receptor in the fruit fly, Drosophila, which, in the adult, triggers an intracellular cascade generating the expression of antimicrobial peptides in response to microbial infection. A series of cell surface TLRs acting as sensors for extracellular infections have been identified (Fig. 9.11) which are activated by microbial elements such as peptidoglycan, lipoproteins, mycobacterial lipoarabinomannan, yeast zymosan and flagellin. Other PRRs displayed by phagocytes include the cell bound ‘C-type (calcium-dependent) lectins’, of which the macrophage mannose receptor is an example, and ‘scavenger receptors’, which recognize a variety of anionic polymers and acetylated low density proteins. Examples of intracellular PAMPs are the unmethylated guanosine-cytosine (CpG) sequences of bacterial DNA and double-stranded RNA from RNA viruses.
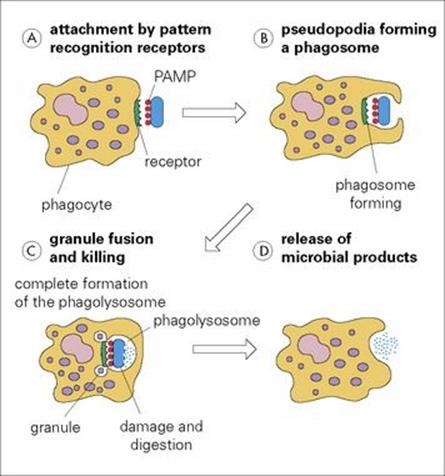
Figure 9.10 Phagocytosis. (A) Phagocytes attach to microorganisms (blue icon) via their cell surface receptors which recognize pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide. (B) If the membrane now becomes activated by the attached infectious agent, the pathogen is taken into a phagosome by pseudopodia, which extend around it. (C) Once inside the cell, the various granules fuse with the phagosome to form a phagolysosome. (D) The infectious agent is then killed by a battery of microbicidal degradation mechanisms, and the microbial products are released.
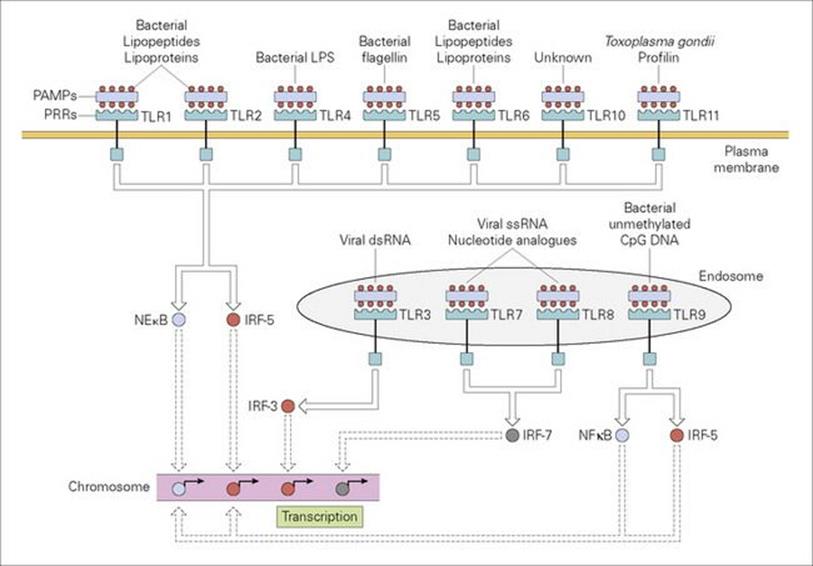
Figure 9.11 Recognition of PAMPs by a subset of pattern recognition receptors (PRRs) termed Toll-like receptors (TLRs). TLRs reside within plasma membrane or endosomal membrane compartments, as shown. All TLRs have multiple N-terminal leucine-rich repeats forming a horseshoe-shaped structure which acts as the PAMP-binding domain. Upon engagement of the TLR ectodomain with an appropriate PAMP (some examples are shown), signals are propagated into the cell that activate the nuclear factor kB (NFkB) and/or interferon regulated factor (IRF) transcription factors, as shown. NFkB and IRF transcription factors then direct the expression of numerous antimicrobial gene products such as cytokines and chemokines, as well as proteins that are involved in altering the activation state of the cell.
The phagocyte is activated through PAMP recognition
The attached microbe may then signal through the phagocyte receptors to initiate the ingestion phase by activating an actin-myosin contractile system, which sends arms of cytoplasm around the particle until it is completely enclosed within a vacuole (phagosome; Fig. 9.12; see Fig. 9.11). Shortly afterwards, the cytoplasmic granules fuse with a phagosome and discharge their contents around the incarcerated microorganism.
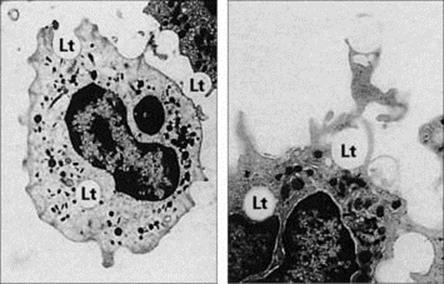
Figure 9.12 Electron micrographic study of phagocytosis. These two micrographs show human phagocytes engulfing latex particles (Lt). (A) × 3000; (B) × 4500.
(Courtesy of C.H.W. Horne.)
The internalized microbe is the target for a fearsome array of killing mechanisms
As phagocytosis is initiated, the attached microbes also signal through one of the PRRs to engineer an appropriate defensive response to the different types of infection through a number of NFκB-mediated responses. This activation of a unique plasma membrane reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase reduces oxygen to a series of powerful microbicidal agents, namely superoxide anion, hydrogen peroxide, singlet oxygen and hydroxyl radicals (Box 9.2; see also Ch. 14). Subsequently, the peroxide, in association with myeloperoxidase, generates a potent halogenating system from halide ions, which is capable of killing both bacteria and viruses.
![]()
Box 9.2 Antimicrobial Mechanisms in Phagocytic Vacuoles
Oxygen-independent antimicrobial mechanisms
|
Cathepsin G and elastase |
|
|
Low molecular weight defensins |
|
|
High molecular weight cationic proteins |
|
|
Bactericidal permeability-increasing protein |
|
|
Lactoferrin |
Complex with iron |
|
Lysozyme |
Splits proteoglycan |
|
Acid hydrolases |
Degrade dead microbes |
Oxygen-dependent antimicrobial mechanisms
Reaction Sequence Generated by NADPH Oxidase:
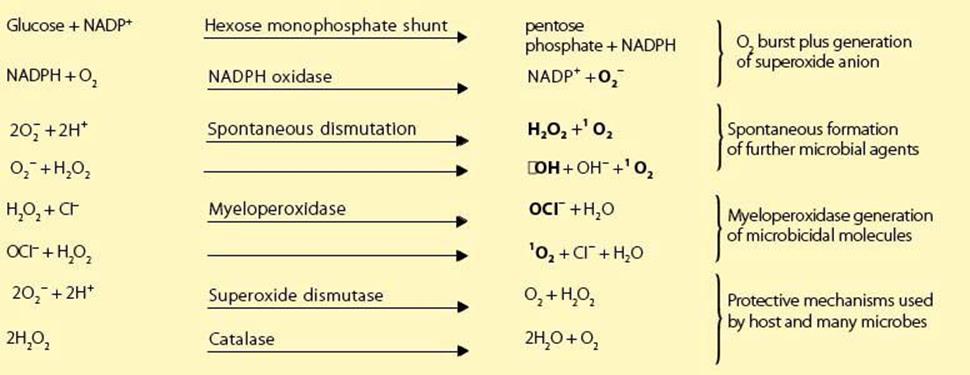
Nitric Oxide Reaction Sequence
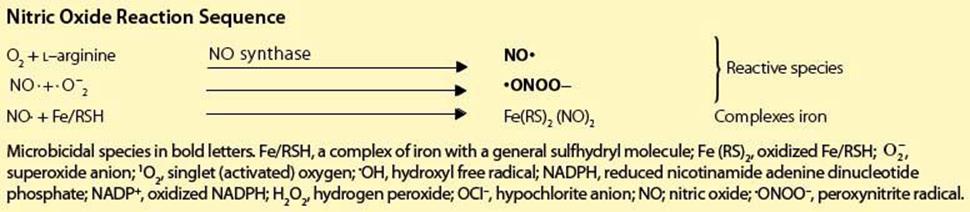
Microbicidal species in bold letters. Fe/RSH, a complex of iron with a general sulfhydryl molecule; Fe (RS)2, oxidized Fe/RSH; ![]() , superoxide anion; 1O2, singlet (activated) oxygen; •OH, hydroxyl free radical; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NADP+, oxidized NADPH; H2O2, hydrogen peroxide; OCI–, hypochlorite anion; NO; nitric oxide; •ONOO–, peroxynitrite radical.
, superoxide anion; 1O2, singlet (activated) oxygen; •OH, hydroxyl free radical; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NADP+, oxidized NADPH; H2O2, hydrogen peroxide; OCI–, hypochlorite anion; NO; nitric oxide; •ONOO–, peroxynitrite radical.
![]()
As superoxide anion is formed, the enzyme superoxide dismutase acts to convert it to molecular oxygen and hydrogen peroxide, but in the process consumes hydrogen ions. Therefore initially there is a small increase in pH, which facilitates the antibacterial function of the families of cationic proteins derived from the phagocytic granules. These molecules damage microbial membranes by the proteolytic action of cathepsin G and by direct adherence to the microbial surface. The defensins have an amphipathic structure which allows them to insert into microbial membranes to form destabilizing voltage-regulated ion channels. These antibiotic peptides reach extraordinarily high concentrations within the phagosome and act as disinfectants against a wide spectrum of bacteria, fungi and enveloped viruses. Other important factors are:
• lactoferrin, which complexes iron to deprive bacteria of essential growth elements
• lysozyme, which splits the proteoglycan cell wall of bacteria
• nitric oxide, which can lead not only to iron seclusion but, together with its derivative, the peroxynitrite radical, can also be directly microbicidal.
The pH now falls so that the dead or dying microorganisms are extensively degraded by acid hydrolytic enzymes, and the degradation products released to the exterior.
NFκB activation can also lead to the release of proinflammatory mediators. These include the antiviral interferons, the small protein cytokines interleukin-1β (IL-1β), IL-6, IL-12 and TNF (TNFα), which activate other cells through binding to specific receptors, and chemokines such as IL-8, which represent a subset of chemoattractant cytokines.
Microbial nucleotide breakdown products of infectious agents that have succeeded in gaining access to the interior of a cell can be recognized by the so-called NOD proteins and the typical CpG DNA motif which binds to the endosomal TLR9. Other endosomal Toll-like receptors, TLR3 and TLR7/8, are responsive to intracellular viral RNA sequences and engender production of antiviral interferon.
Phagocytes are mobilized and targeted onto the microorganism by chemotaxis
Phagocytosis cannot occur unless the bacterium first attaches to the surface of the phagocyte, and clearly this cannot happen unless both have become physically close to each other. There is therefore a need for a mechanism that mobilizes phagocytes from afar and targets them onto the bacterium. Many bacteria produce chemical substances, such as formyl methionyl peptides, which directionally attract leukocytes, a process known as ‘chemotaxis’. However, this is a relatively weak signalling system, and evolution has provided the body with a far more effective ‘magnet’ that uses a complex series of proteins collectively termed ‘complement’.
Activation of the complement system
Complement resembles blood clotting, fibrinolysis and kinin formation in being a major triggered enzyme cascade system. Such systems are characterized by their ability to produce a rapid, highly amplified response to a trigger stimulus mediated by a cascade phenomenon in which the product of one reaction is the enzymic catalyst of the next. The most abundant and most central component is C3 (complement components are designated by the letter ‘C’ followed by a number), and the cleavage of this molecule is at the heart of all complement-mediated phenomena.
In normal plasma, C3 undergoes spontaneous activation at a very slow rate to generate the split product C3b. This is able to complex with another complement component, factor B, which is then acted upon by a normal plasma enzyme, factor D, to produce the C3-splitting enzyme ![]() This C3 convertase can then split new molecules of C3 to give C3a (a small fragment) and further C3b. This represents a positive feedback circuit with potential for runaway amplification; however, the overall process is restricted to a tick-over level by powerful regulatory mechanisms, which break the unstable soluble-phase C3 convertase into inactive cleavage products (Fig. 9.13).
This C3 convertase can then split new molecules of C3 to give C3a (a small fragment) and further C3b. This represents a positive feedback circuit with potential for runaway amplification; however, the overall process is restricted to a tick-over level by powerful regulatory mechanisms, which break the unstable soluble-phase C3 convertase into inactive cleavage products (Fig. 9.13).
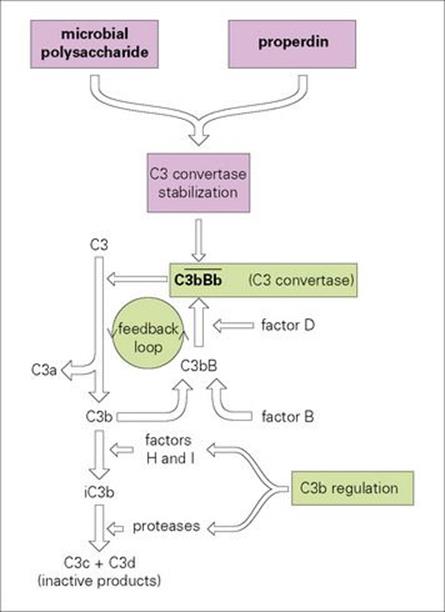
Figure 9.13 Activation of complement by microorganisms. C3b is formed by the spontaneous breakdown of C3 complexes with factor B to form C3bB which is split by factor D to produce a C3 convertase ![]() capable of further cleaving C3. The convertase is heavily regulated by factors H and I but can be stabilized on the surface of microbes and properdin. The horizontal bar indicates an enzymically active complex. iC3b, inactive C3b.
capable of further cleaving C3. The convertase is heavily regulated by factors H and I but can be stabilized on the surface of microbes and properdin. The horizontal bar indicates an enzymically active complex. iC3b, inactive C3b.
In the presence of certain molecules, such as the carbohydrates on the surface of many bacteria, the C3 convertase can become attached and stabilized against breakdown. Under these circumstances, there is active generation of new C3 convertase molecules, and what is known as the ‘alternative’ complement pathway can swing into full tempo (see Ch. 10).
Complement synergizes with phagocytic cells to produce an acute inflammatory response
Activation of the alternative complement pathway with the consequent splitting of very large numbers of C3 molecules has important consequences for the orchestration of an integrated antimicrobial defense strategy (Fig. 9.14). Large numbers of C3b produced in the immediate vicinity of the microbial membrane bind covalently to that surface and act as opsonins (molecules that make the particle they coat more susceptible to engulfment by phagocytic cells; see below). This C3b, together with the C3 convertase, acts on the next component in the sequence, C5, to produce a small fragment, C5a which, together with C3a, has a direct effect on mast cells to cause their degranulation (Fig. 9.15). This results in the release not only of mediators of vascular permeability, but also of factors chemotactic for polymorphs (Table 9.2). The circulating equivalent of the tissue mast cell, the basophil, is shown in Figure 9.16.
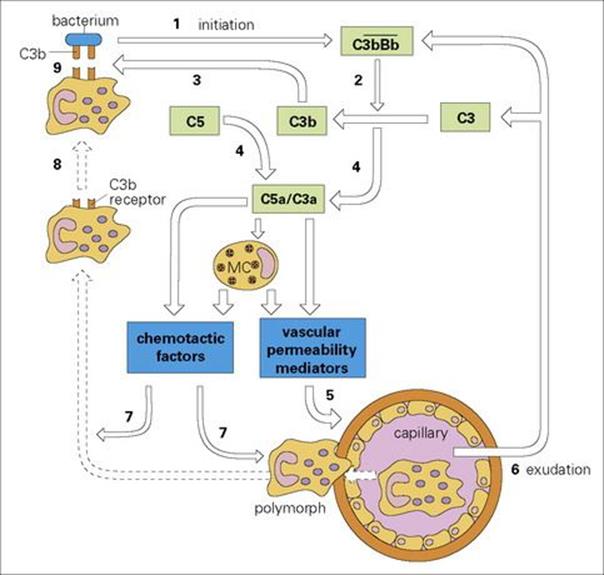
Figure 9.14 The defensive strategy of the acute inflammatory reaction initiated by bacterial activation of the alternative complement pathway. Activation of the ![]() C3 convertase by the bacterium (1) leads to the generation of C3b (2) (which binds to the bacterium (3)), C3a and C5a (4), which recruit mast cell (MC) mediators. These in turn cause capillary dilation (5), exudation of plasma proteins (6), and chemotactic attraction (7) and adherence of polymorphs to the C3b-coated bacterium (8). Note that C5a itself is also chemotactic. The polymorphs are then activated for phagocytosis and the final kill (9).
C3 convertase by the bacterium (1) leads to the generation of C3b (2) (which binds to the bacterium (3)), C3a and C5a (4), which recruit mast cell (MC) mediators. These in turn cause capillary dilation (5), exudation of plasma proteins (6), and chemotactic attraction (7) and adherence of polymorphs to the C3b-coated bacterium (8). Note that C5a itself is also chemotactic. The polymorphs are then activated for phagocytosis and the final kill (9).
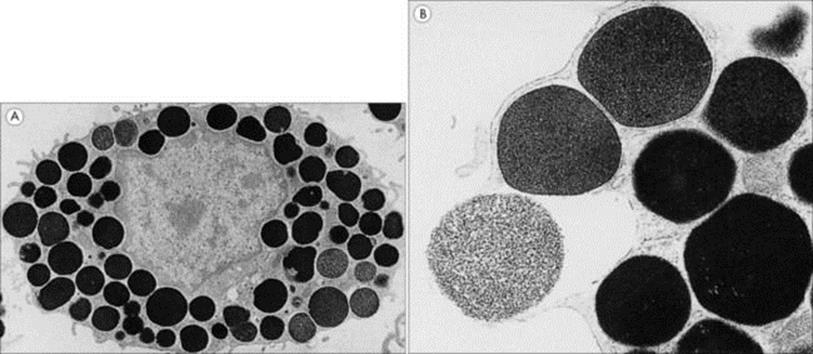
Figure 9.15 Electron micrographs of rat peritoneal mast cells. These show (A) the resting cell with its electron-dense granules (× 6000) and (B) a granule in the process of exocytosis (× 30 000).
(Courtesy of T.S.C. Orr.)
Table 9.2 The major inflammatory mediators that control blood supply and vascular permeability or modulate cell movement
|
Inflammatory mediators |
||
|
Mediator |
Main source |
Actions |
|
Histamine |
Mast cells, basophils |
Increased vascular permeability, smooth muscle contraction, chemokinesis |
|
5-hydroxytryptamine (5HT – serotonin) |
Platelets, mast cells (rodent) |
Increased vascular permeability, smooth muscle contraction |
|
Platelet activating factor (PAF) |
Basophils, neutrophils, macrophages |
Mediator release from platelets, increased vascular permeability, smooth muscle contraction, neutrophil activation |
|
IL-8 (CXCL8) |
Mast cells, endothelium, monocytes and lymphocytes |
Polymorph and monocyte localization |
|
C3a |
Complement C3 |
Mast cell degranulation, smooth muscle contraction |
|
C5a |
Complement C5 |
Mast cell degranulation, neutrophil and macrophage chemotaxis, neutrophil activation, smooth muscle contraction, increased capillary permeability |
|
Bradykinin |
Kinin system (kininogen) |
Vasodilation, smooth muscle contraction, increased capillary permeability, pain |
|
Fibrinopeptides and fibrin breakdown products |
Clotting system |
Increased vascular permeability, neutrophil and macrophage chemotaxis |
|
Prostaglandin E2 (PGE2) |
Cyclo-oxygenase pathway, mast cells |
Vasodilation, potentiates increased vascular permeability produced by histamine and bradykinin |
|
Leukotriene B4 (LTB4) |
Lipoxygenase pathway, mast cells |
Neutrophil chemotaxis, synergizes with PGE2 in increasing vascular permeability |
|
Leukotriene D4 (LTD4) |
Lipoxygenase pathway |
Smooth muscle contraction, increasing vascular permeability |
Other mediators are generated from the coagulation process. Chemotaxis refers to directed migration of granulocytes up the concentration gradient of the mediator, whereas chemokinesis describes randomly increased motility of these cells.
(Reproduced from Male D, Brostoff J, Roth DB, Roitt I. Immunology, 7th edition, 2006. Mosby Elsevier, with permission.)
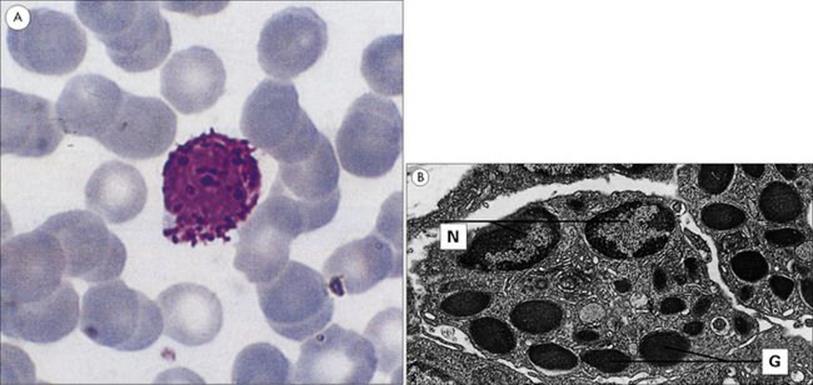
Figure 9.16 Morphology of the basophil. (A) This blood smear shows a typical basophil with its deep violet-blue granules. Wright’s stain (× 1500). (B) Electron micrograph showing the ultrastructure of the basophil. Basophils in guinea pig skin showing the nuclei (N) and characteristic randomly distributed granules (G) (× 6000).
(Courtesy of D. McLaren.)
The vascular permeability mediators increase the permeability of capillaries by modifying the intercellular forces between the endothelial cells of the vessel wall. This allows the exudation of fluid and plasma components, including more complement, to the site of the infection. These mediators (Table 9.2) also up-regulate molecules such as intercellular adhesion molecule-1 (ICAM-1) and endothelial cell leukocyte adhesion molecule-1 (ELAM-1), which bind to specific complementary molecules on the polymorphs and encourage them to stick to the walls of the capillaries, a process termed ‘margination’.
The chemotactic factors, on the other hand, provide a chemical gradient which attracts marginated polymorphonuclear leukocytes from their intravascular location, through the walls of the blood vessels, and eventually leads them to the site of the C3b-coated bacteria that initiated the whole activation process. Polymorphs have a well-defined receptor for C3b on their surface, and as a result, the opsonized bacteria adhere very firmly to the surface of these newly arrived cells.
The processes of capillary dilation (erythema), exudation of plasma proteins and of fluid (oedema) due to hydrostatic and osmotic pressure changes, and the accumulation of neutrophils are collectively termed the ‘acute inflammatory response’, and result in a highly effective way of focusing phagocytic cells onto complement-coated microbial targets.
It also seems clear that the macrophage can be stimulated by certain bacterial toxins such as the lipopolysaccharides (LPS), by the action of C5a, and by the phagocytosis of C3b-coated bacteria, to secrete other potent mediators of acute inflammation, independently of the mast cell-directed pathway (Fig. 9.17).
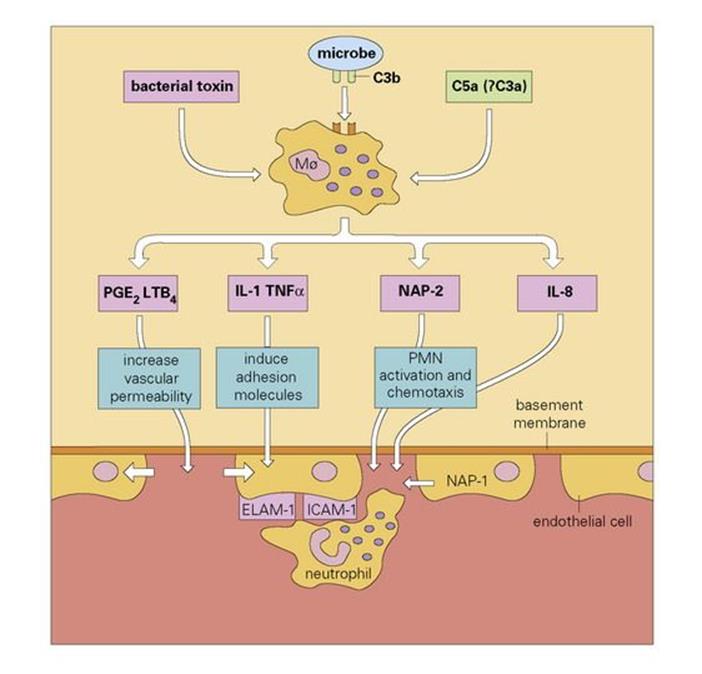
Figure 9.17 A role for the macrophage (Mø) in the initiation of acute inflammation. Stimulation induces macrophage secretion of mediators. Blood neutrophils stick to the adhesion molecules on the endothelial cell and use them to provide traction as they force their way between the cells, through the basement membrane (with the help of secreted elastase) and up the chemotactic gradient. During this process they become progressively activated by neutrophil activating peptide-2 (NAP-2). PGE2, prostaglandin E2; LTB4, leukotriene B4; IL-1, interleukin-1; PMN, polymorphonuclear neutrophil; TNFα, tumour necrosis factor alpha; ELAM-1, endothelial cell leukocyte adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1.
C9 molecules form the ‘membrane attack complex’, which is involved in cell lysis
We have already introduced the idea that following the activation of C3 the next component to be cleaved is C5; the larger C5b fragment that results becomes membrane bound. This subsequently binds components C6, C7 and C8, which form a complex capable of inducing a critical conformational change in the terminal component C9. The unfolded C9 molecules become inserted into the lipid bilayer and polymerize to form an annular ‘membrane attack complex’ (MAC) (Figs 9.18, 9.19). This behaves as a transmembrane channel that is fully permeable to electrolytes and water; because of the high internal colloid osmotic pressure of cells, there is a net influx of sodium (Na+) and this frequently leads to lysis.
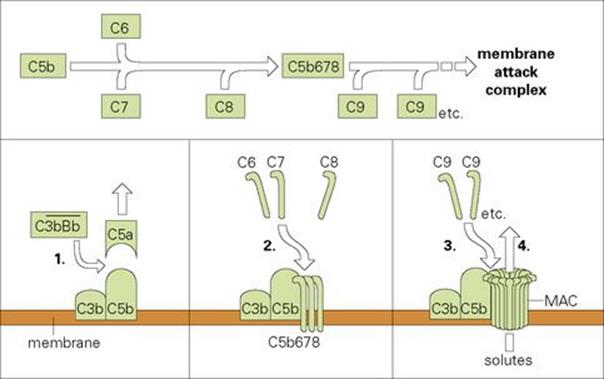
Figure 9.18 Assembly of the C5b-9 membrane attack complex (MAC). (1) Recruitment of a further C3b into the C3bBb enzymic complex generates a C5 convertase which cleaves C5a from C5 and leaves the remaining C5b attached to the membrane. (2) Once C5b is membrane bound, C6 and C7 attach themselves to form the stable complex C5b67, which interacts with C8 to yield C5b678. (3) This unit has some effect in disrupting the membrane, but primarily causes the polymerization of C9 to form tubules traversing the membrane. The resulting tubule is referred to as a MAC. (4) Disruption of the membrane by this structure permits the free exchange of solutes, which are primarily responsible for cell lysis.
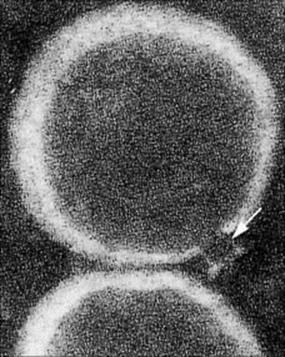
Figure 9.19 Electron micrograph of the MAC. The funnel-shaped lesion (arrowed) is due to a human C5b–9 complex that has been reincorporated into lecithin liposomal membranes (× 234 000).
(Courtesy of J. Tranum-Jensen and S. Bhakdi.)
Acute phase proteins
Certain proteins in the plasma, collectively termed ‘acute phase proteins’, increase in concentration in response to early ‘alarm’ mediators such as the cytokines interleukin-1 (IL-1), IL-6 and tumour necrosis factor (TNF), released as a result of infection or tissue injury. Many acute phase reactants such as mannose binding lectin and C-reactive protein (CRP) increase dramatically during inflammation (Fig. 9.20). Like the professional phagocytes, both use pattern recognition receptors to bind to molecular patterns on the pathogen (PAMPs), to generate defensive effector functions (Fig. 9.21). Other acute phase reactants show more moderate rises, usually less than fivefold (see Table 9.3). In general, these proteins are thought to have defensive roles.
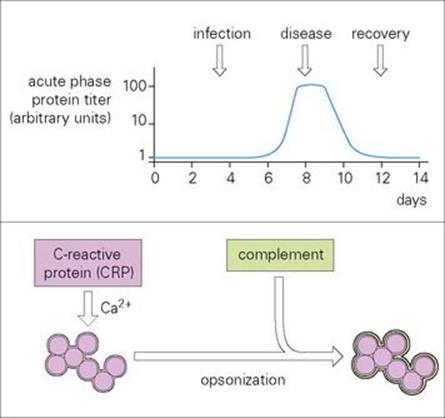
Figure 9.20 Acute phase proteins, here exemplified by C-reactive protein (CRP), are serum proteins that increase rapidly in concentration (sometimes up to 100-fold) following infection (graph). They are important in innate immunity to infection. CRP recognizes and binds in a calcium (Ca2 +)-dependent fashion to molecular groups found on a wide variety of bacteria and fungi. In particular, it uses its pattern recognition to bind the phosphocholine moiety of pneumococci. The CRP acts as an opsonin and activates complement with all the associated sequelae. Mannose binding protein reacts not only with mannose but several other sugars, enabling it to bind to a wide variety of Gram-negative and -positive bacteria, yeasts, viruses and parasites, subsequently activating the complement system and phagocytic cells. The structurally related ficolins typically recognize PAMPs containing N-acetylglucosamine and can also activate the lectin complement pathway.
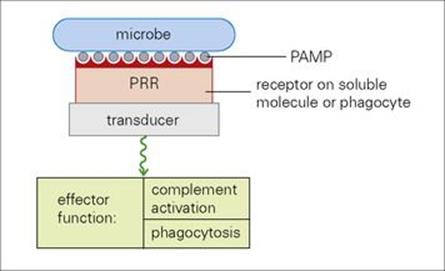
Figure 9.21 A major defensive strategy in which soluble factors, such as CRP (C reactive protein) and mannose binding protein, and professional phagocytes use their pattern recognition receptors (PRR) to bind to the pathogen-associated molecular patterns (PAMPs) on the microbial surface and signal through their transducer structures to initiate appropriate effector functions.
Table 9.3 Acute phase proteins produced in response to infection in the human
|
Acute phase reactant |
Function |
|
Dramatic increases in concentration |
|
|
C-reactive protein |
Fixes complement, opsonizes |
|
Mannose binding lectin |
Fixes complement, opsonizes |
|
α1 acid glycoprotein |
Transports protein |
|
Serum amyloid A protein |
Complexes chondroitin sulphate |
|
Moderate increases in concentration |
|
|
α1 proteinase inhibitors |
Inhibit bacterial proteases |
|
α1 anti-chymotrypsin |
Inhibits bacterial proteases |
|
Surfactant protein A |
Binds influenza virus haemagglutinin |
|
C3, C9, factor B |
Increase complement function |
|
Ceruloplasmin |
O2 scavenger |
|
Fibrinogen |
Coagulation |
|
Angiotensin |
Blood pressure |
|
Haptoglobin |
Binds haemoglobin |
|
Fibronectin |
Cell attachment |
Other extracellular antimicrobial factors
There are many microbicidal agents that operate at short range within phagocytic cells, but also appear in various body fluids in sufficient concentration to have direct inhibitory effects on infectious agents. For example, lysozyme is present in fluids such as tears and saliva in amounts capable of acting against the proteoglycan wall of susceptible bacteria. Similarly, lactoferrin may appear in the blood in sufficient concentration to complex iron and deprive bacteria of this important growth factor. Whether agents that normally act over a short range, such as reactive oxygen metabolites or TNF (a cytotoxic molecule produced by macrophages and other cell types), can reach concentrations in the body fluids that are adequate to allow them to act at a distance from the cell producing them will be discussed in Chapter 14, particularly when considering the mechanisms by which the blood-borne forms of parasites such as malaria are attacked.
Interferons are a family of broad spectrum antiviral molecules
Interferons (IFNs) are widespread throughout the animal kingdom and are again discussed further in Chapter 14. They were first recognized by the phenomenon of viral interference, in which a cell infected with one virus is found to be resistant to superinfection by a second unrelated virus. Leukocytes produce many different α-interferons (IFNα), while fibroblasts and probably all cell types synthesize IFNβ. A third type (IFNγ) is not a component of the innate immune system and will be discussed in Chapter 10 as a member of the important cytokine family.
When cells are infected by a virus, they synthesize and secrete IFNs α and β, which bind to specific receptors on nearby uninfected cells. The bound IFN exerts its antiviral effect by facilitating the synthesis of two new enzymes, which interfere with the machinery used by the virus for its own replication. The mechanism of action of IFN is discussed more fully in Chapter 14; the net result is to set up a cordon of infection-resistant cells around the site of virus infection, so restraining its spread (Fig. 9.22). IFN is highly effective in vivo, as supported by experiments in which mice injected with an antiserum to murine IFN were found to be killed by several hundred times less virus than was needed to kill the controls. It should be emphasized, however, that IFN seems to play a significant role in recovery from, rather than prevention of, viral infections.
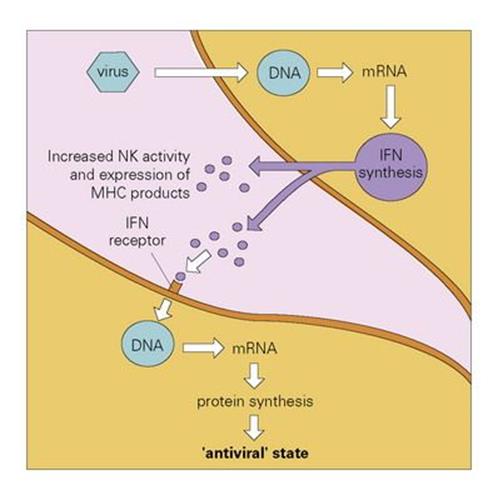
Figure 9.22 The action of interferon (IFN). Virus infecting a cell induces the production of IFNα/β. This is released and binds to IFN receptors on other cells. The IFN induces the production of antiviral proteins, which are activated if virus enters the second cell, and increased synthesis of surface MHC molecules which enhance susceptibility to cytotoxic T cells (cf. Ch. 10). NK, natural killer; MHC, major histocompatibility complex.
Extracellular killing
Natural killer cells attach to virally infected cells, allowing them to be differentiated from normal cells
There is a widely held view that viruses represent fragments of the genome of multicellular organisms that have achieved the ability to exist in an extracellular state. The small number of genes present in the viral genome, however, does not include those required for viral replication. Accordingly, it is essential for viruses to penetrate the cells of an infected host in order to subvert the cells’ replicative machinery towards viral replication. Clearly, it is in the interests of the host to try to kill such infected cells before the virus has had a chance to reproduce. Natural killer (NK) cells are cytotoxic cells that appear to have evolved to carry out just such a task. These are large granular lymphocytes (LGLs) (Fig. 9.23) that recognize virus-infected or stressed cells and allow them to be differentiated from normal cells; this clever discrimination is mediated by activating receptors on the NK cells such as NKG2D that recognize ligands on the infected cell that are related to MHC Class I molecules, and inhibitory receptors which bind to MHC Class I molecules on normal cells, generating signals that counteract those from the activating receptors. Activation of the NK cell results in the extracellular release of its granule contents into the space between the target and effector cells. These contents include perforin molecules, which resemble C9 in many respects, especially in their ability to insert into the membrane of the target cell and polymerize to form annular transmembrane pores, like the MAC. This permits the entry of another granule protein, granzyme B, which leads to death of the target cell by apoptosis (programmed cell death), a process mediated by a cascade of proteolytic enzymes termed caspases, which terminates with the ultimate fragmentation of DNA by a Ca-dependent endonuclease (Fig. 9.24).
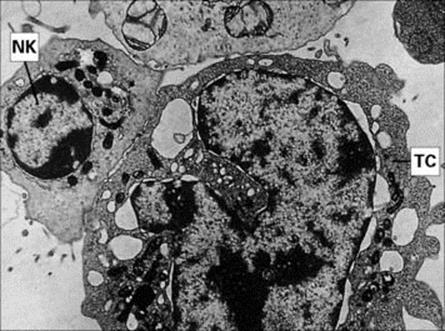
Figure 9.23 Electron micrograph of an NK cell killing a tumour cell (TC). NK cells bind to and kill IgG antibody-coated (see Fig. 10.13), and non-coated, tumour cells. It is essential for the membranes of the two cells to be closely apposed in order for the NK cell to deliver the ‘kiss of death’ (× 4500).
(Courtesy of P. Lydyard.)
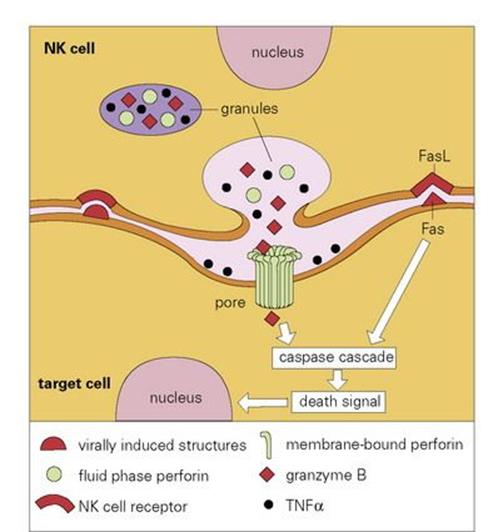
Figure 9.24 Schematic model of lysis of virally infected target cell by a natural killer (NK) cell. As the NK cell receptors bind to the surface of the virally infected cell, and if signals from activation receptors exceed those from the inhibitory receptors that recognize normal MHC Class I molecules, there is exocytosis of granules and release of cytolytic mediators into the intercellular cleft. A calcium (Ca2 +)-dependent conformational change in the perforin enables it to insert and polymerize within the membrane of the target cell to form a transmembrane pore, which allows entry of granzyme B into the target cell, where it causes programmed cell death (apoptosis). A back-up cytolytic system using engagement of the Fas receptor with its ligand (FasL), can also trigger apoptosis as can binding of granule-derived tumour necrosis factor alpha (TNFα) to its receptor. Unlike the PRR-mediated activation of phagocytes by intracellular components – so-called danger-associated molecular patterns (DAMPs) – released on necrotic cell-death typically caused by tissue trauma, burns and other non-physiological stimuli, cells undergoing apoptoticdeath do not activate the immune system because they express surface molecules such as phosphatidyl serine which mark them out for phagocytic removal before they release their intracellular DAMPs.
Subsidiary mechanisms that can activate the caspase pathway include engagement of Fas on the target cell by the NK Fas ligand, and binding of tumour necrosis factor (TNF) released from the NK granules to surface receptors. TNF was first recognized as a product of activated macrophages known to be capable of killing certain other cells, particularly some tumour cells.
Yet a further mode of cytotoxicity can be turned on by the activated macrophage, involving the direct ‘burning’ of the surface of another cell by means of a stream of reactive oxygen intermediates, produced at the macrophage membrane by the respiratory oxygen burst, as discussed previously (see Box 9.2).
Eosinophils act against large parasites
It takes little imagination to realize that professional phagocytes are far too small to be capable of physically engulfing large parasites such as helminths. An alternative strategy, such as killing by an extracellular broadside of the type discussed above would seem to be a more appropriate form of defence. Eosinophils appear to have evolved to fulfil this role. These polymorphonuclear relatives of the neutrophil have distinctive cytoplasmic granules, which stain strongly with acidic dyes (Fig. 9.25) and have a characteristic ultrastructural appearance. A major basic protein (MBP) has been identified in the core of the granule, while the matrix has been shown to contain an eosinophilic cationic protein, a peroxidase and a perforin-like molecule. Eosinophils have surface receptors for C3b and when activated generate copious amounts of active oxygen metabolites.
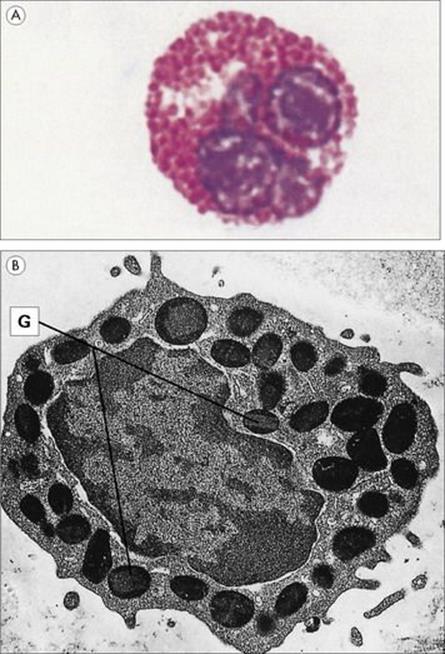
Figure 9.25 The eosinophil granulocyte is capable of extracellular killing of parasites (e.g. worms) by releasing its granule contents. (A) Morphology of the eosinophil. This blood smear enriched for granulocytes shows an eosinophil with its multilobed nucleus and heavily stained cytoplasmic granules. Leishman’s stain (× 1800). (Courtesy of P. Lydyard.) (B) Electron micrograph showing the ultrastructure of a guinea pig eosinophil. The mature eosinophil contains granules (G) with central crystalloids (× 8000).
(Courtesy of D. McLaren.)
Many helminths can activate the alternative complement pathway but, although resistant to C9 attack, their coating with C3b allows adherence to the eosinophils through their C3b surface receptors. Once activated, the eosinophil launches its extracellular ammunition, which includes the release of major basic proteins and the cationic protein to damage the parasite membrane, with a possibility of a further ‘chemical burn’ from the oxygen metabolites and ‘leaky pore’ formation by the perforins.
![]()
 Key Facts
Key Facts
• The innate system of immune defence consists of a formidable barrier to entry and second-line defence by phagocytes and circulating soluble factors. Colonization of the body by normally non-pathogenic (‘opportunistic’) microorganisms occurs whenever there is a hereditary or acquired deficiency in any of these functions.
• The main phagocytic cells are polymorphonuclear neutrophils and macrophages. They adhere to the surface of the microbe by receptors which recognize pathogen-associated molecular patterns (PAMPs). This activates the engulfment process so that the organisms are taken inside the cell in a phagocytic vacuole which fuses with cytoplasmic granules. A formidable array of oxygen-dependent and oxygen-independent microbicidal mechanisms then comes into play.
• The complement system, a multicomponent triggered enzyme cascade, is used to attract phagocytic cells to the microbes and engulf them.
• The most abundant complement component, C3, is split by a convertase enzyme formed from its own cleavage product C3b and factor B and stabilized against breakdown caused by factors H and I through association with the microbial surface. As it is formed, C3b becomes covalently linked to the microorganism.
• The next most abundant component, C5, is activated to yield a small peptide, C5a, while residual C5b binds to the surface of the microorganism and assembles the terminal components C6–9 into a membrane attack complex (MAC), which is freely permeable to solutes and can lead to osmotic lysis. In addition, C5a is a potent chemotactic agent for polymorphs and greatly increases capillary permeability.
• C3a and C5a act on mast cells, causing the release of further mediators such as histamine, LTB4 and TNFα, with effects on capillary permeability and adhesiveness and neutrophil chemotaxis. They also activate neutrophils, which bind to the C3b-coated microbes by their surface C3b receptors and then ingest them.
• The influx of polymorphs and increase in vascular permeability constitute the potent antimicrobial acute inflammatory response.
• Inflammation can also be initiated by tissue macrophages, which subserve a similar role to that of the mast cell since signalling by bacterial toxins C5a or by C3b-coated bacteria adhering to surface complement receptors on tissue macrophages causes the release of TNFα, LTB4, PGE2, the neutrophil chemotactic factor, IL-8, and a neutrophil-activating peptide.
• Other humoral defences include the acute phase proteins such as CRP, and the IFNs, which can block viral replication.
• Virally infected cells can be killed by NK cells, following increased recognition by activation receptors that overcomes inhibitory signals from normal MHC Class I recognition.
• Extracellular killing can also be effected by C3b-bound eosinophils, which may be responsible for the failure of many large parasites to establish a foothold in potential hosts.
• It is probably true to say that engulfment and killing by phagocytic cells is the mechanism used to dispose of the majority of microbes, and the mobilization and activation of these cells by orchestrated responses such as the acute inflammatory response (Fig. 9.26) is a key feature of innate immunity. However, not every organism is readily susceptible to phagocytosis or even to killing by complement or lysozyme, and this brings us to the role of the adaptive immune response, which is explored in Chapter 10.
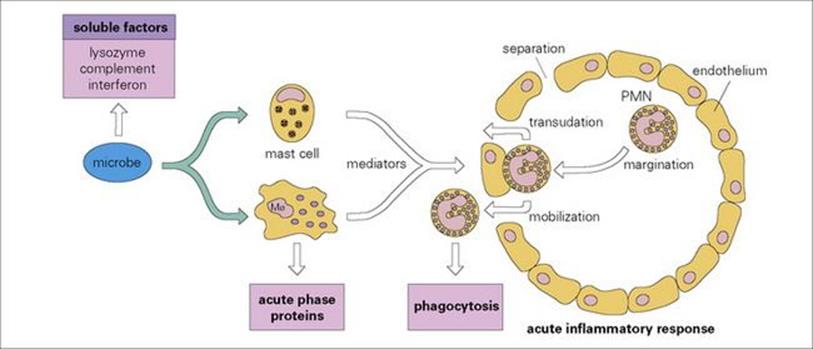
Figure 9.26 Mobilization of defensive components of innate immunity. Microbes, either through complement activation or through direct effects on macrophages, release mediators which increase capillary permeability to allow transudation of plasma bactericidal molecules, and chemotactically attract plasma polymorphs from the bloodstream to the infection site. PMN, polymorphonuclear neutrophil.
![]()