Medical Microbiology
Section 3 The conflicts
15 Spread and replication
Introduction
An infection may be a surface infection or a systemic infection
Many successful microorganisms multiply in epithelial cells at the site of entry on the body surface, but fail to spread to deeper structures or through the body. Local spread takes place readily on a fluid-covered mucosal surface, often aided by ciliary action, and large-scale movements of fluid spread the infection to more distant areas on the surface. This is obvious in the gastrointestinal tract. In the upper respiratory tract, high ‘winds’ (coughing, sneezing) can splatter infectious agents onto new areas of mucosa, or into the openings of sinuses or the middle ear, while the gentler downward trickle of mucus during sleep may seed an infectious agent into the lower respiratory tract. As a result, large areas of the body surface can be involved within a few days, with shedding to the exterior. There is not enough time for a primary immune response to be generated, and therefore non-adaptive responses – interferon, natural killer cells – are more important in controlling the infection. These surface infections therefore show a ‘hit-and-run’ pattern.
In contrast, other microorganisms spread systemically through the body via lymph or blood. They often undergo a complex or stepwise invasion of various tissues before reaching the final site of replication and shedding to the exterior (e.g. measles, typhoid). Surface and systemic infections and their consequences are compared in Figure 15.1.
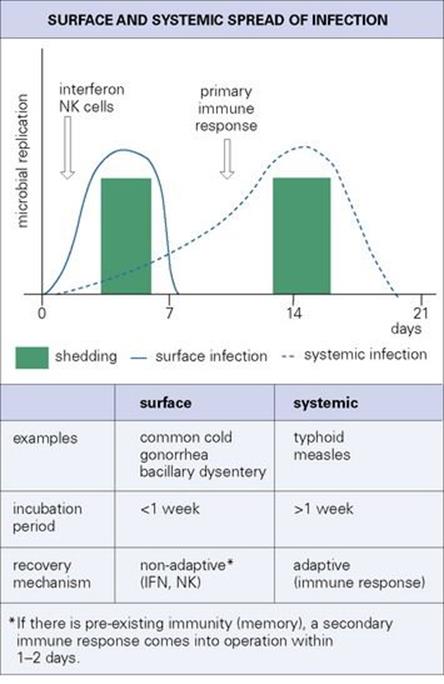
Figure 15.1 Surface and systemic infections. IFN, interferon; NK, natural killer.
Features of surface and systemic infections
A variety of factors determine whether an infection is a surface or a systemic infection
What prevents surface infections from spreading more deeply? Why do the microbes that cause systemic infections leave the relatively safe haven of the body surface to spread through the body, where they will bear the full onslaught of host defences? These are important questions. For instance, what are the factors that persuade meningococci residing harmlessly on the nasal mucosa to invade deeper tissues, reach the blood and meninges, and cause meningitis (see Ch. 24)? The answer is not known.
Temperature is one factor that can restrict microbes to body surfaces. Rhinovirus infections, for instance, are restricted to the upper respiratory tract because they are temperature sensitive, replicating efficiently at 33°C, but not at the temperatures encountered in the lower respiratory tract (37°C). Mycobacterium leprae is also temperature sensitive, which accounts for its replication being more or less limited to nasal mucosa, skin and superficial nerves.
The site of budding is a factor that can restrict viruses to body surfaces. Influenza and parainfluenza viruses invade surface epithelial cells of the lung, but are liberated by budding from the free (external) surface of the epithelial cell, not from the basal layer from where they could spread to deeper tissues (Fig. 15.2).
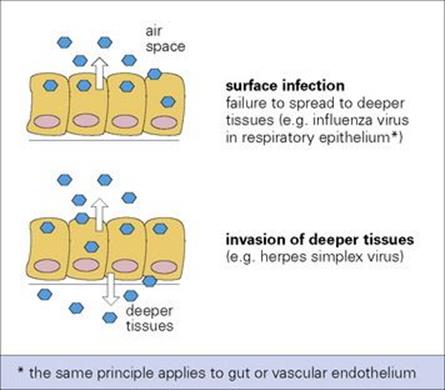
Figure 15.2 Topography of virus release from epithelial surfaces can determine the pattern of infection.
Many microorganisms are obliged to spread systemically because they fail to spread and multiply at the site of initial infection, the body surface. In the case of measles or typhoid, there is, for unknown reasons, next to no replication at the site of initial respiratory or intestinal infection. Only after spreading through the body systemically are large numbers of microorganisms delivered back to the same surfaces, where they multiply and are shed to the exterior. Other microorganisms need to spread systemically because they have committed themselves to infection by one route, while major replication and shedding occurs at a different site. The microbe must reach the replication site, and there is then no need for extensive replication at the site of initial infection. For instance, mumps and hepatitis A viruses infect via the respiratory and alimentary routes, respectively, but must spread through the body to invade and multiply in salivary glands (mumps) and liver (hepatitis A).
In systemic infections, there is a stepwise invasion of different tissues of the body
This stepwise invasion is illustrated in Figure 15.3, and such infections include measles (Fig. 15.4) and typhoid (Fig. 15.5). Although the final sites of multiplication may be essential for microbial shedding and transmission (e.g. measles), they are sometimes completely unnecessary from this point of view (e.g. meningococcal meningitis, paralytic poliomyelitis). These microbes are not shed to the exterior after multiplying in the meninges or spinal cord.
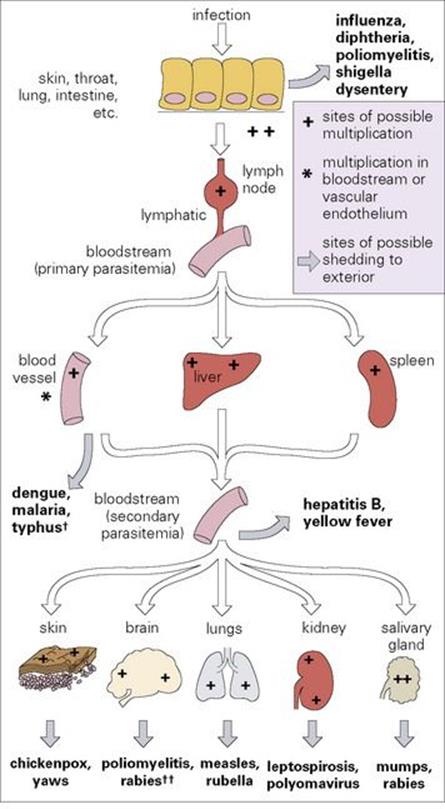
Figure 15.3 The spread of infection throughout the body. Bone marrow and muscle are possible sources for secondary parasitaemia in addition to blood vessels, liver and spleen. †In dengue, malaria and typhus, multiplication occurs in blood cells or vascular endothelium. ††Poliovirus invades the brain and spinal cord from the blood, but is not shed from these sites, whereas rabies invades and later travels from brain to salivary glands via peripheral nerves.
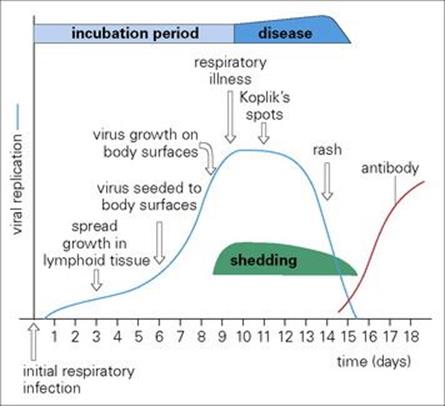
Figure 15.4 The pathogenesis of measles. Virus invades body surfaces from the blood, traversing blood vessels to reach surface epithelium first in the respiratory tract where there are only 1–2 layers of epithelial cells and then in mucosae (Koplik’s spots) and finally in the skin (rash).
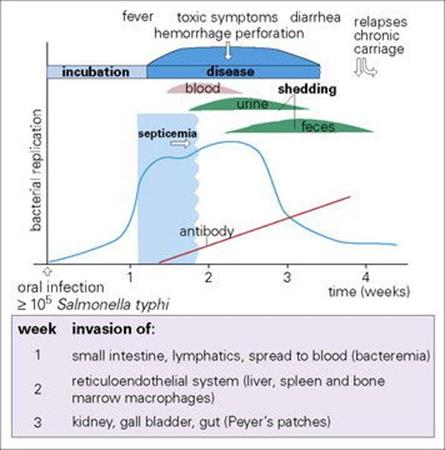
Figure 15.5 The pathogenesis of typhoid fever.
For the microbe, systemic spread is fraught with obstacles, and a major encounter with immune and other defences is inevitable. Microorganisms have therefore been forced to develop strategies for bypassing or countering these defences (see Ch. 16).
Rapid replication is essential for surface infections
The rate of replication of the infecting microorganism is of central importance, and doubling times vary from 20 min to several days (Table 15.1). Hit-and-run (surface) infections need to replicate rapidly, whereas a microorganism that divides every few days (e.g. Mycobacterium tuberculosis) is likely to cause a slowly evolving disease with a long incubation period. Microorganisms nearly always multiply faster in vitro than they do in the intact host, as might be expected if host defences are performing a useful function. In the host, microorganisms are phagocytosed and killed and the supply of nutrients may be limited. The net increase in numbers is slower than in laboratory cultures where microbes are not only free from attack by host defences, but also every effort has been made to supply them with optimal nutrients, susceptible cells, and so on.
Table 15.1 Replication rates of different microorganisms
|
Microorganisms |
Situation |
Mean doubling time |
|
Most viruses |
In cella |
< 1 h |
|
Many bacteria, e.g. Escherichia coli, staphylococci |
In vitro |
20–30 min |
|
Salmonella typhimurium |
In vitro |
30 min |
|
Mycobacterium tuberculosis |
In vitro |
24 h |
|
Mycobacterium lepraeb |
In vivo |
2 weeks |
|
Treponema pallidumb |
In vivo |
30 h |
|
Plasmodium falciparum |
In vitro/in vivo (erythrocyte or hepatic cell) |
8 h |
a But some viruses show greatly delayed replication or delayed spread from cell to cell.
b But cannot be cultivated in vitro.
Mechanisms of spread through the body
Spread to lymph and blood
Invading microbes encounter a variety of defences on entering the body
After traversing the epithelium and its basement membrane at the body surface, invading microbes face the following defences:
• tissue fluids containing antimicrobial substances (antibody, complement).
• local macrophages (histiocytes). Subcutaneous and submucosal macrophages are a threat to microbial survival.
• the physical barrier of local tissue structure. Local tissues consist of various cells in a hydrated gel matrix; although viruses can spread by stepwise invasion of cells, invasion is more difficult for bacteria, and those that spread effectively sometimes possess special spreading factors (e.g. streptococcal hyaluronidase).
• the lymphatic system. The rich network of the lymphatic system soon conveys microorganisms to the battery of phagocytic and immunologic defences awaiting them in the local lymph node (Fig. 15.6). Macrophages, strategically placed in the marginal and other lymph sinuses, constitute an efficient filtering system for lymph.
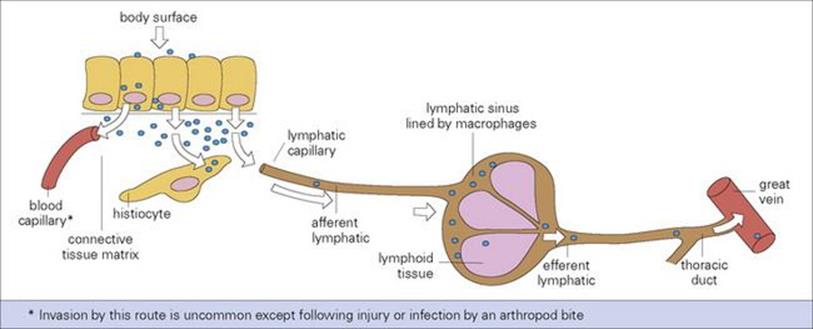
Figure 15.6 Microbial invasion and spread to lymph and blood. Microbes (or other particles) beneath surface epithelium readily enter local lymphatics.
The infection may be halted at any stage, but by multiplying locally or in lymph nodes and by evading phagocytosis, the microorganism can ultimately reach the bloodstream. Therefore, a minor injury to the skin, followed by a red streak (inflamed lymphatic) and a tender, swollen local lymph node are classic signs of streptococcal invasion. Most bacteria cause a great deal of inflammation when they invade in this way. In the early stages, lymph flow increases, but eventually, if there is enough inflammation and tissue damage in the node itself, the flow of the lymph may cease. In contrast, viruses and other intracellular microorganisms often invade lymph and blood silently and asymptomatically during the incubation period; this is facilitated when they infect monocytes or lymphocytes without initially damaging them.
Spread from blood
The fate of microorganisms in the blood depends upon whether they are free or associated with circulating cells
Viruses or small numbers of bacteria can enter the blood without causing a general body disturbance. For instance, transient bacteraemia are fairly common in normal individuals (e.g. they may occur after defecation or brushing teeth), but the bacteria are usually filtered out and destroyed in macrophages lining the liver and spleen sinusoids. Under certain circumstances, the same bacteria have a chance to localize in less well-defended sites, such as congenitally abnormal heart valves in the case of viridans streptococci causing infective endocarditis, or in the ends of growing bones in the case of Staphylococcus aureus osteomyelitis.
If microorganisms are free in the blood, they are exposed to body defences such as antibodies and phagocytes. However, if they are associated with circulating cells, these cells can protect them from host defences and carry them around the body. For example, many viruses, such as Epstein–Barr virus (EBV) and rubella, and intracellular bacteria (Listeria, Brucella) are present in lymphocytes or monocytes and, if not damaged or destroyed, these ‘carrying cells’ protect and transport them. Malaria infects erythrocytes.
On entering the blood, microorganisms are exposed to macrophages of the reticuloendothelial system (see Ch. 9). Here, in the sinusoids, where blood flows slowly, they are often phagocytosed and destroyed. But certain microorganisms survive and multiply in these cells (Salmonella typhi,Leishmania donovani, yellow fever virus). The microorganism may then:
• spread to adjacent hepatic cells in the liver (hepatitis viruses), or splenic lymphoid tissues (measles virus)
• re-invade the blood (S. typhi, hepatitis viruses).
Each circulating microorganism invades characteristic target organs and tissues
If uptake by reticuloendothelial macrophages is not complete within a short time, or if large numbers of microorganisms are present in the blood, there is an opportunity for localization elsewhere in the vascular system. Why each circulating microorganism invades characteristic target organs and tissues (Table 15.2) is not completely understood, but may be due to:
• specific receptors for the microorganism, leading to localization on the vascular endothelium of certain target organs
• random localization in organs throughout the body, only some of them being suitable for subsequent colonization and replication
• accumulation of circulating microbes in sites where there is local inflammation, because of the slower flow and sticky endothelium in inflamed vessels.
Table 15.2 Circulating microorganisms that invade organs via small blood vessels
|
Microbe |
Disease |
Principal organs invadeda |
|
Viruses |
||
|
Hepatitis B |
Hepatitis B |
Liver |
|
Rubella |
Congenital rubella |
Placenta (fetus) |
|
Varicella-zoster virus |
Chickenpox |
Skin, respiratory tract |
|
Polio |
Poliomyelitis |
Brain, spinal cord |
|
Mumps |
Mumps |
Parotid, mammary glands |
|
Bacteria |
||
|
Rickettsia rickettsi |
Rocky Mountain spotted fever |
Skin |
|
Treponema pallidum |
Secondary syphilis |
Skin, mucosae |
|
Neisseria meningitidis |
Meningitis |
Meninges |
|
Protozoa |
||
|
Trypanosoma cruzi |
Chagas disease |
Heart, skeletal muscle |
|
Plasmodium spp. |
Malaria |
Liver |
|
Helminths |
||
|
Schistosoma spp. (larvae) |
Schistosomiasis |
Veins of bladder, bowel |
|
Ascaris lumbricoides (larvae) |
Ascariasis |
Lung |
|
Ancylostoma duodenale (larvae) |
Hookworm |
Lung |
a In liver, sinusoids; elsewhere, capillaries, venules.
After localization and organ invasion, the replicating microbe is shed from the body if the organ has a surface with access to the outside world (see Fig. 15.3). It may also be shed back into the bloodstream, either directly or via the lymphatic system.
Spread via nerves
Certain viruses spread via peripheral nerves from peripheral parts of the body to the central nervous system and vice versa
Tetanus toxin reaches the central nervous system (CNS) by this route. Rabies, herpes simplex virus (HSV) and varicella-zoster virus (VZV) travel in axons (see Chs 13 and 24) and although the rate is slow, being accounted for by axonal flow (up to 10 mm/h), this movement is important in the pathogenesis of these infections. Rabies not only reaches the CNS largely by peripheral nerves, but takes the same route from the CNS when it invades the salivary glands. Few, if any, host defences are in a position to control this type of viral spread once nerves are invaded. Routes of invasion of the CNS are illustrated in Figure 15.7.
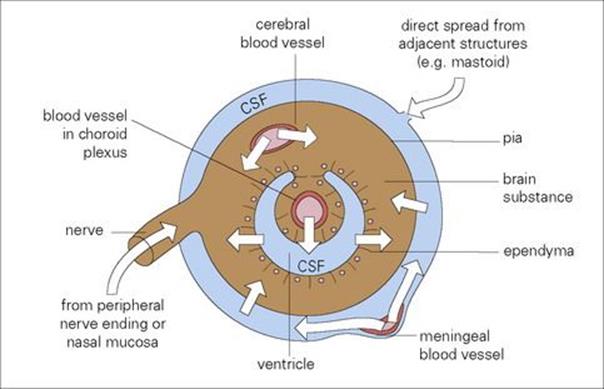
Figure 15.7 Routes of microbial invasion of the central nervous system. CSF, cerebrospinal fluid.
An uncommon route of spread to the CNS is via olfactory nerves with axons terminating on olfactory mucosa. For instance, certain free-living amoebae (e.g. Naegleria spp.) found in sludge at the bottom of freshwater pools may take this route and cause meningoencephalitis in swimmers (see Ch. 24). Viruses and bacteria in the nasopharynx (e.g. meningococci, poliovirus) generally spread to the CNS via the blood.
Spread via cerebrospinal fluid
Once microorganisms have crossed the blood–cerebrospinal barrier, they spread rapidly in the cerebrospinal fluid spaces
Such microorganisms can then invade neural tissues (echoviruses, mumps virus) as well as multiply locally (Neisseria meningitidis, Haemophilus influenzae, Streptococcus pneumoniae) and possibly infect ependymal and meningeal cells.
Spread via other routes
Rapid spread from one visceral organ to another can take place via the pleural or peritoneal cavity
Both the pleural and peritoneal cavities are lined by macrophages, as if in expectation of such invasion, and the peritoneal cavity contains an antimicrobial armoury, consisting of the omentum (the ‘abdominal policeman’), and many lymphocytes, macrophages and mast cells. Injury or disease in an abdominal organ provides a source of infection for peritonitis, as do chest wounds or lung infections for pleurisy.
Genetic determinants of spread and replication
The pathogenicity of a microorganism is determined by the interplay of a variety of factors
These factors are referred to in Chapters 12 and 16. A distinction is sometimes made between pathogenicity and virulence: virulence implies a quantitative measure of pathogenicity. For instance, it can be expressed as the number of organisms necessary to cause death in 50% of individuals: lethal dose 50 (LD50). Nearly all pathogenicity factors are controlled by host and microbial genes. It has long been known that there are host genetic influences on susceptibility to infectious disease, and that mutations in microorganisms affect their pathogenicity. A number of these genetic factors have been revealed by the application of molecular genetics techniques, and as a result it is increasingly possible to identify the specific gene products involved. Progress has also been made, though with greater difficulty, in understanding the mode of action of these gene products.
Genetic determinants in the host
The ability of a microorganism to infect and cause disease in a given host is influenced by the genetic constitution of the host
At a relatively gross level, some human pathogens either do not infect other species or infect only closely related primates (e.g. measles, trachoma, typhoid, hepatitis B, warts), whereas others infect a very wide range of hosts (rabies, anthrax). Also, within a given host species, there are genetic determinants of susceptibility. The best examples are found in animals, but there are examples for human disease (see below).
One example at the molecular level is the sickle cell gene and susceptibility to malaria. Malaria merozoites (see Ch. 27) parasitize red blood cells and metabolize haemoglobin, freeing haem and using globin as a source of amino acids. The sickle cell gene causes a substitution of the amino acid valine for glutamic acid at one point in the b-polypeptide chain of the haemoglobin molecule. The new haemoglobin (haemoglobin S) becomes insoluble when reduced, and precipitates inside the red cell envelope, distorting the cell into the shape of a sickle. In homozygous individuals, there are two of these genes and the individual has the disease sickle cell anaemia, because the red cells are so fragile they sickle under normal circumstances. But in the heterozygote (sickle cell trait), the gene is less harmful, and provides resistance to severe forms of falciparum malaria, which ensures its selection in endemic malarial regions. The gene would be eliminated from populations after 10–20 generations unless it conferred some advantage. Restriction endonuclease analyses of the gene in Indian and West African populations have revealed that it arose independently in these malarious countries. Homozygotes, however, show increasing susceptibility to other infections, particularly Strep. pneumoniae, as a result of splenic dysfunction following repeated splenic infarcts.
Other examples are individuals whom are nonsecretors of ABO blood groups, due to homozygosity for a fucosyl-transferase 2 (FUT2) variant that is critical for AB antigen synthesis, who are completely resistant to norovirus infections that cause diarrhea. Almost total resistance to HIV-1 infection and new variant Creutzfeldt–Jakob disease is seen in individuals homozygous for a 32-base pair deletion in the CCR5 chemokine receptor gene and those homozygous for valine at codon 129 of the prion protein gene, respectively.
Susceptibility often operates at the level of the immune response
A poor immune response to a given infection can lead to increased susceptibility to disease, whereas an immune response that is too vigorous may lead to immunopathologic disease (see Ch. 17). Of particular importance are the major histocompatibility complex (MHC) genes on chromosome 6, coding for MHC class II (HLA DP, DQ, DR) antigens and controlling specific immune responses (see Chs 10 and 11). For example, susceptibility to leprosy (see Ch. 26) is strongly influenced by MHC class II genes. People with the HLA DR3 antigen are more susceptible to tuberculoid leprosy, whereas those with HLA DQ1 are more susceptible to lepromatous leprosy.
Studies of identical twins (Box 15.1) provide evidence that genetic determinants affect susceptibility to tuberculosis. The present-day European population shows considerable resistance to this disease. During the great epidemics of pulmonary tuberculosis in Europe in the seventeenth, eighteenth and nineteenth centuries, genetically susceptible individuals were weeded out. In 1850, mortality rates in Boston, New York, London, Paris and Berlin were over 500/100 000, but with improvements in living conditions these fell to 180/100 000 by 1900, and they have fallen even more since then. However, previously unexposed populations, especially in Africa and the Pacific Islands, show much greater susceptibility to respiratory tuberculosis. In the Plains Indians living in the Qu’Appelle Valley reservation in Saskatchewan, Canada, in 1886, tuberculosis spread through the body to infect glands, bones, joints and meninges, giving a death rate of 9000/100 000.
![]()
Box 15.1  Lessons in microbiology
Lessons in microbiology
Genetically determined susceptibility to infection
There are several classic examples of susceptibility to infectious disease determined by unidentified but presumably genetic factors in the human host.
The Lubeck disaster due to vaccination with virulent tubercle bacilli
In Lubeck, Germany, between December 1929 and April 1930, three oral doses of living virulent tubercle bacilli, instead of attenuated (vaccine) bacilli were inadvertently given to 251 infants < 10 days old. There were 72 deaths, 135 developed clinical tuberculosis but recovered and were alive and well 12 years later, while 44 became tuberculin-positive but remained well. Each received the same inoculum, and it seems likely that the differences in outcome were largely due to genetic factors in the host. This disaster was a setback for early BCG enthusiasts. Dr George Deycke, in whose laboratory the contaminated batch of BCG had been produced (but never tested for virulence before use), was tried, found guilty of manslaughter and injury by negligence, and sent to prison, together with the Director of Lubeck Health Office.
A military misfortune due to contamination of yellow fever vaccine with hepatitis B virus
In 1942, > 45 000 US military personnel were vaccinated against yellow fever, but were inadvertently injected at the same time with hepatitis B virus present as a contaminant in the human serum used to stabilize the vaccine. There were 914 clinical cases of hepatitis, of which 580 were mild, 301 moderate and 33 severe. Even with a given batch of vaccine, the incubation period varied in the range 10–20 weeks. Serologic tests were not then available, so the number of subclinical infections is unknown. In this case, both physiologic and genetic influences on susceptibility may have played a part.
Identical twins are affected similarly by respiratory tuberculosis
A study of tuberculosis in twins when at least one twin had the disease showed that, for identical twins, the other twin was affected in 87% of cases. With non-identical twins, the equivalent figure was only 26%. In addition, the identical twins had a similar type of clinical disease.
![]()
Genetic determinants in the microbe
Virulence is often coded for by more than one microbial gene
Virulence is determined by numerous factors such as adhesion, penetration into cells, antiphagocytic activity, production of toxins and interaction with the immune system. Consequently, different genes and gene products are involved in different ways and at different stages in pathogenesis.
Under natural circumstances, microorganisms are constantly undergoing genetic change (i.e. mutations). The single-stranded RNA viruses in particular show very high mutation rates. Mutations affecting surface antigens undergo rapid selection in the host under immune pressure (antibody, cell-mediated immunity), as in the case of the rapidly evolving M proteins of streptococci, and the capsid proteins of picornaviruses. In addition, genetic changes in bacteria are often due to acquisition or loss of genetic elements such as integrins, pathogenicity islands, transposons and plasmids (see Chs 2 and 33).
Changes in the virulence of a microorganism take place during artificial culture in the laboratory. For instance, in the classic procedure for obtaining a live vaccine (see Ch. 34), a microorganism is repeatedly grown (passaged) in vitro, and this generally leads to reduced pathogenicity in the host. The new strain is then referred to as ‘attenuated’ (Table 15.3).
Table 15.3 Examples of attenuation of pathogens following repeated passage in vitro
|
Pathogen |
Passage |
Attenuated (live) product |
|
Mycobacterium bovis |
10 years of repeated passage in glycerin-bile-potato medium |
Bacille Calmette–Guérin (BCG) vaccine |
|
Rubella virus |
27 passages in human diploid cells |
Rubella vaccine (Wistar RA 27/3) |
Our understanding of the genetic basis for microbial pathogenicity has advanced due to genetic manipulation techniques such as cloning and site-specific mutagenesis. For instance, by introducing or deleting/inactivating genome segments, the virulence genes can be identified. Examples are shown in Table 15.4. Major advances in genomics including rapid DNA sequencing and sophisticated computer programs for DNA analysis have also made major contributions to our understanding of virulence genes and conditions affecting their expression. This has allowed sequencing of the entire genome for many infectious agents (bacteria and viruses). This information has facilitated the assignment of virulence functions to specific loci and greatly improved our understanding of the way microorganisms sense and respond to the host environment (e.g. two-component regulation and quorum sensing; Figs 2.10 and 2.11, respectively).
Table 15.4 of microbial pathogenicity
|
Microorganism |
Gene or gene product |
Effect on virulence |
|
Streptococcus pyogenes |
M protein; 60 mm long coiled coil extending from bacterial cell wall with N terminal hypervariable domain |
Antiphagocytic role; inhibits opsonization; exact mechanism unknown |
|
Amino acid sequence overlap with host components (myosin, tropomyosin, keratin, etc.) |
Autoimmune complication |
|
|
Yersinia enterocolitica |
Invasion (inv) gene codes for 92 kDa protein on bacterial surface |
Required for uptake of bacteria into epithelial cells and macrophages of Peyer’s patches |
|
Shigella spp. |
Ipa B (invasion plasmid antigen B) gene |
Mediates lysis of vacuolar membrane and escape of bacteria into cytoplasm of colonic epithelial cellsa |
|
Leishmania donovani |
Arg-Gly-Asp sequence of gp63 surface protein |
Binds to C3b receptor on macrophage and thereby infects this cell |
|
Neisseria gonorrhoeae |
Genes for proteins of pili (fimbriae) and for certain outer membrane proteins |
Proteins mediate attachment to mucosal cells, independent control of each gene makes N. gonorrhoeae the ‘master chameleon’, altering its surface antigens to evade host immune responses |
|
Herpes simplex virus type 1 |
Gene for envelope glycoprotein C (gC) |
gC acts as receptor for C3b, blocking the classical pathway and enabling virus or virus-infected cell to resist lysis by complement plus antibody |
|
Helicobacter pyloriCagA |
Cag pathogenicity island encoding 40 genes including cagA gene |
Transportation of protein into stomach epithelial cells activates the epidermal growth factor receptor (EGFR) resulting in altered cellular signal transduction and gene expression associated with ulcer formation |
a Shigellae enter the gut wall via M cells in Peyer’s patches, then invade colonic epithelial cells from basolateral surfaces. In this, as in other bacterial infections, invasiveness depends upon coordinated expression of many different genes.
Other factors affecting spread and replication
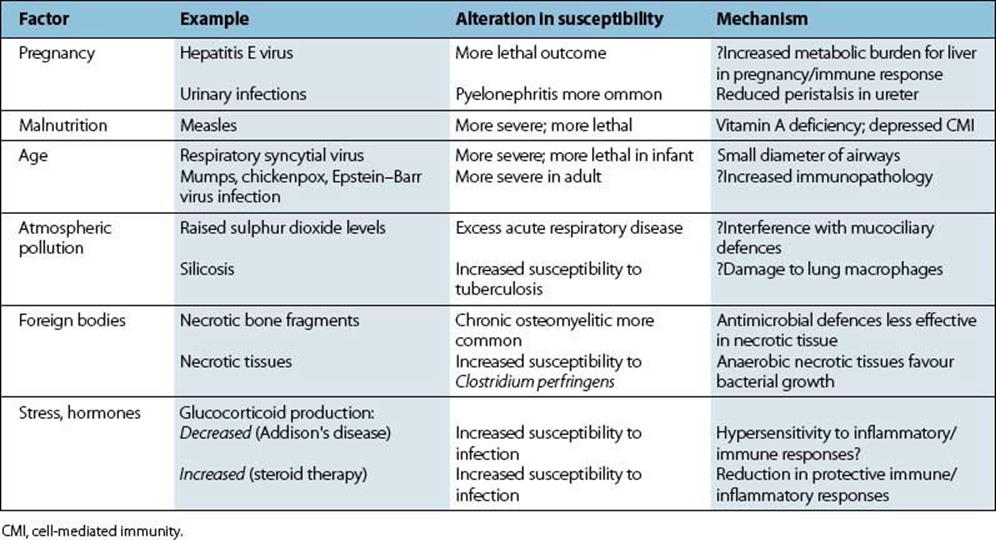
Various other factors have an influence on susceptibility to infectious disease (Table 15.5). In most cases, it is not known whether this involves differences in microbial spread and replication or differences in host immune and inflammatory responses. Infections in hosts with immunologic and other defects are described in Chapter 33.
Table 15.5 Host factors influencing susceptibility to infectious diseases
The brain can influence immune responses
When stress (a loosely used word) is associated with malnutrition or crowding, it may be difficult to disentangle the separate influences of these various factors on susceptibility to infection, as in the case of tuberculosis. The brain can, however, influence immune responses, acting via the hypothalamus, pituitary and adrenal cortex. It has long been known that glucocorticoids, which have powerful actions on immune cells, are needed for resistance to infection and trauma. A shortage of glucocorticoids, as in Addison’s disease, or an excess, as with steroid therapy, results in increased susceptibility to infection (Table 15.5). In addition, the brain, the endocrine and the immune systems often use the same molecular messengers: cytokines, peptide hormones, neurotransmitters. Neural cells, for instance, have receptors for interferons and for interleukins IL-1, IL-2, IL-3, and IL-6, and thymic lymphocytes can produce prolactin and growth hormone. Immune–neuroendocrine cross-talk now has molecular respectability and provides an acceptable basis for the influence of the brain on immunity and infectious disease.
![]()
 Key Facts
Key Facts
• Infections restricted to the body surfaces (e.g. common cold, shigella dysentery) have shorter incubation periods than systemic infections (e.g. measles, typhoid), and adaptive (immune) host responses tend to be less important.
• Microbes with a slow growth rate (e.g. M. tuberculosis) tend to cause diseases which evolve slowly.
• Spread through the body takes place primarily via lymph and blood. The fate of circulating microbes depends upon whether they are free or present in circulating blood cells.
• Uptake by reticuloendothelial cells in liver and spleen focuses infection into these organs, but specific localization in the vascular bed of other organs (e.g. mumps virus in salivary glands, meningococci in meninges) is not understood.
• Viruses can spread in either direction along nerve axons, and this is important in the pathogenesis of recurrent herpes simplex virus infection, zoster and rabies.
• Pathogenicity and virulence are strongly influenced by genetic factors in the host (e.g. tuberculosis in identical twins) and by genetic factors in the microbe (e.g. sickle cell trait in falciparum malaria).
• Our understanding of virulence has been greatly enhanced by advances in molecular biology which have allowed sequence analysis of entire microbial genomes and a clearer view of microbial response to the host environment.
![]()