Lippincott’s Illustrated Reviews: Biochemistr, Sixth Edition (2014)
UNIT III: Lipid Metabolism
Chapter 17. Phospholipid, Glycosphingolipid, and Eicosanoid Metabolism
I. OVERVIEW OF PHOSPHOLIPIDS
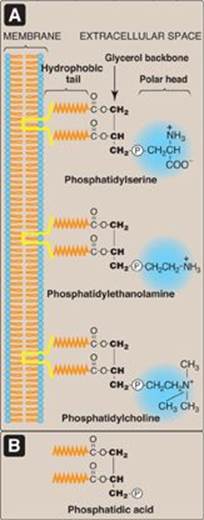
Phospholipids are polar, ionic compounds composed of an alcohol that is attached by a phosphodiester bond to either diacylglycerol (DAG) or sphingosine. Like fatty acids, phospholipids are amphipathic in nature. That is, each has a hydrophilic head, which is the phosphate group plus whatever alcohol is attached to it (for example, serine, ethanolamine, and choline, highlighted in blue in Figure 17.1A), and a long, hydrophobic tail containing fatty acids or fatty acid–derived hydrocarbons (shown in orange in Figure 17.1A). Phospholipids are the predominant lipids of cell membranes. In membranes, the hydrophobic portion of a phospholipid molecule is associated with the nonpolar portions of other membrane constituents, such as glycolipids, proteins, and cholesterol. The hydrophilic (polar) head of the phospholipid extends outward, interacting with the intracellular or extracellular aqueous environment (see Figure 17.1A). Membrane phospholipids also function as a reservoir for intracellular messengers, and, for some proteins, phospholipids serve as anchors to cell membranes. Nonmembrane phospholipids serve additional functions in the body, for example, as components of lung surfactant and essential components of bile, where their detergent properties aid in the solubilization of cholesterol.
Figure 17.1 A. Structures of some glycerophospholipids. B. Phosphatidic acid. ![]() = phosphate (an anion).
= phosphate (an anion).
II. STRUCTURE OF PHOSPHOLIPIDS
There are two classes of phospholipids: those that have glycerol (from glucose) as a backbone and those that have sphingosine (from serine and palmitate). Both classes are found as structural components of membranes, and both play a role in the generation of lipid-signaling molecules.
A. Glycerophospholipids
Phospholipids that contain glycerol are called glycerophospholipids (or phosphoglycerides). Glycerophospholipids constitute the major class of phospholipids and are the predominant lipids in membranes. All contain (or are derivatives of) phosphatidic acid (PA), which is DAG with a phosphate group on carbon 3 (Figure 17.1B). PA is the simplest phosphoglyceride and is the precursor of the other members of this group.
1. Glycerophospholipids from phosphatidic acid and an alcohol: The phosphate group on PA can be esterified to another compound containing an alcohol group (see Figure 17.1). For example:
|
Serine |
+ PA → phosphatidylserine (PS) |
|
Ethanolamine |
+ PA → phosphatidylethanolamine (PE) (cephalin) |
|
Choline |
+ PA → phosphatidylcholine (PC) (lecithin) |
|
Inositol |
+ PA → phosphatidylinositol (PI) |
|
Glycerol |
+ PA → phosphatidylglycerol (PG) |
Figure 17.2 Structure of cardiolipin (diphosphatidylglycerol). ![]() = phosphate.
= phosphate.
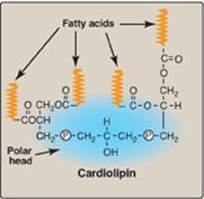
2. Cardiolipin: Two molecules of PA esterified through their phosphate groups to an additional molecule of glycerol is called cardiolipin, or diphosphatidylglycerol (Figure 17.2). Cardiolipin is found in membranes in bacteria and eukaryotes. In eukaryotes, cardiolipin is virtually exclusive to the inner mitochondrial membrane, where it maintains the structure and function of certain respiratory complexes of the electron transport chain. [Note: Cardiolipin is antigenic and is recognized by antibodies raised against Treponema pallidum, the bacterium that causes syphilis.]
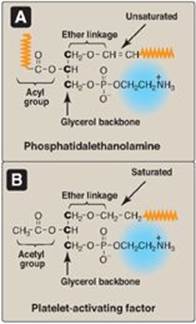
3. Plasmalogens: When the fatty acid at carbon 1 of a glycerophospholipid is replaced by an unsaturated alkyl group attached by an ether (rather than by an ester) linkage to the core glycerol molecule, an ether phosphoglyceride known as a plasmalogen is produced. For example, phosphatidalethanolamine, which is abundant in nerve tissue (Figure 17.3A), is the plasmalogen that is similar in structure to phosphatidylethanolamine. Phosphatidalcholine (abundant in heart muscle) is the other quantitatively significant ether lipid in mammals. [Note: Plasmalogens use “al” rather than “yl” in their names.]
4. Platelet-activating factor: A second example of an ether glycerophospholipid is platelet-activating factor (PAF), which has a saturated alkyl group in an ether link to carbon 1 and an acetyl residue (rather than a fatty acid) at carbon 2 of the glycerol backbone (Figure 17.3B). PAF is synthesized and released by a variety of cell types. It binds to surface receptors, triggering potent thrombotic and acute inflammatory events. For example, PAF activates inflammatory cells and mediates hypersensitivity, acute inflammatory, and anaphylactic reactions. It causes platelets to aggregate and activate, and neutrophils and alveolar macrophages to generate superoxide radicals to kill bacteria (see p. 148). It also lowers blood pressure. [Note: PAF is one of the most potent bioactive molecules known, causing effects at concentrations as low as 10-11 mol/l.]
Figure 17.3 The ether glycerophospholipids. A. The plasmalogen phosphatidalethanolamine. B. Platelet-activating factor. (![]() is a long, hydrophobic hydrocarbon chain.).
is a long, hydrophobic hydrocarbon chain.).
B. Sphingophospholipids: sphingomyelin
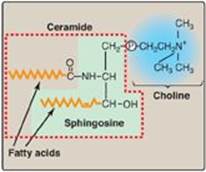
The backbone of sphingomyelin is the amino alcohol sphingosine, rather than glycerol (Figure 17.4). A long-chain fatty acid is attached to the amino group of sphingosine through an amide linkage, producing a ceramide, which can also serve as a precursor of glycolipids (see p. 209). The alcohol group at carbon 1 of sphingosine is esterified to phosphorylcholine, producing sphingomyelin, the only significant sphingophospholipid in humans. Sphingomyelin is an important constituent of the myelin sheath of nerve fibers. [Note: The myelin sheath is a layered, membranous structure that insulates and protects neuronal fibers of the central nervous system (CNS).]
III. PHOSPHOLIPID SYNTHESIS
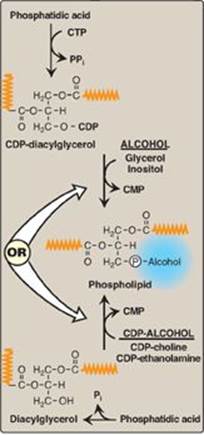
Glycerophospholipid synthesis involves either the donation of PA from cytidine diphosphate (CDP)-diacylglycerol to an alcohol or the donation of the phosphomonoester of the alcohol from CDP-alcohol to 1,2-diacylglycerol (Figure 17.5). In both cases, the CDP-bound structure is considered an “activated intermediate,” and cytidine monophosphate (CMP) is released as a side product of glycerophospholipid synthesis. A key concept in phosphoglyceride synthesis, therefore, is activation, either of DAG or the alcohol to be added, by linkage with CDP. [Note: This is similar in principle to the activation of sugars by their attachment to uridine diphosphate (UDP) (see p. 126).] The fatty acids esterified to the glycerol alcohol groups can vary widely, contributing to the heterogeneity of this group of compounds, with saturated fatty acids typically found at carbon 1 and unsaturated ones at carbon 2. Most phospholipids are synthesized in the smooth endoplasmic reticulum (ER). From there, they are transported to the Golgi apparatus and then to membranes of organelles or the plasma membrane or are secreted from the cell by exocytosis. [Note: Ether lipid synthesis from dihydroxyacetone phosphate occurs in peroxisomes.]
Figure 17.4 Structure of sphingomyelin, showing sphingosine (in green box) and ceramide components (in dashed box). ![]() = phosphate.
= phosphate.
A. Phosphatidic acid
PA is the precursor of many other phosphoglycerides. The steps in its synthesis from glycerol phosphate and two fatty acyl coenzyme A (CoA) molecules were illustrated in Figure 16.14, p. 189, in which PA is shown as a precursor of triacylglycerol.
Essentially all cells except mature erythrocytes can synthesize phospholipids, whereas triacylglycerol synthesis occurs essentially only in liver, adipose tissue, lactating mammary glands, and intestinal mucosal cells.
B. Phosphatidylcholine and phosphatidylethanolamine
PC and PE are the most abundant phospholipids in most eukaryotic cells. The primary route of their synthesis uses choline and ethanolamine obtained either from the diet or from the turnover of the body’s phospholipids. [Note: In the liver, PC also can be synthesized from PS and PE (see below).]
1. Synthesis from preexisting choline and ethanolamine: These synthetic pathways involve the phosphorylation of choline or ethanolamine by kinases, followed by conversion to the activated form, CDP-choline or CDP-ethanolamine. Finally, choline-phosphate or ethanolamine-phosphate is transferred from the nucleotide (leaving CMP) to a molecule of DAG (see Figure 17.5).
Figure 17.5 Phospholipid synthesis requires activation of either diacylglycerol or an alcohol by linkage to cytidine diphosphate (CDP). CMP = cytidine monophosphate; CTP = cytidine triphosphate; Pi = inorganic phosphate; PPi = pyrophosphate. (![]() is a fatty acid hydrocarbon chain.)
is a fatty acid hydrocarbon chain.)
a. Significance of choline reutilization: The reutilization of choline is important because, whereas humans can synthesize choline de novo, the amount made is insufficient for our needs. Thus, choline is an essential dietary nutrient with an Adequate Intake (see p. 358) of 550 mg for men and 425 mg for women. [Note: Choline is also used for the synthesis of acetylcholine, a neurotransmitter.]
b. Role of phosphatidylcholine in lung surfactant: The pathway described above is the principal pathway for the synthesis of dipalmitoylphosphatidylcholine (DPPC, or dipalmitoyl lecithin). In DPPC, positions 1 and 2 on the glycerol are occupied by palmitate. DPPC, made and secreted by type II pneumocytes, is a major lipid component of lung surfactant, which is the extracellular fluid layer lining the alveoli. Surfactant serves to decrease the surface tension of this fluid layer, reducing the pressure needed to reinflate alveoli, thereby preventing alveolar collapse (atelectasis). [Note: Surfactant is a complex mixture of lipids (90%) and proteins (10%), with DPPC being the major component for reducing surface tension.] Respiratory distress syndrome (RDS) in preterm infants is associated with insufficient surfactant production and/or secretion and is a significant cause of all neonatal deaths in Western countries.
Lung maturity of the fetus can be gauged by determining the ratio of DPPC to sphingomyelin, usually written as the L (for lecithin)-to-S ratio, in amniotic fluid. A value of two or above is evidence of maturity, because it reflects the major shift from sphingomyelin to DPPC synthesis that occurs in the pneumocytes at about 32 weeks of gestation.
Lung maturation can be accelerated by giving the mother glucocorticoids shortly before delivery to induce expression of specific genes. Postnatal administration of natural or synthetic surfactant (by intratracheal instillation) is also used. [Note: Acute respiratory distress syndrome (ARDS), seen in all age groups, is the result of alveolar damage (due to infection, injury, or aspiration) that causes fluid to accumulate in the alveoli, impeding the exchange of oxygen and carbon dioxide.]
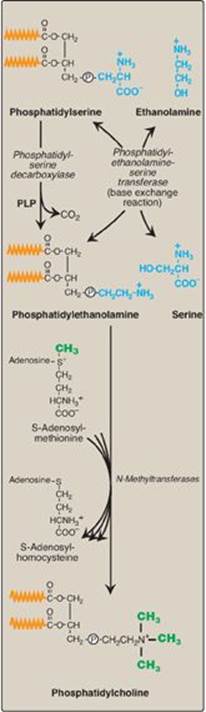
2. Synthesis of phosphatidylcholine from phosphatidylserine in the liver: The liver requires a mechanism for producing PC, even when free choline levels are low, because it exports significant amounts of PC in the bile and as a component of serum lipoproteins. To provide the needed PC, PS is decarboxylated to PE by PS decarboxylase, an enzyme requiring pyridoxal phosphate (PLP) as a coenzyme. PE then undergoes three methylation steps to produce PC, as illustrated in Figure 17.6. S-adenosylmethionine is the methyl group donor (see p. 264).
C. Phosphatidylserine
PS synthesis in mammalian tissues is provided by the base exchange reaction, in which the ethanolamine of PE is exchanged for free serine (see Figure 17.6). This reaction, although reversible, is used primarily to produce the PS required for membrane synthesis.
Figure 17.6 Synthesis of phosphatidylcholine from phosphatidylserine in the liver. (![]() is a fatty acid hydrocarbon chain.)
is a fatty acid hydrocarbon chain.) ![]() = phosphate; PLP = pyridoxal phosphate.
= phosphate; PLP = pyridoxal phosphate.
D. Phosphatidylinositol
PI is synthesized from free inositol and CDP-diacylglycerol as shown in Figure 17.5. PI is an unusual phospholipid in that it most frequently contains stearic acid on carbon 1 and arachidonic acid on carbon 2 of the glycerol. PI, therefore, serves as a reservoir of arachidonic acid in membranes and, thus, provides the substrate for prostaglandin synthesis when required (see p. 213 for a discussion of these compounds). [Note: There is asymmetry in the phospholipid composition of the cell membrane. PS and PI, for example, are found primarily on the inner leaflet. Asymmetry is achieved by enzymes known as “flippases” and “floppases.”]
Figure 17.7 Structure of phosphatidylinositol 4,5-bisphosphate. Cleavage by phospholipase C produces inositol 1,4,5-trisphosphate and diacylglycerol. (![]() is a fatty acid hydrocarbon chain.)
is a fatty acid hydrocarbon chain.) ![]() = phosphate.
= phosphate.
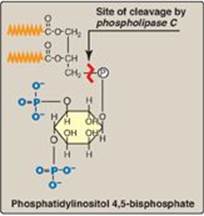
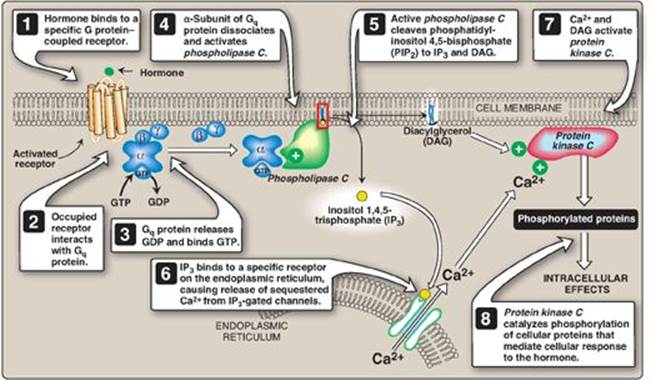
1. Role in signal transmission across membranes: The phosphorylation of membrane-bound PI produces polyphosphoinositides such as phosphatidylinositol 4,5-bisphosphate ([PIP2] Figure 17.7). The hydrolytic cleavage of PIP2 by phospholipase C occurs in response to the binding of a variety of neurotransmitters, hormones, and growth factors to G protein–coupled receptors (such as the α-1 adrenergic receptor) on the cell membrane and activation of the Gq alpha subunit (Figure 17.8). The products of this cleavage, inositol 1,4,5-trisphosphate (IP3) and DAG, mediate the mobilization of intracellular calcium and the activation of protein kinase C, which act synergistically to evoke specific cellular responses. Signal transmission across the membrane is thus accomplished.
Figure 17.8 Role of inositol trisphosphate and diacylglycerol in intracellular signaling. GTP = guanosine triphosphate; GDP = guanosine diphosphate.
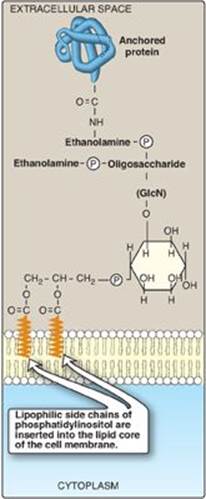
2. Role in membrane protein anchoring: Specific proteins can be covalently attached through a carbohydrate bridge to membrane-bound PI (Figure 17.9). Alkaline phosphatase, a digestive enzyme found on the surface of the small intestine that attacks organic phosphates, is an example of a protein attached to such a glycosyl phosphatidylinositol (GPI) anchor. [Note: GPI-linked proteins are also found in a variety of parasitic protozoans, such as, trypanosomes and leishmania.] Being attached to a membrane lipid (rather than being an integral part of the membrane) allows GPI-anchored proteins increased lateral mobility on the surface of the plasma membrane. The protein can be cleaved from its anchor by the action of phospholipase C (see Figure 17.8), releasing DAG. [Note: A deficiency in the synthesis of GPI in hematopoietic cells results in the hemolytic disease paroxysmal nocturnal hemoglobinuria.]
E. Phosphatidylglycerol and cardiolipin
Phosphatidylglycerol occurs in relatively large amounts in mitochondrial membranes and is a precursor of cardiolipin (diphosphatidyglycerol). It is synthesized by a two-step reaction from CDP-diacylglycerol and glycerol 3-phosphate. Cardiolipin (see Figure 17.2) is synthesized by the transfer of diacylglycerophosphate from CDP-diacylglycerol to a preexisting molecule of phosphatidylglycerol.
Figure 17.9 Example of a glycosyl phosphatidylinositol (GPI) membrane protein anchor. GlcN = glucosamine; ![]() = phosphate.
= phosphate.
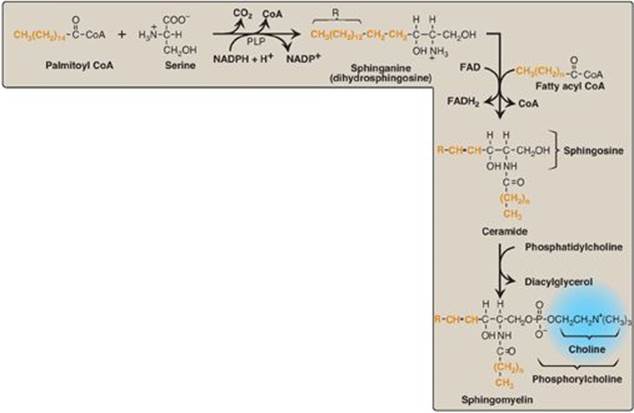
F. Sphingomyelin
Sphingomyelin, a sphingosine-based phospholipid, is a major structural lipid in the membranes of nerve tissue. The synthesis of sphingomyelin is shown in Figure 17.10. Briefly, palmitoyl CoA condenses with serine, as CoA and the carboxyl group (as CO2) of serine are lost. [Note: This reaction, like the decarboxylation reactions involved in the synthesis of PE from PS and of regulators from amino acids (for example, the catecholamines from tyrosine, see p. 286) requires pyridoxal phosphate (a derivative of vitamin B6) as a coenzyme (see p. 378).] The product is reduced in a nicotinamide adenine dinucleotide phosphate (NADPH)-requiring reaction to sphinganine (dihydrosphingosine), which is acylated at the amino group with one of a variety of long-chain fatty acids and then desaturated to produce a ceramide, the immediate precursor of sphingomyelin (and other sphingolipids, as described on p. 208).
Ceramides play a key role in maintaining the skin’s water-permeability barrier. Decreased ceramide levels are associated with a number of skin diseases.
Phosphorylcholine from PC is transferred to the ceramide, producing sphingomyelin and DAG. [Note: Sphingomyelin of the myelin sheath contains predominantly longer-chain fatty acids such as lignoceric acid and nervonic acid, whereas gray matter of the brain has sphingomyelin that contains primarily stearic acid.]
Figure 17.10 Synthesis of sphingomyelin. PLP = pyridoxal phosphate; NADP(H) = nicotinamide adenine dinucleotide phosphate; FAD(H2) = flavin adenine dinucleotide; CoA = coenzyme A.
IV. DEGRADATION OF PHOSPHOLIPIDS
The degradation of phosphoglycerides is performed by phospholipases found in all tissues and pancreatic juice (for a discussion of phospholipid digestion, see p. 175). A number of toxins and venoms have phospholipase activity, and several pathogenic bacteria produce phospholipases that dissolve cell membranes and allow the spread of infection. Sphingomyelin is degraded by the lysosomal phospholipase, sphingomyelinase (see below).
A. Phosphoglycerides
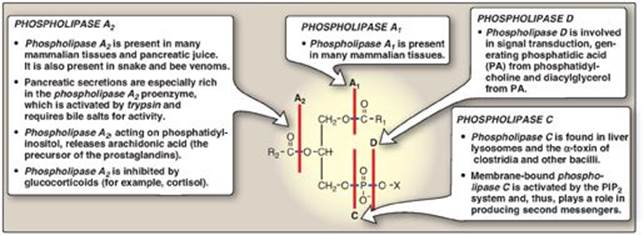
Phospholipases hydrolyze the phosphodiester bonds of phosphoglycerides, with each enzyme cleaving the phospholipid at a specific site. The major phospholipases are shown in Figure 17.11. [Note: Removal of the fatty acid from carbon 1 or 2 of a phosphoglyceride produces a lysophosphoglyceride, which is the substrate for lysophospholipases.] Phospholipases release molecules that can serve as second messengers (for example, DAG and IP3) or that are the substrates for synthesis of messengers (for example, arachidonic acid). Phospholipases are responsible not only for degrading phospholipids, but also for “remodeling” them. For example, phospholipases A1 and A2remove specific fatty acids from membrane-bound phospholipids, which can be replaced with different fatty acids using fatty acyl CoA transferase. This mechanism is used as one way to create the unique lung surfactant DPCC (see p. 204) and to insure that carbon 2 of PI (and sometimes of PC) is bound to arachidonic acid. [Note: Barth syndrome, a rare X-linked disorder characterized by cardiomyopathy, muscle weakness, and neutropenia, is the result of defects in cardiolipin remodeling.]
Figure 17.11 Degradation of glycerophospholipids by phospholipases. PIP2 = phosphatidylinositol 4,5-bisphosphate; R1 and R2 = fatty acids; X = an alcohol.
B. Sphingomyelin
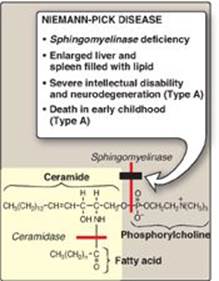
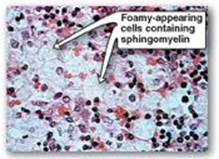
Sphingomyelin is degraded by sphingomyelinase, a lysosomal enzyme that hydrolytically removes phosphorylcholine, leaving a ceramide. The ceramide is, in turn, cleaved by ceramidase into sphingosine and a free fatty acid (Figure 17.12). [Note: The ceramide and sphingosine released regulate signal transduction pathways, in part by influencing the activity of protein kinase C and, thus, the phosphorylation of its protein substrates. They also promote apoptosis.] Niemann-Pick disease (Types A and B) is an autosomal-recessive disease caused by the inability to degrade sphingomyelin due to a deficiency of sphingomyelinase, a type of phospholipase C. In the severe infantile form (Type A, which shows less than 1% of normal enzymic activity), the liver and spleen are the primary sites of lipid deposits and are, therefore, greatly enlarged. The lipid consists primarily of the sphingomyelin that cannot be degraded (Figure 17.13). Infants with this lysosomal storage disease experience rapid and progressive neurodegeneration as a result of deposition of sphingomyelin in the CNS, and they die in early childhood. A less severe variant (Type B, which shows 5% or more of normal activity) with a later age of onset and a longer survival time causes little to no damage to neural tissue, but lungs, spleen, liver, and bone marrow are affected, resulting in a chronic form of the disease. Although Niemann-Pick disease occurs in all ethnic groups, Type A occurs with greater frequency in the Ashkenazi Jewish population.
Figure 17.12 Degradation of sphingomyelin. [Note: Type B is the nonneuropathic form. It has a later age of onset and a longer survival time than Type A.]
V. OVERVIEW OF GLYCOLIPIDS
Glycolipids are molecules that contain both carbohydrate and lipid components. Like the phospholipid sphingomyelin, glycolipids are derivatives of ceramides in which a long-chain fatty acid is attached to the amino alcohol sphingosine. They are, therefore, more precisely called glycosphingolipids. [Note: Ceramides, then, are the precursors of both phosphorylated and glycosylated sphingolipids.] Like the phospholipids, glycosphingolipids are essential components of all membranes in the body, but they are found in greatest amounts in nerve tissue. They are located in the outer leaflet of the plasma membrane, where they interact with the extracellular environment. As such, they play a role in the regulation of cellular interactions (for example, adhesion and recognition), growth, and development.
Membrane glycosphingolipids associate with cholesterol and GPI-anchored proteins to form lipid rafts, laterally mobile microdomains of the plasma membrane that function to organize and regulate signaling and trafficking functions of membranes.
Figure 17.13 Accumulation of lipids in spleen cells from a patient with Niemann-Pick disease.
Glycosphingolipids are antigenic and are the source of blood group antigens, various embryonic antigens specific for particular stages of fetal development, and some tumor antigens. [Note: The carbohydrate portion of a glycolipid is the antigenic determinant.] They also are used as cell surface receptors for cholera and tetanus toxins as well as for certain viruses and microbes. Genetic disorders associated with an inability to properly degrade the glycosphingolipids result in lysosomal accumulation of these compounds. [Note: Changes in the carbohydrate portion of glycosphingolipids (and glycoproteins) are characteristic of transformed cells (cells with dysregulated growth).]
Figure 17.14 Structure of a neutral glycosphingolipid, galactocerebroside. (![]() is a hydrophobic hydrocarbon chain.)
is a hydrophobic hydrocarbon chain.)

VI. STRUCTURE OF GLYCOSPHINGOLIPIDS
The glycosphingolipids differ from sphingomyelin in that they do not contain phosphate, and the polar head function is provided by a monosaccharide or oligosaccharide attached directly to the ceramide by an O-glycosidic bond (Figure 17.14). The number and type of carbohydrate moieties present determine the type of glycosphingolipid.
A. Neutral glycosphingolipids
The simplest neutral (uncharged) glycosphingolipids are the cerebrosides. These are ceramide monosaccharides that contain either a molecule of galactose (forming ceramide-galactose or galactocerebroside, the most common cerebroside found in myelin, as shown in Figure 17.14) or glucose (forming ceramide-glucose or glucocerebroside, which serves primarily as an intermediate in the synthesis and degradation of the more complex glycosphingolipids). [Note: Members of a group of galacto- or glucocerebrosides may also differ from each other in the type of fatty acid attached to the sphingosine.] As their name implies, cerebrosides are found predominantly in the brain and peripheral nervous tissue, with high concentrations in the myelin sheath. Ceramide oligosaccharides (or globosides) are produced by attaching additional monosaccharides to a glucocerebroside, for example, ceramide-glucose-galactose (also known as lactosylceramide). The additional monosaccharides can include substituted sugars such as N-acetylgalactosamine.
B. Acidic glycosphingolipids
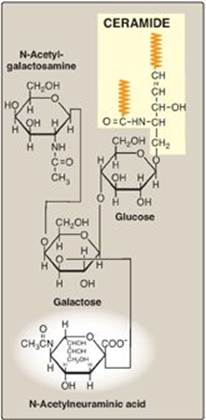
Acidic glycosphingolipids are negatively charged at physiologic pH. The negative charge is provided by N-acetylneuraminic acid ([NANA], a sialic acid, as shown in Figure 17.15) in gangliosides, or by sulfate groups in sulfatides.
1. Gangliosides: These are the most complex glycosphingolipids and are found primarily in the ganglion cells of the CNS, particularly at the nerve endings. They are derivatives of ceramide oligosaccharides and contain one or more molecules of NANA. The notation for these compounds is G (for ganglioside) plus a subscript M, D, T, or Q to indicate whether there is one (mono), two (di), three (tri), or four (quatro) molecules of NANA in the ganglioside, respectively. Additional numbers and letters in the subscript designate the monomeric sequence of the carbohydrate attached to the ceramide. (See Figure 17.15 for the structure of GM2.) Gangliosides are of medical interest because several lipid storage disorders involve the accumulation of NANA-containing glycosphingolipids in cells (see Figure 17.20, p. 212).
Figure 17.15 Structure of the ganglioside GM2. (![]() is a hydrophobic hydrocarbon chain.)
is a hydrophobic hydrocarbon chain.)
2. Sulfatides: These sulfoglycosphingolipids are sulfated galactocerebrosides that are negatively charged at physiologic pH. Sulfatides are found predominantly in the brain and kidneys.
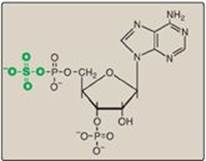
Figure 17.16 Structure of 3ʹ-phosphoadenosine-5ʹ-phosphosulfate.
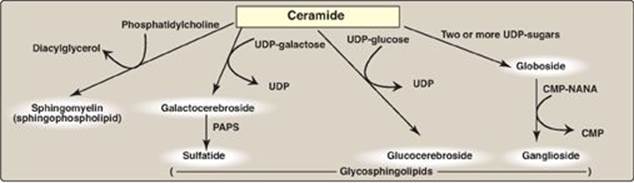
VII. SYNTHESIS AND DEGRADATION OF GLYCOSPHINGOLIPIDS
Synthesis of glycosphingolipids occurs primarily in the Golgi by sequential addition of glycosyl monomers transferred from UDP-sugar donors to the acceptor molecule. The mechanism is similar to that used in glycoprotein synthesis (see p. 166).
A. Enzymes involved in synthesis
The enzymes involved in the synthesis of glycosphingolipids are glycosyltransferases that are specific for the type and location of the glycosidic bond formed. [Note: These enzymes can recognize both glycosphingolipids and glycoproteins as substrates.]
B. Addition of sulfate groups
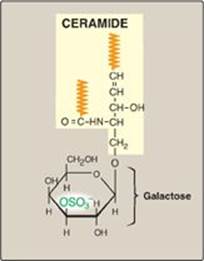
A sulfate group from the sulfate carrier 3ʹ-phosphoadenosine-5ʹ-phosphosulfate ([PAPS], Figure 17.16) is added by a sulfotransferase to the 3ʹ-hydroxyl group of the galactose in a galactocerebroside, forming the sulfatide galactocerebroside 3-sulfate (Figure 17.17). [Note: PAPS is also the sulfur donor in glycosaminoglycan synthesis (see p. 162) and steroid hormone catabolism (see p. 240).] An overview of the synthesis of sphingolipids is shown in Figure 17.18.
C. Degradation of glycosphingolipids
Glycosphingolipids are internalized by endocytosis as described for the glycosaminoglycans. All of the enzymes required for the degradative process are present in lysosomes, which fuse with the endocytotic vesicles. The lysosomal enzymes hydrolytically and irreversibly cleave specific bonds in the glycosphingolipid. As seen with the glycosaminoglycans (see p. 163) and glycoproteins (see p. 170), degradation is a sequential process following the rule “last on, first off,” in which the last group added during synthesis is the first group removed in degradation. [Note: Defects in the degradation of the polysaccharide chains in these three glycoconjugates, therefore, result in lysosomal storage diseases.]
Figure 17.17 Structure of galactocerebroside 3-sulfate. (![]() is a hydrophobic hydrocarbon chain.)
is a hydrophobic hydrocarbon chain.)
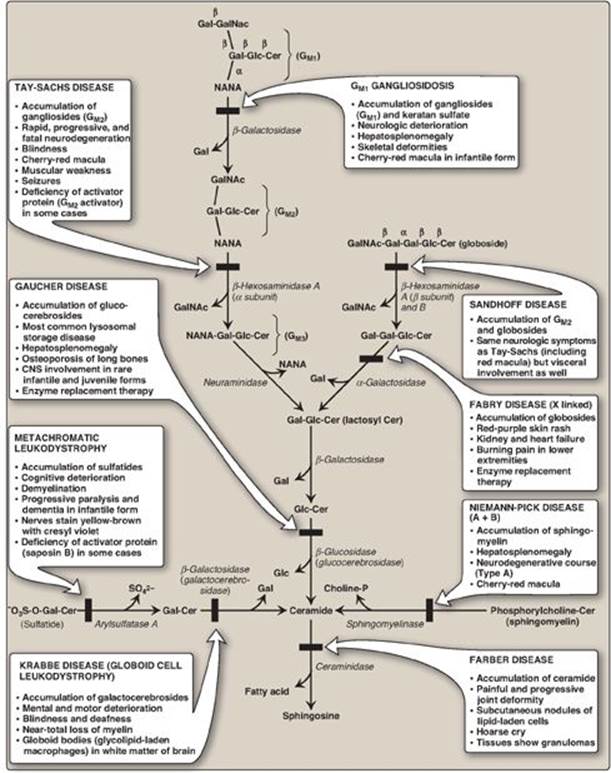
D. Sphingolipidoses
In a normal individual, synthesis and degradation of glycosphingolipids are balanced, so that the amount of these compounds present in membranes is constant. If a specific lysosomal acid hydrolase required for degradation is partially or totally missing, a sphingolipid accumulates. Lysosomal lipid storage diseases caused by these deficiencies are called sphingolipidoses. The result of a specific acid hydrolase deficiency may be seen dramatically in nerve tissue, where neurologic deterioration can lead to early death. Figure 17.20 provides an outline of the pathway of sphingolipid degradation and descriptions of some sphingolipidoses. [Note: Some sphingolipidoses can also result from defects in lysosomal activator proteins (for example, the saposins) that facilitate access of the hydrolases to short carbohydrate chains as degradation proceeds.]
Figure 17.18 Overview of sphingolipid synthesis. UDP = uridine diphosphate; CMP = cytidine monophosphate; NANA = N-acetylneuraminic acid; PAPS = 3ʹ-phosphoadenosine-5ʹ-phosphosulfate.
1. Common properties: A specific lysosomal hydrolytic enzyme is deficient in the classic form of each disorder. Therefore, usually only a single sphingolipid (the substrate for the deficient enzyme) accumulates in the involved organs in each disease. [Note: The rate of biosynthesis of the accumulating lipid is normal.] The disorders are progressive and, although many are fatal in childhood, extensive phenotypic variability is seen leading to the designation of different clinical types, such as Types A and B in Niemann-Pick disease. Genetic variability is also seen because a given disorder can be caused by any one of a variety of mutations within a single gene. The sphingolipidoses are autosomal-recessive diseases, except for Fabry disease, which is X linked. The incidence of the sphingolipidoses is low in most populations, except for Gaucher and Tay-Sachs diseases, which, like Niemann-Pick disease, show a high frequency in the Ashkenazi Jewish population. [Note: Tay-Sachs also has a high frequency in Irish American, French Canadian, and Louisiana Cajun populations.]
Figure 17.19 Aspirated bone marrow cells from a patient with Gaucher disease.
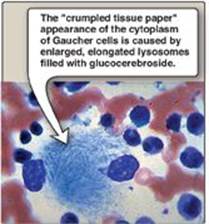
2. Diagnosis and treatment: A specific sphingolipidosis can be diagnosed by measuring enzyme activity in cultured fibroblasts or peripheral leukocytes or by analysis of DNA (see p. 473). Histologic examination of the affected tissue is also useful. [Note: Shell-like inclusion bodies are seen in Tay-Sachs, and a wrinkled tissue paper appearance of the cytosol is seen in Gaucher disease (Figure 17.19).] Prenatal diagnosis, using cultured amniocytes or chorionic villi, is available. Gaucher disease, in which macrophages become engorged with glucocerebroside, and Fabry disease, in which globosides accumulate in the vascular endothelial lysosomes of the brain, heart, kidneys, and skin, are treated by recombinant human enzyme replacement therapy, but the monetary cost is extremely high. Gaucher has also been treated by bone marrow transplantation (because macrophages are derived from hematopoietic stem cells) and by substrate reduction therapy through pharmacologic reduction of glucosylcer-amide, the substrate for the deficient enzyme.
Figure 17.20 Degradation of sphingolipids showing the lysosomal enzymes affected in related genetic diseases, the sphingolipidoses. All of the diseases are autosomal recessive except Fabry disease, which is X linked, and all can be fatal in early life. Cer = ceramide; Gal = galactose; Glc = glucose; GalNAc = N-acetylgalactosamine; NANA = N-acetylneuraminic acid; CNS = central nervous system. SO42- = sulfate.
VIII. EICOSANOIDS: PROSTAGLANDINS AND RELATED COMPOUNDS
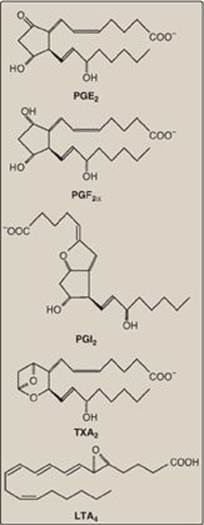
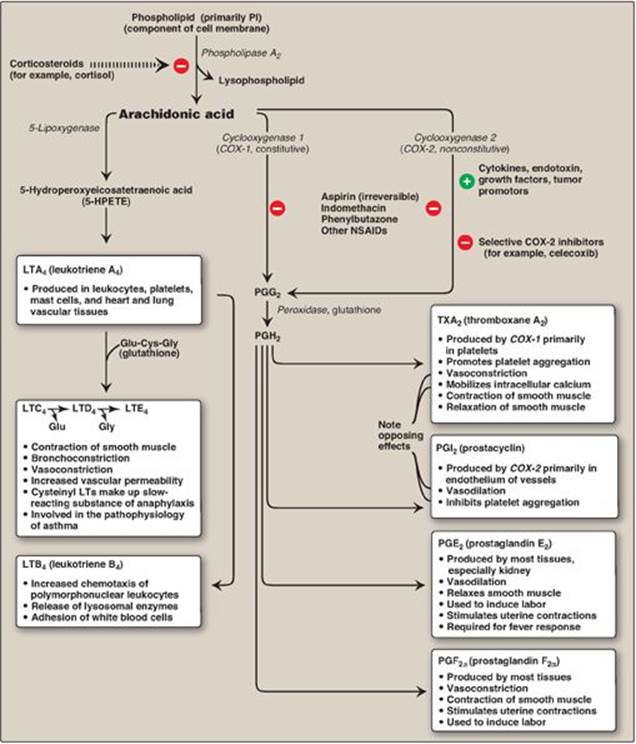
Prostaglandins, and the related compounds thromboxanes and leukotrienes, are collectively known as eicosanoids to reflect their origin from polyunsaturated fatty acids with 20 carbons (eicosa = 20). They are extremely potent compounds that elicit a wide range of responses, both physiologic (inflammatory response) and pathologic (hypersensitivity). They ensure gastric integrity and renal function, regulate smooth muscle contraction (intestine and uterus are key sites) and blood vessel diameter, and maintain platelet homeostasis. Although they have been compared to hormones in terms of their actions, eicosanoids differ from endocrine hormones in that they are produced in very small amounts in almost all tissues rather than in specialized glands. They also act locally rather than after transport in the blood to distant sites, as occurs with endocrine hormones such as insulin. Eicosanoids are not stored, and they have an extremely short half-life, being rapidly metabolized to inactive products. Their biologic actions are mediated by plasma membrane G protein–coupled receptors (see p. 94), which are different in different organ systems, and typically result in changes in cyclic adenosine monophosphate production. Examples of prostaglandins and related structures are shown in Figure 17.21.
A. Synthesis of prostaglandins and thromboxanes
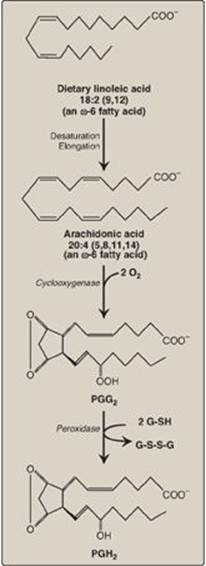
Arachidonic acid, an ω-6 fatty acid containing 20 carbons and four double bonds (an eicosatetraenoic fatty acid), is the immediate precursor of the predominant type of prostaglandins in humans (series 2 or those with two double bonds, as shown in Figure 17.22). It is derived by the elongation and desaturation of the essential fatty acid linoleic acid, also an ω-6 fatty acid. Arachidonic acid is incorporated into membrane phospholipids (typically PI) at carbon 2, from which it is released by phospholipase A2 in response to a variety of signals (Figure 17.23). [Note: Series 1 prostaglandins contain one double bond and are derived from an ω-6 eicosatrienoic fatty acid, dihomo-γ-linolenic acid, whereas series 3 contain three double bonds and are derived from eicosapentaenoic acid (EPA), an ω-3 fatty acid. See p. 363.]
Figure 17.21 Examples of prostaglandin structures. [Note: Prostaglandins are named as follows: PG plus a third letter (for example, A, D, E, or F), which designates the type and arrangement of functional groups in the molecule. The subscript number indicates the number of double bonds in the molecule. PGI2 is known as prostacyclin. Thromboxanes are designated by TX and leukotrienes by LT.]
1. Synthesis of PGH2: The first step in prostaglandin synthesis is the oxidative cyclization of free arachidonic acid to yield PGH2 by prostaglandin endoperoxide synthase (PGH synthase). This enzyme is an ER membrane-bound protein that has two catalytic activities: fatty acid cyclooxygenase (COX), which requires two molecules of O2, and peroxidase, which is dependent on reduced glutathione (see p. 148). PGH2 is converted to a variety of prostaglandins and thromboxanes, as shown in Figure 17.23, by cell-specific synthases.
a. Isozymes of PGH synthase: Two isozymes of PGH synthase, usually denoted as COX-1 and COX-2, are known. COX-1 is made constitutively in most tissues and is required for maintenance of healthy gastric tissue, renal homeostasis, and platelet aggregation. COX-2 is inducible in a limited number of tissues in response to products of activated immune and inflammatory cells. [Note: The increase in prostaglandin synthesis subsequent to the induction of COX-2 mediates the pain, heat, redness, and swelling of inflammation and the fever of infection.]
2. Inhibition of prostaglandin synthesis: The synthesis of prostaglandins can be inhibited by a number of unrelated compounds. For example, cortisol (a steroidal anti-inflammatory agent) inhibits phospholipase A2 activity (see Figure 17.23) and, therefore, the precursor of the prostaglandins, arachidonic acid, is not made available from membrane phospholipids. Aspirin, indomethacin, and phenylbutazone (all nonsteroidal anti-inflammatory drugs [NSAIDs]) inhibit both COX-1 and COX-2 and, thus, prevent the synthesis of the parent prostaglandin, PGH2. [Note: Systemic inhibition of COX-1, with subsequent damage to the stomach and the kidneys and impaired clotting of blood, is the basis of aspirin’s toxicity.] Aspirin (but not other NSAIDs) also induces synthesis of lipoxins and resolvins, lipid mediators with anti-inflammatory effects that are made from arachidonic acid and EPA, respectively. Inhibitors specific for COX-2 (the coxibs, for example, celecoxib) were designed to reduce pathologic inflammatory processes mediated by COX-2 while maintaining the physiologic functions of COX-1. However, their use has been associated with increased risk of heart attacks attacks, likely as a result of decreased PGI2 synthesis (see below), and some have been withdrawn from the market.
Figure 17.22 Oxidation and cyclization of arachidonic acid by the two catalytic activities (cyclooxygenase and peroxidase) of prostaglandin endoperoxide synthase. G-SH = reduced glutathione; G-S-S-G = oxidized glutathione.
B. Synthesis of leukotrienes
Arachidonic acid is converted to a variety of linear hydroperoxy (–OOH) acids by a separate pathway involving a family of lipoxygenases (LOXs). For example, 5-lipoxygenase converts arachidonic acid to 5-hydroperoxy-6,8,11,14 eicosatetraenoic acid ([5-HPETE], see Figure 17.23). 5-HPETE is converted to a series of leukotrienes containing four double bonds, the nature of the final products varying according to the tissue. Leukotrienes are mediators of allergic response and inflammation. Their synthesis is not inhibited by NSAIDs. [Note: Aspirin-induced asthma is a response to overproduction of leukotrienes with NSAID use.] Inhibitors of 5-lipoxygenase and leukotriene receptor antagonists are used in the treatment of asthma.
C. Role of prostaglandins in platelet homeostasis
Thromboxane A2 (TXA2) is produced by COX-1 in activated platelets. It promotes adherence and aggregation of circulating platelets and contraction of vascular smooth muscle, thereby promoting formation of blood clots (thrombi). (See online Chapter 34.) Prostacyclin (PGI2), produced by COX-2 in vascular endothelial cells, inhibits platelet aggregation and stimulates vasodilation and, so, impedes thrombogenesis. The opposing effects of TXA2and PGI2 limit thrombi formation to sites of vascular injury. [Note: Aspirin has an antithrombogenic effect. It inhibits TXA2 synthesis by COX-1 in platelets and PGI2 synthesis by COX-2 in endothelial cells through irreversible acetylation of these isozymes (Figure 17.24). The inhibition of COX-1 cannot be overcome in platelets, which lack nuclei. However, the inhibition of COX-2 can be overcome in endothelial cells, because they have a nucleus and, therefore, can generate more of the enzyme. This difference is the basis of low-dose aspirin therapy used to lower the risk of stroke and heart attacks by decreasing formation of thrombi.]
Figure 17.23 Overview of the biosynthesis and function of some important prostaglandins (PGs), leukotrienes (LTs), and a thromboxane (TX) from arachidonic acid. [Note: The arachidonic acid in the membrane phospholipid was derived from the ω-6 essential fatty acid, linoleic, also an ω-6 fatty acid.] PI = phosphatidylinositol; NSAIDs = nonsteroidal anti-inflammatory drugs; Glu = glutamate; Cys = cysteine; Gly = glycine.
IX. CHAPTER SUMMARY
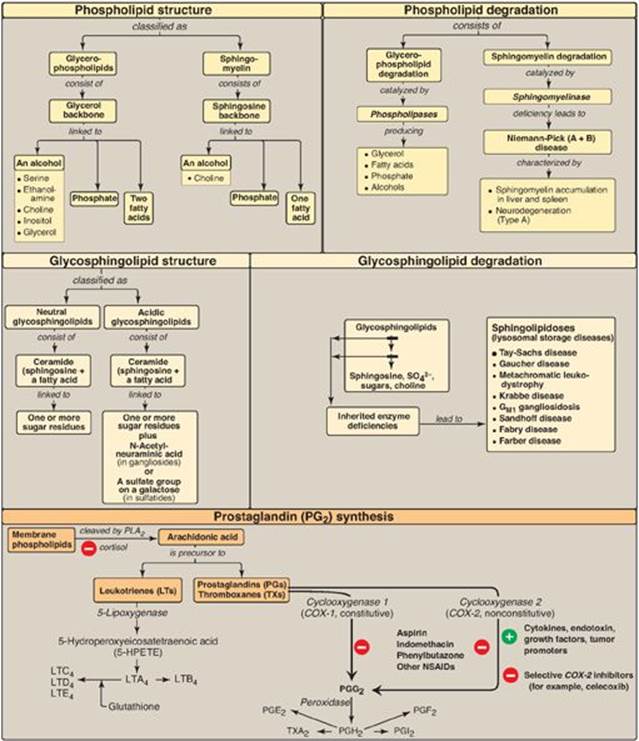
Phospholipids are polar, ionic compounds composed of an alcohol (for example, choline or ethanolamine) attached by a phosphodiester bond to either diacylglycerol (DAG), producing phosphatidylcholine or phosphatidylethanolamine, or to the amino alcohol sphingosine (Figure 17.25). Addition of a long-chain fatty acid to sphingosine produces a ceramide. Addition of a phosphorylcholine produces the phospholipid sphingomyelin. Phospholipids are the predominant lipids of cell membranes. Nonmembrane phospholipids serve as components of lung surfactant and bile. Dipalmitoylphosphatidylcholine, also called dipalmitoyl lecithin, is the major lipid component of lung surfactant. Insufficient surfactant production causes respiratory distress syndrome. Phosphatidylinositol (PI) serves as a reservoir for arachidonic acid in membranes. The phosphorylation of membrane-bound PI produces phosphatidylinositol 4,5-bisphosphate (PIP2). This compound is degraded by phospholipase C in response to the binding of a variety of neurotransmitters, hormones, and growth factors to membrane G protein–coupled receptors. The products of this degradation, inositol 1,4,5-trisphosphate (IP3) and DAG mediate the mobilization of intracellular calcium and the activation of protein kinase C, which act synergistically to evoke cellular responses. Specific proteins can be covalently attached via a carbohydrate bridge to membrane-bound phosphatidylinositol (glycosyl phosphatidylinositol, or GPI), forming a GPI anchor. A deficiency in the synthesis of GPI in hematopoietic cells results in a hemolytic disease, paroxysmal nocturnal hemoglobinuria. The degradation of phosphoglycerides is performed by phospholipases found in all tissues and pancreatic juice. Sphingomyelin is degraded to a ceramide plus phosphorylcholine by the lysosomal enzyme sphingomyelinase, a deficiency of which causes Niemann-Pick (A + B) disease. Glycosphingolipids are derivatives of ceramides to which carbohydrates have been attached. When one sugar molecule is added to the ceramide, a cerebroside is produced. If an oligosaccharide is added, a globoside is produced. If an acidic N-acetylneuraminic acid molecule is added, a ganglioside is produced. Glycosphingolipids are found predominantly in cell membranes of the brain and peripheral nervous tissue, with high concentrations in the myelin sheath. They are antigenic. Glycolipids are degraded in the lysosomes by acid hydrolases. A deficiency of any one of these enzymes produces a sphingolipidosis, in which a characteristic sphingolipid accumulates. Prostaglandins(PGs), thromboxanes (TXs), and leukotrienes (LTs) are produced in very small amounts in almost all tissues, act locally, and have an extremely short half-life. They serve as mediators of the inflammatory response. Arachidonic acid is the immediate precursor of the predominant class of PGs in humans (those with two double bonds). It is derived by the elongation and desaturation of the essential fatty acid linoleic acid and is stored in the membrane as a component of a phospholipid, generally PI. Arachidonic acid is released from the phospholipid by phospholipase A2 (inhibited by cortisol). Synthesis of the PGs and TXs begins with the oxidative cyclization of free arachidonic acid to yield PGH2 by prostaglandin endoperoxide synthase (PGH synthase), an endoplasmic reticulum membrane protein that has two catalytic activities: fatty acid cyclooxygenase (COX) and peroxidase. There are two isozymes of PGH synthase: COX-1 (constitutive) and COX-2 (nonconstitutive). Aspirin irreversibly inhibits both. Opposing effects of PGI2 and TXA2 limit clot formation. LTs are linear molecules produced from arachidonic acid by the 5-lipoxygenase pathway. They mediate allergic response and are not inhibited by aspirin or other NSAIDs.
Figure 17.24 Irreversible acetylation of cyclooxygenase (COX)-1 and COX-2 by aspirin.
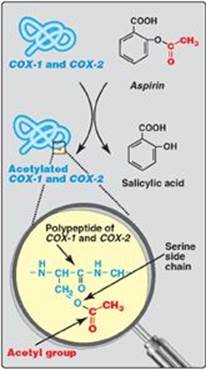
Figure 17.25 Key concept map for complex lipids. PLA2 = phospholipase A2; SO42- = sulfate ion; NSAIDs = nonsteroidal anti-inflammatory drugs.
Study Questions
Choose the ONE best answer.
17.1 Aspirin-induced asthma (AIA) is a severe reaction to nonsteroidal anti-inflammatory drugs (NSAIDs) characterized by bronchoconstriction 30 minutes to several hours after ingestion. Which of the following statements best explains the symptoms seen in patients with AIA? NSAIDs:
A. inhibit the activity of the cystic fibrosis transmembrane conductance regulator protein, resulting in thickened secretions that block airways.
B. inhibit cyclooxygenase but not lipoxygenase, resulting in the flow of arachidonic acid to leukotriene synthesis.
C. activate the cyclooxygenase activity of PGH synthase, resulting in increased synthesis of prostaglandins that promote vasodilation.
D. activate phospholipases, resulting in decreased amounts of dipalmytoylphosphatidylcholine and alveolar collapse (atelectasis).
Correct answer = B. Nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit cyclooxygenase but not lipoxygenase, so any arachidonic acid available is used for the synthesis of bronchoconstricting leukotrienes. NSAIDs have no effect on the cystic fibrosis transmembrane conductance regulator protein protein, defects in which are the cause of cystic fibrosis. Steroids, not NSAIDs, inhibit phospholipase A2. Cyclooxygenase is inhibited by NSAIDs, not activated. NSAIDs have no effect on phospholipases.
17.2 An infant, born at 28 weeks of gestation, rapidly gave evidence of respiratory distress. Clinical laboratory and imaging (X-ray) results supported the diagnosis of infant respiratory distress syndrome. Which of the following statements about this syndrome is true?
A. It is unrelated to the baby’s premature birth.
B. It is a consequence of too few type II pneumocytes.
C. The lecithin/sphingomyelin ratio in the amniotic fluid is likely to be greater than two.
D. The concentration of dipalmitoylphosphatidylcholine in the amniotic fluid would be expected to be lower than that of a full-term baby.
E. It is an easily treated disorder with low mortality.
F. It is treated by administering surfactant to the mother just before she gives birth.
Correct answer = D. Dipalmitoylphosphatidylcholine (DPPC, or dipalmitoyl lecithin) is the lung surfactant found in mature, healthy lungs. Respiratory distress syndrome (RDS) can occur in lungs that make too little of this compound. If the lecithin/sphingomyelin ratio in amniotic is greater than two, a newborn’s lungs are considered to be sufficiently mature (premature lungs would be expected to have a ratio lower than two). The RDS would not be due to too few type II pneumocytes, which would simply be secreting sphingomyelin rather than DPPC at 28 weeks of gestation. The mother is given a glucocorticoid, not surfactant, prior to giving birth. Surfactant would be administered to the baby postnatally to reduce surface tension.
17.3 A 10-year-old boy was evaluated for burning sensations in his feet and clusters of small, red-purple spots on his skin. Laboratory studies revealed protein in his urine. Enzymic analysis revealed a deficiency of α-galactosidase, and enzyme replacement therapy was recommended. The most likely diagnosis is:
A. Fabry disease.
B. Farber disease.
C. Gaucher disease.
D. Krabbe disease.
E. Niemann-Pick disease.
Correct answer = A. Fabry disease, a deficiency of α-galactosidase, is the only X-linked sphingolipidosis. It is characterized by pain in the extremities, a red-purple skin rash, and kidney and cardiac complications. Protein in his urine indicates kidney damage. Enzyme replacement therapy is available.
17.4 Current medical advice for individuals experiencing chest pain is to call emergency medical services and chew a regular-strength, noncoated aspirin. What is the basis for recommending aspirin?
Aspirin has an antithrombogenic effect: It prevents formation of blood clots that could occlude heart vessels. Aspirin inhibits thromboxane A2 synthesis by cyclooxygenase–1 in platelets through irreversible acetylation, thereby inhibiting platelet activation and vasoconstriction. Chewing a noncoated aspirin increases the rate of its absorption.