Lippincott’s Illustrated Reviews: Biochemistr, Sixth Edition (2014)
UNIT IV: Nitrogen Metabolism
Chapter 22. Nucleotide Metabolism
I. OVERVIEW
Ribonucleoside and deoxyribonucleoside phosphates (nucleotides) are essential for all cells. Without them, neither ribonucleic acid (RNA) nor deoxyribonucleic acid (DNA) can be produced, and, therefore, proteins cannot be synthesized or cells proliferate. Nucleotides also serve as carriers of activated intermediates in the synthesis of some carbohydrates, lipids, and conjugated proteins (for example, uridine diphosphate [UDP]-glucose and cytidine diphosphate [CDP]-choline) and are structural components of several essential coenzymes, such as coenzyme A, flavin adenine dinucleotide (FAD[H2]), nicotinamide adenine dinucleotide (NAD[H]), and nicotinamide adenine dinucleotide phosphate (NADP[H]). Nucleotides, such as cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), serve as second messengers in signal transduction pathways. In addition, nucleotides play an important role as “energy currency” in the cell. Finally, nucleotides are important regulatory compounds for many of the pathways of intermediary metabolism, inhibiting or activating key enzymes. The purine and pyrimidine bases found in nucleotides can be synthesized de novo or can be obtained through salvage pathways that allow the reuse of the preformed bases resulting from normal cell turnover. [Note: Little of the purines and pyrimidines supplied by diet are utilized and are degraded instead.]
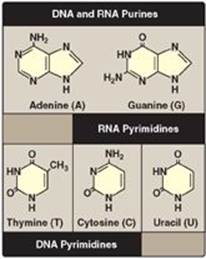
Figure 22.1 Purines and pyrimidines commonly found in DNA and RNA.
II. NUCLEOTIDE STRUCTURE
Nucleotides are composed of a nitrogenous base; a pentose monosaccharide; and one, two, or three phosphate groups. The nitrogen-containing bases belong to two families of compounds: the purines and the pyrimidines.
A. Purine and pyrimidine structures
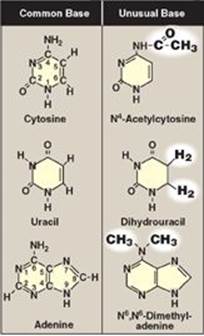
Both DNA and RNA contain the same purine bases: adenine (A) and guanine (G). Both DNA and RNA contain the pyrimidine cytosine (C), but they differ in their second pyrimidine base: DNA contains thymine (T), whereas RNA contains uracil (U). T and U differ in that only T has a methyl group (Figure 22.1). Unusual (modified) bases are occasionally found in some species of DNA and RNA (for example, in some viral DNA) and in transfer RNA (tRNA). Base modifications include methylation, glycosylation, acetylation, and reduction. Some examples of unusual bases are shown in Figure 22.2. [Note: The presence of an unusual base in a nucleotide sequence may aid in its recognition by specific enzymes or protect it from being degraded by nucleases.]
Figure 22.2 Examples of unusual bases.
B. Nucleosides
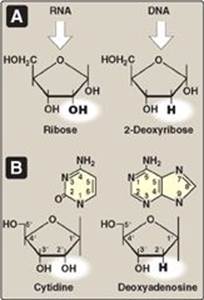
The addition of a pentose sugar to a base through a glycosidic bond produces a nucleoside. If the sugar is ribose, a ribonucleoside is produced, and if the sugar is 2-deoxyribose, a deoxyribonucleoside is produced (Figure 22.3A). The ribonucleosides of A, G, C, and U are named adenosine, guanosine, cytidine, and uridine, respectively. The deoxyribonucleosides of A, G, C, and T have the added prefix, “deoxy-” (for example, deoxyadenosine). [Note: The compound deoxythymidine is often simply called thymidine, with the “deoxy-” prefix being understood, because it is incorporated into DNA only.] The carbon and nitrogen atoms in the rings of the base and the sugar are numbered separately (Figure 22.3B). Note that the carbons in the pentose are numbered 1ʹ to 5. Thus, when the 5-carbon of a nucleoside (or nucleotide) is referred to, a carbon atom in the pentose, rather than an atom in the base, is being specified.
C. Nucleotides
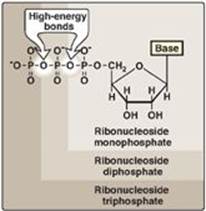
The addition of one or more phosphate groups to a nucleoside produces a nucleotide. The first phosphate group is attached by an ester linkage to the 5ʹ-OH of the pentose, forming a nucleoside 5ʹ-phosphate or a 5ʹ-nucleotide. The type of pentose is denoted by the prefix in the names “5ʹ-ribonucleotide” and “5ʹ-deoxyribonucleotide.” If one phosphate group is attached to the 5ʹ-carbon of the pentose, the structure is a nucleoside monophosphate, like adenosine monophosphate [AMP] also called adenylate). If a second or third phosphate is added to the nucleoside, a nucleoside diphosphate (for example, adenosine diphosphate [ADP] or triphosphate (for example, adenosine triphosphate [ATP]) results (Figure 22.4). The second and third phosphates are each connected to the nucleotide by a “high-energy” bond. [Note: The phosphate groups are responsible for the negative charges associated with nucleotides and cause DNA and RNA to be referred to as “nucleic acids.”]
Figure 22.3 A. Pentoses found in nucleic acids. B. Examples of the numbering systems for purine- and pyrimidinecontaining nucleosides.
III. SYNTHESIS OF PURINE NUCLEOTIDES
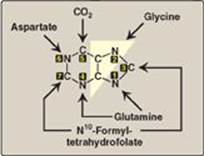
The atoms of the purine ring are contributed by a number of compounds, including amino acids (aspartate, glycine, and glutamine), CO2, and N10-formyltetrahydrofolate (Figure 22.5). The purine ring is constructed primarily in the liver by a series of reactions that add the donated carbons and nitrogens to a preformed ribose 5-phosphate. (See p. 147 for the synthesis of ribose 5-phosphate by the pentose phosphate pathway.)
A. Synthesis of 5-phosphoribosyl-1-pyrophosphate
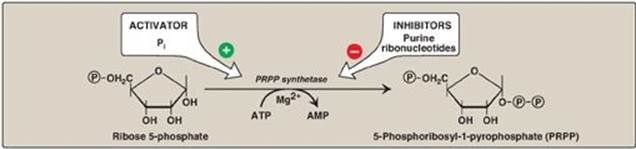
5-Phosphoribosyl-1-pyrophosphate (PRPP) is an “activated pentose” that participates in the synthesis and salvage of purines and pyrimidines. Synthesis of PRPP from ATP and ribose 5-phosphate is catalyzed by PRPP synthetase(Figure 22.6). This X-linked enzyme is activated by inorganic phosphate and inhibited by purine nucleotides (end-product inhibition). [Note: The sugar moiety of PRPP is ribose, and, therefore, ribonucleotides are the end products of de novo purine synthesis. When deoxyribonucleotides are required for DNA synthesis, the ribose sugar moiety is reduced (see p. 297).]
Figure 22.4 Ribonucleoside monophosphate, diphosphate, and triphosphate.
B. Synthesis of 5-phosphoribosylamine
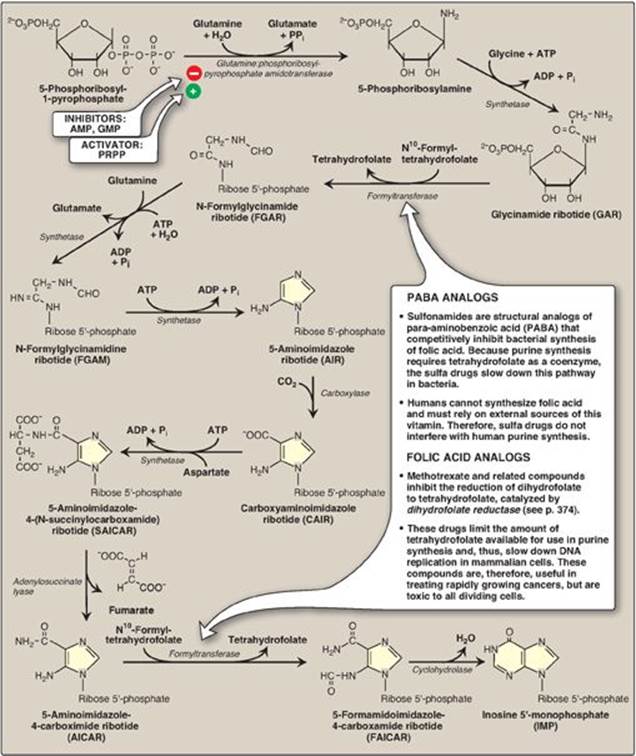
Synthesis of 5-phosphoribosylamine from PRPP and glutamine is shown in Figure 22.7. The amide group of glutamine replaces the pyrophosphate group attached to carbon 1 of PRPP. This is the committed step in purine nucleotide biosynthesis. The enzyme, glu-tamine:phosphoribosylpyrophosphate amidotransferase, is inhibited by the purine 5ʹ-nucleotides AMP and guanosine monophosphate ([GMP] also called guanylate), the end products of the pathway. The rate of the reaction is also controlled by the intracellular concentration of PRPP. [Note: The concentration of PRPP is normally far below the Michaelis constant (Km) for the amidotransferase. Therefore, any small change in the PRPP concentration causes a proportional change in rate of the reaction (see p. 59).]
Figure 22.5 Sources of the individual atoms in the purine ring. The order in which the atoms are added is shown by the numbers in the black boxes (see Figure 22.7).
C. Synthesis of inosine monophosphate, the “parent” purine nucleotide
The next nine steps in purine nucleotide biosynthesis leading to the synthesis of inosine monophosphate ([IMP] whose base is hypoxanthine) are illustrated in Figure 22.7. Four steps in this pathway require ATP as an energy source, and two steps in the pathway require N10-formyltetrahydrofolate as a one-carbon donor (see p. 267). [Note: Hypoxanthine is found in tRNA (see p. 437).]
Figure 22.6 Synthesis of PRPP, showing the activator and inhibitors of the reaction. [Note: This is not the committed step of purine synthesis because PRPP is used in other pathways.] ![]() = phosphate; Pi = inorganic phosphate; AMP = adenosine monophosphate.
= phosphate; Pi = inorganic phosphate; AMP = adenosine monophosphate.
D. Synthetic inhibitors of purine synthesis
Some synthetic inhibitors of purine synthesis (for example, the sulfonamides), are designed to inhibit the growth of rapidly dividing microorganisms without interfering with human cell functions (see Figure 22.7). Other purine synthesis inhibitors, such as structural analogs of folic acid (for example, methotrexate), are used pharmacologically to control the spread of cancer by interfering with the synthesis of nucleotides and, therefore, of DNA and RNA (see Figure 22.7).
Figure 22.7 De novo synthesis of purine nucleotides, showing the inhibitory effect of some structural analogs. AMP = adenosine monophosphate; ADP = adenosine diphosphate; GMP = guanosine monophosphate; PRPP = 5-phosphoribosyl- 1-pyrophosphate; Pi = inorganic phosphate; PPi = pyrophosphate.
Inhibitors of human purine synthesis are extremely toxic to tissues, especially to developing structures such as in a fetus, or to cell types that normally replicate rapidly, including those of bone marrow, skin, gastrointestinal (GI) tract, immune system, or hair follicles. As a result, individuals taking such anticancer drugs can experience adverse effects, including anemia, scaly skin, GI tract disturbance, immunodeficiencies, and hair loss.
E. Synthesis of adenosine and guanosine monophosphate
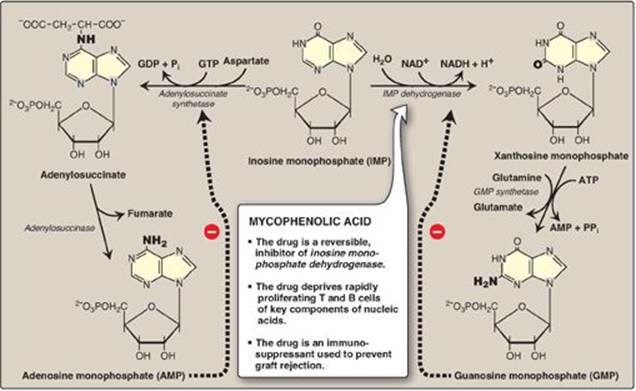
The conversion of IMP to either AMP or GMP uses a two-step, energy-requiring pathway (Figure 22.8). Note that the synthesis of AMP requires guanosine triphosphate (GTP) as an energy source, whereas the synthesis of GMP requires ATP. Also, the first reaction in each pathway is inhibited by the end product of that pathway. This provides a mechanism for diverting IMP to the synthesis of the purine present in lesser amounts. If both AMP and GMP are present in adequate amounts, the de novo pathway of purine synthesis is turned off at the amidotransferase step.
Figure 22.8 Conversion of IMP to AMP and GMP showing feedback inhibition. [Note: AMP is also called adenylate. GMP is also called guanylate.] NAD(H) = nicotinamide adenine dinucleotide; GDP = guanosine diphosphate; GTP = guanosine triphosphate; AMP = adenosine monophosphate; Pi = inorganic phosphate; PPi = pyrophosphate.
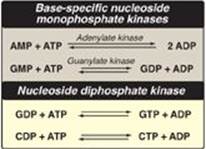
F. Conversion of nucleoside monophosphates to nucleoside diphosphates and triphosphates
Nucleoside diphosphates are synthesized from the corresponding nucleoside monophosphates by base-specific nucleoside monophosphate kinases (Figure 22.9). [Note: These kinases do not discriminate between ribose or deoxyribose in the substrate.] ATP is generally the source of the transferred phosphate because it is present in higher concentrations than the other nucleoside triphosphates. Adenylate kinase is particularly active in the liver and in muscle, where the turnover of energy from ATP is high. Its function is to maintain equilibrium among the adenine nucleotides (AMP, ADP, and ATP). Nucleoside diphosphates and triphosphates are interconverted by nucleoside diphosphate kinase, an enzyme that, unlike the monophosphate kinases, has broad substrate specificity.
Figure 22.9 Conversion of nucleoside monophosphates to nucleoside diphosphates and triphosphates.
AMP = adenosine monophosphate;
ADP = adenosine diphosphate;
GMP = guanosine monophosphate;
GDP = guanosine diphosphate;
GTP = guanosine triphosphate;
CDP = cytidine diphosphate;
CTP = cytidine triphosphate.
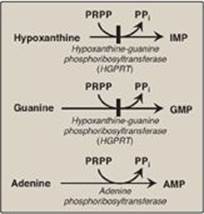
G. Salvage pathway for purines
Purines that result from the normal turnover of cellular nucleic acids, or the small amount that is obtained from the diet and not degraded, can be converted to nucleoside triphosphates and used by the body. This is referred to as the “salvage pathway” for purines. [Note: Salvage is particularly important in the brain.]
1. Salvage of purine bases to nucleotides: Two enzymes are involved: adenine phosphoribosyltransferase (APRT) and hypoxanthine-guanine phosphoribosyltransferase (HGPRT). Both enzymes use PRPP as the source of the ribose 5-phosphate group (Figure 22.10). The release of pyrophosphate and its subsequent hydrolysis by pyrophosphatase makes these reactions irreversible. [Note: Adenosine is the only purine nucleoside to be salvaged. It is phosphorylated to AMP by adenosine kinase.]
Figure 22.10 Salvage pathways of purine nucleotide synthesis. [Note: Virtually complete deficiency of HGPRT results in Lesch-Nyhan syndrome. Partial deficiencies of HGPRT are known. As the amount of functional enzyme increases, the severity of the symptoms decreases.]
IMP = inosine monophosphate;
GMP = guanosine monophosphate;
AMP = adenosine monophosphate;
PRPP = 5-phosphoribosyl-1- pyrophosphate;
PPi = pyrophosphate.
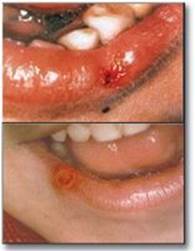
2. Lesch-Nyhan syndrome: Lesch-Nyhan is a rare, X-linked recessive disorder associated with a virtually complete deficiency of HGPRT. The deficiency results in an inability to salvage hypoxanthine or guanine, from which excessive amounts of uric acid, the end product of purine degradation, are then produced (see p. 298). In addition, the lack of this salvage pathway causes increased PRPP levels and decreased IMP and GMP levels. As a result, glutamine:phosphoribosylpyrophosphate amidotransferase (the regulated step in purine synthesis) has excess substrate and decreased inhibitors available, and de novo purine synthesis is increased. The combination of decreased purine reutilization and increased purine synthesis results in increased degradation of purines and the production of large amounts of uric acid, making Lesch-Nyhan a heritable cause of hyperuricemia. In patients with Lesch-Nyhan syndrome, the hyperuricemia frequently results in the formation of uric acid stones in the kidneys (urolithiasis) and the deposition of urate crystals in the joints (gouty arthritis) and soft tissues. In addition, the syndrome is characterized by motor dysfunction, cognitive deficits, and behavioral disturbances that include self-mutilation (for example, biting of lips and fingers) as shown in Figure 22.11).
IV. SYNTHESIS OF DEOXYRIBONUCLEOTIDES
The nucleotides described thus far all contain ribose (ribonucleotides). The nucleotides required for DNA synthesis, however, are 2ʹ-deoxyribonucleotides, which are produced from ribonucleoside diphosphates by the enzyme ribonucleotide reductase during the S-phase of the cell cycle (see p. 407). [Note: The same enzyme acts on pyrimidine ribonucleotides.]
Figure 22.11 Lesions on the lips of Lesch-Nyhan patients caused by self-mutilation.
A. Ribonucleotide reductase
Ribonucleotide reductase (ribonucleoside diphosphate reductase) is composed of two nonidentical dimeric subunits, R1 and R2, and is specific for the reduction of purine nucleoside diphosphates (ADP and GDP) and pyrimidine nucleoside diphosphates (CDP and UDP) to their deoxy forms (dADP, dGDP, dCDP, and dUDP). The immediate donors of the hydrogen atoms needed for the reduction of the 2ʹ-hydroxyl group are two sulfhydryl groups on the enzyme itself, which, during the reaction, form a disulfide bond (Figure 22.12).
Figure 22.12 Conversion of ribonucleotides to deoxyribonucleotides. NADP(H) = nicotinamide adenine dinucleotide phosphate; dATP = deoxyadenosine triphosphate.
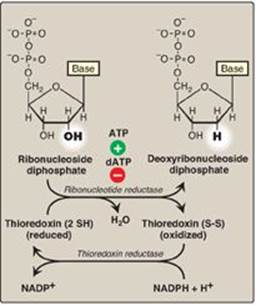
1. Regeneration of reduced enzyme: In order for ribonucleotide reductase to continue to produce deoxyribonucleotides, the disulfide bond created during the production of the 2ʹ-deoxy carbon must be reduced. The source of the reducing equivalents for this purpose is thioredoxin, a peptide coenzyme of ribonucleotide reductase. Thioredoxin contains two cysteine residues separated by two amino acids in the peptide chain. The two sulfhydryl groups of thioredoxin donate their hydrogen atoms to ribonucleotide reductase, forming a disulfide bond in the process (see p. 19).
2. Regeneration of reduced thioredoxin: Thioredoxin must be converted back to its reduced form in order to continue to perform its function. The necessary reducing equivalents are provided by NADPH + H+, and the reaction is catalyzed by thioredoxin reductase (see Figure 22.12).
B. Regulation of deoxyribonucleotide synthesis
Ribonucleotide reductase is responsible for maintaining a balanced supply of the deoxyribonucleotides required for DNA synthesis. To achieve this, the regulation of the enzyme is complex. In addition to the catalytic (active) site, there are allosteric sites on the enzyme involved in regulating its activity (Figure 22.13).
1. Activity sites: The binding of dATP to allosteric sites (known as the activity sites) on the enzyme inhibits the overall catalytic activity of the enzyme and, therefore, prevents the reduction of any of the four nucleoside diphosphates. This effectively prevents DNA synthesis and explains the toxicity of increased levels of dATP seen in conditions such as adenosine deaminase deficiency (see p. 301). In contrast, ATP bound to these sites activates the enzyme.
Figure 22.13 Regulation of ribonucleotide reductase. dATP = deoxyadenosine triphosphate; dTTP = deoxythymidine triphosphate; dGTP = deoxyguanosine triphosphate.
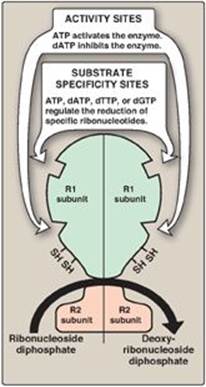
2. Substrate specificity sites: The binding of nucleoside triphosphates to additional allosteric sites (known as the substrate specificity sites) on the enzyme regulates substrate specificity, causing an increase in the conversion of different species of ribonucleotides to deoxyribonucleotides as they are required for DNA synthesis. For example, deoxythymidine triphosphate binding at the specificity sites causes a conformational change that allows reduction of GDP to dGDP at the catalytic site.
The drug hydroxyurea (hydroxycarbamide) inhibits ribonucleotide reductase, thereby inhibiting the generation of substrates for DNA synthesis. Hydroxyurea is an antineoplastic agent and is used in the treatment of cancers such as melanoma. Hydroxyurea is also used in the treatment of sickle cell disease (see p. 36). However, the increase in fetal hemoglobin seen with hydroxyurea is not due to its effect on ribonucleotide reductase.
V. DEGRADATION OF PURINE NUCLEOTIDES
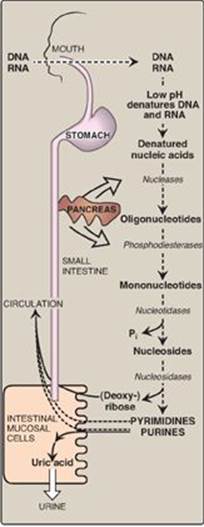
Degradation of dietary nucleic acids occurs in the small intestine, where a family of pancreatic enzymes hydrolyzes the nucleic acids to nucleotides. Inside the intestinal mucosal cells, purine nucleotides are sequentially degraded by specific enzymes to nucleosides and free bases, with uric acid as the end product of this pathway. [Note: Purine nucleotides from de novo synthesis are degraded in the liver primarily. The free bases are sent out from liver and salvaged by peripheral tissues.]
A. Degradation of dietary nucleic acids in the small intestine
Ribonucleases and deoxyribonucleases, secreted by the pancreas, hydrolyze dietary RNA and DNA to oligonucleotides. Oligonucleotides are further hydrolyzed by pancreatic phosphodiesterases, producing a mixture of 3ʹ- and 5ʹ-mononucleotides. In the intestinal mucosal cells, a family of nucleotidases removes the phosphate groups hydrolytically, releasing nucleosides that are further degraded by nucleosidases (nucleoside phosphorylases) to free bases plus (deoxy) ribose 1-phosphate. Dietary purine bases are not used to any appreciable extent for the synthesis of tissue nucleic acids. Instead, they are generally converted to uric acid in intestinal mucosal cells. Most of the uric acid enters the blood and is eventually excreted in the urine. A summary of this pathway is shown in Figure 22.14. [Note: Mammals other than primates express urate oxidase (uricase) which cleaves the purine ring, generating allantoin. Modified recombinant urate oxidase is now used clinically to lower urate levels.]
B Formation of uric acid
A summary of the steps in the production of uric acid and genetic diseases associated with deficiencies of specific degradative enzymes are shown in Figure 22.15. [Note: The bracketed numbers refer to specific reactions in the figure.]
[1] An amino group is removed from AMP to produce IMP by AMP deaminase or from adenosine to produce inosine (hypoxanthine-ribose) by adenosine deaminase.
[2] IMP and GMP are converted into their nucleoside forms (inosine and guanosine) by the action of 5ʹ-nucleotidase.
[3] Purine nucleoside phosphorylase converts inosine and guanosine into their respective purine bases, hypoxanthine and guanine. [Note: A mutase interconverts ribose 1- and ribose 5-phosphate.]
[4] Guanine is deaminated to form xanthine.
[5] Hypoxanthine is oxidized by xanthine oxidase to xanthine, which is further oxidized by xanthine oxidase to uric acid, the final product of human purine degradation. Uric acid is excreted primarily in the urine.
C. Diseases associated with purine degradation
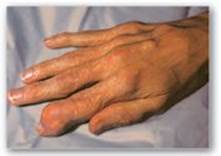
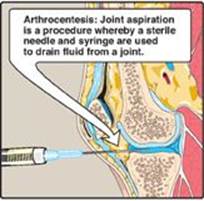
1. Gout: Gout is a disorder initiated by high levels of uric acid (the end product of purine catabolism) in blood (hyperuricemia), as a result of either the overproduction or underexcretion of uric acid. The hyperuricemia can lead to the deposition of monosodium urate (MSU) crystals in the joints and an inflammatory response to the crystals, causing first acute and then progressing to chronic gouty arthritis. Nodular masses of MSU crystals (tophi) may be deposited in the soft tissues, resulting in chronic tophaceous gout (Figure 22.16). Formation of uric acid stones in the kidney (urolithiasis) may also be seen. [Note: Hyperuricemia, while necessary, is not sufficient to cause gout, but gout is always preceded by hyperuricemia. Hyperuricemia is typically asymptomatic but may be indicative of comorbid conditions such as hypertension.] The definitive diagnosis of gout requires aspiration and examination of synovial fluid (Figure 22.17) from an affected joint (or material from a tophus) using polarized light microscopy to confirm the presence of needle-shaped MSU crystals (Figure 22.18).
a. Underexcretion of uric acid: In over 90% of individuals, hyperuricemia is caused by underexcretion of uric acid. Underexcretion can be primary, due to as-yet-unidentified inherent excretory defects, or secondary to known disease processes that affect how the kidney handles urate (for example, in lactic acidosis, lactate increases renal urate reabsorption, thereby decreasing its excretion) and to environmental factors such as the use of drugs (for example, thiazide diuretics) or exposure to lead (saturnine gout).
Figure 22.14 Digestion of dietary nucleic acids. [Note: Much of the metabolism of the mononucleotides occurs within the intestinal mucosal cells.] Pi = inorganic phosphate.
b. Overproduction of uric acid: A less common cause of hyperuricemia is from the overproduction of uric acid. Primary hyperuricemia is, for the most part, idiopathic (having no known cause). However, several identified mutations in the gene for X-linked PRPP synthetase result in the enzyme having an increased maximal velocity (Vmax) (see p. 58) for the production of PRPP, a lower Km (see p. 59) for ribose 5-phosphate, or a decreased sensitivity to purine nucleotides, its allosteric inhibitors (see p. 62). In each case, increased availability of PRPP increases purine production, resulting in elevated levels of plasma uric acid. Lesch-Nyhan syndrome (see p. 296) also causes hyperuricemia as a result of the decreased salvage of hypoxanthine and guanine and the subsequent increased availability of PRPP. Secondary hyperuricemia is typically the consequence of increased availability of purines (for example, in patients with myeloproliferative disorders or who are undergoing chemotherapy and so have a high rate of cell turnover). Hyperuricemia can also be the result of seemingly unrelated metabolic diseases, such as von Gierke disease (see Figure 11.8 on p. 130) or hereditary fructose intolerance (see p. 138).
Figure 22.15 The degradation of purine nucleotides to uric acid, illustrating some of the genetic diseases associated with this pathway. [Note: The numbers in brackets refer to the corresponding numbered citations in the text.] BMT = bone marrow transplantation; ERT = enzyme replacement therapy; Pi = inorganic phosphate.
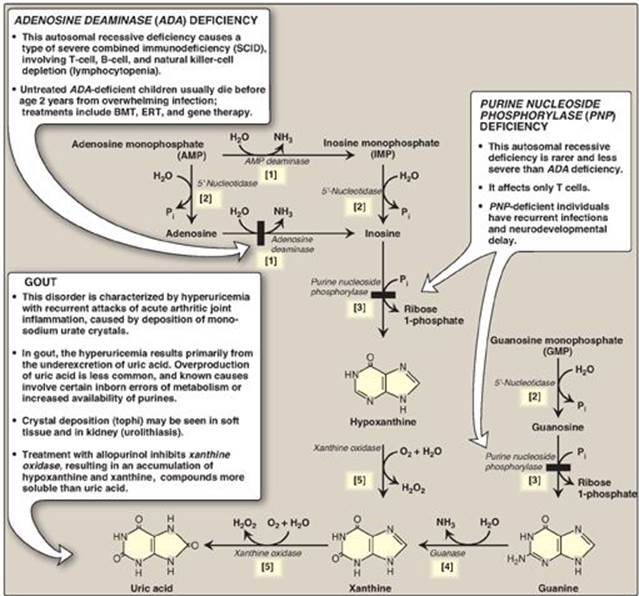
Figure 22.16 Tophaceous gout.
A diet rich in meat, seafood (particularly shellfish), and ethanol is associated with increased risk of gout, whereas a diet rich in low-fat dairy products is associated with a decreased risk.
c. Treatment of gout: Acute attacks of gout are treated with anti-inflammatory agents. Colchicine; steroidal drugs, such as prednisone; and nonsteroidal drugs, such as indomethacin, are used. [Note: Colchicine prevents formation of microtubules, thereby decreasing the movement of neutrophils into the affected area. Like the other anti-inflammatory drugs, it has no effect on uric acid levels.] Long-term therapeutic strategies for gout involve lowering the uric acid level below its saturation point (6.5 mg/dl), thereby preventing the deposition of urate crystals. Uricosuric agents, such as probenecid or sulfinpyrazone, that increase renal excretion of uric acid, are used in patients who are “underexcretors” of uric acid. Allopurinol, a structural analog of hypoxanthine, inhibits uric acid synthesis and is used in patients who are “overproducers” of uric acid. Allopurinol is converted in the body to oxypurinol, which inhibits xanthine oxidase (XO), resulting in an accumulation of hypoxanthine and xanthine (see Figure 22.15), compounds more soluble than uric acid and, therefore, less likely to initiate an inflammatory response. In patients with normal levels of HGPRT, the hypoxanthine can be salvaged, reducing the levels of PRPP and, therefore, de novo purine synthesis. Febuxostat, a non-purine inhibitor of XO, is also available. [Note: Uric acid levels in the blood normally are close to the saturation point. One reason for this may be the strong antioxidant effects of uric acid.]
Figure 22.17 Analysis of joint fluid can help to define causes of joint swelling or arthritis, such as infection, gout, and rheumatoid disease.
2. Adenosine deaminase deficiency: Adenosine deaminase (ADA) is expressed in a variety of tissues, but, in humans, lymphocytes have the highest activity of this cytoplasmic enzyme. A deficiency of ADA results in an accumulation of adenosine, which is converted to its ribonucleotide or deoxyribonucleotide forms by cellular kinases. As dATP levels rise, ribonucleotide reductase is inhibited, thereby preventing the production of all deoxyribose-containing nucleotides (see p. 297). Consequently, cells cannot make DNA and divide. [Note: The dATP and adenosine that accumulate in ADA deficiency lead to developmental arrest and apoptosis of lymphocytes.] In its most severe form, this autosomal-recessive disorder causes a type of severe combined immunodeficiency disease (SCID), involving a decrease in T cells, B cells, and natural killer cells. It is estimated that, in the United States, ADA deficiency accounts for approximately 14% of all cases of SCID. Treatments include bone marrow transplantation, enzyme replacement therapy, and gene therapy. Without appropriate treatment, children with this disorder usually die from infection by age 2 years. [Note: Purine nucleoside phosphorylase deficiency results in a less severe immunodeficiency primarily involving T cells.]
Figure 22.18 Gout can be diagnosed by the presence of negatively birefringent monosodium urate crystals in aspirated synovial fluid examined by polarized-light microscopy. Here, crystals are within polymorphonuclear leukocytes.

VI. PYRIMIDINE SYNTHESIS AND DEGRADATION
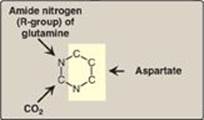
Unlike the synthesis of the purine ring, which is constructed on a preexisting ribose 5-phosphate, the pyrimidine ring is synthesized before being attached to ribose 5-phosphate, which is donated by PRPP. The sources of the atoms in the pyrimidine ring are glutamine, CO2, and aspartate (Figure 22.19).
Figure 22.19 Sources of the individual atoms in the pyrimidine ring.
A. Synthesis of carbamoyl phosphate
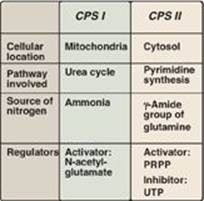
The regulated step of this pathway in mammalian cells is the synthesis of carbamoyl phosphate from glutamine and CO2, catalyzed by carbamoyl phosphate synthetase (CPS) II. CPS II is inhibited by uridine triphosphate (the end product of this pathway, which can be converted into the other pyrimidine nucleotides), and is activated by PRPP. [Note: Carbamoyl phosphate, synthesized by CPS I, is also a precursor of urea (see p. 253). Defects in ornithine transcarbamylase of the urea cycle promote pyrimidine synthesis due to increased availability of carbamoyl phosphate. A comparison of the two enzymes is presented in Figure 22.20.]
Figure 22.20 Summary of the differences between carbamoyl phosphate synthetase (CPS) I and II. PRPP = 5-phosphoribosyl-1-pyrophosphate; UTP = uridine triphosphate.
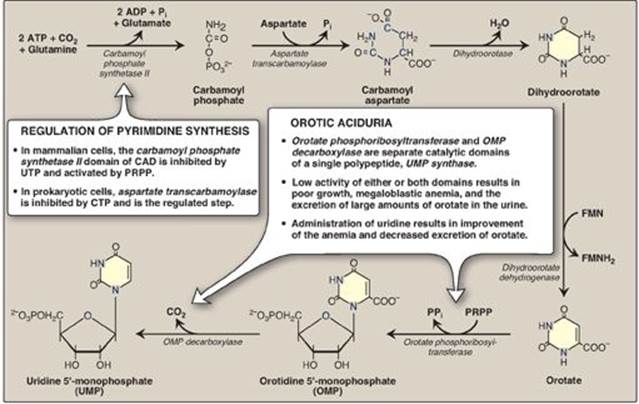
B. Synthesis of orotic acid
The second step in pyrimidine synthesis is the formation of carbamoylaspartate, catalyzed by aspartate transcarbamoylase. The pyrimidine ring is then closed by dihydroorotase. The resulting dihydroorotate is oxidized to produce orotic acid (orotate) as shown in Figure 22.21. The enzyme that produces orotate, dihydroorotate dehydrogenase, is a flavoprotein associated with the inner mitochondrial membrane. All other enzymes in pyrimidine biosynthesis are cytosolic. [Note: The first three enzymic activities in this pathway (CPS II, aspartate transcarbamoylase, and dihydroorotase) are actually three different catalytic domains of a single polypeptide known as CAD from the first letter in the name of each domain. (See p. 19 for a discussion of domains.) This is an example of a multfunctional or multicatalytic polypeptide that facilitates the ordered synthesis of an important compound. Synthesis of the purine nucleotide IMP also involves multifunctional proteins.]
C. Formation of a pyrimidine nucleotide
The completed pyrimidine ring is converted to the nucleotide orotidine monophosphate (OMP) in the second stage of pyrimidine nucleotide synthesis (see Figure 22.21). PRPP is again the ribose 5-phosphate donor. The enzyme orotate phosphoribosyltransferase produces OMP and releases pyrophosphate, thereby making the reaction biologically irreversible. [Note: Both purine and pyrimidine synthesis require glutamine, aspartic acid, and PRPP as essential precursors.] OMP, the parent pyrimidine mononucleotide, is converted to uridine monophosphate (UMP) by orotidylate decarboxylase, which removes the carboxyl group. Orotate phosphoribosyltransferase and orotidylate decarboxylase are also catalytic domains of a single polypeptide chain called UMP synthase. Orotic aciduria (a very rare disorder) may be caused by a deficiency of one or both activities of this bifunctional enzyme, resulting in orotic acid in the urine (see Figure 22.21). UMP is sequentially phosphorylated to UDP and UTP. [Note: The UDP is a substrate for ribonucleotide reductase, which generates dUDP. The dUDP is phosphorylated to dUTP, which is rapidly hydrolyzed to dUMP by UTP diphosphatase (dUTPase). dUTPase, then, plays an important role in reducing availability of dUTP as a substrate for DNA synthesis, thereby preventing erroneous incorporation of uracil into DNA.]
Figure 22.21 De novo pyrimidine synthesis. ADP = adenosine diphosphate; Pi = inorganic phosphate; FMN(H2) = flavin mononucleotide; CTP = cytidine triphosphate; PRPP = 5-phosphoribosyl-1-pyrophosphate; PPi = pyrophosphate.
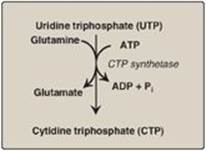
D. Synthesis of cytidine triphosphate
Cytidine triphosphate (CTP) is produced by amination of UTP by CTP synthetase (Figure 22.22), with glutamine providing the nitrogen. [Note: Some CTP is dephosphorylated to cytidine diphosphate (CDP), which is a substrate for ribonucleotide reductase. The dCDP product can be phosphorylated to dCTP for DNA synthesis or dephosphoprylated to dCMP that is deaminated to dUMP.]
Figure 22.22 Synthesis of CTP from UTP. [Note: CTP, required for RNA synthesis, is converted to dCTP for DNA synthesis.] ADP = adenosine diphosphate; Pi = inorganic phosphate.
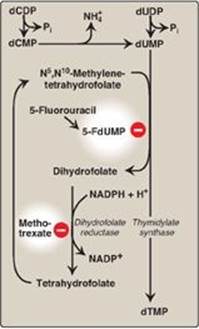
E. Synthesis of deoxythymidine monophosphate
dUMP is converted to deoxythymidine monophosphate (dTMP) by thymidylate synthase, which uses N5,N10-methylene tetrahydrofolate as the source of the methyl group (see p. 267 for a discussion of this coenzyme). This is an unusual reaction in that tetrahydrofolate (THF) contributes not only a one-carbon unit but also two hydrogen atoms from the pteridine ring, resulting in the oxidation of THF to dihydrofolate ([DHF] Figure 22.23). Inhibitors of thymidylate synthase include thymine analogs such as 5-fluorouracil, which serve as antitumor agents. 5-Fluorouracil is metabolically converted to 5-fluorodeoxyuridine monophosphate (5-FdUMP), which becomes permanently bound to the inactivated thymidylate synthase, making the drug a “suicide” inhibitor (see p. 60). DHF can be reduced to THF by dihydrofolate reductase (see Figure 28.3, p. 374), an enzyme that is inhibited by folate analogs such as methotrexate. By decreasing the supply of THF, these drugs not only inhibit purine synthesis (see Figure 22.7), but, by preventing methylation of dUMP to dTMP, they also decrease the availability of this essential component of DNA. DNA synthesis is inhibited and cell growth slowed. Thus, these drugs are used to decrease the growth rate of cancer cells. [Note: Acyclovir (a purine analog) and AZT (3ʹ-azido-3ʹ-deoxythymidine, a pyrimidine analog) are used to treat infections of herpes simplex virus and human immunodeficiency virus, respectively. Each inhibits the viral DNA polymerase.]
Figure 22.23 Synthesis of dTMP from dUMP, illustrating sites of action of antineoplastic drugs.
F. Degradation and salvage of pyrimidines
Unlike the purine ring, which is not cleaved in humans, the pyrimidine ring is opened and degraded to highly soluble products, β-alanine (from the degradation of CMP and UMP) and β-aminoisobutyrate (from TMP degradation), with the production of NH3 and CO2. Pyrimidine bases can be salvaged to nucleosides, which are phosphorylated to nucleotides. However, their high solubility makes pyrimidine salvage less significant clinically than purine salvage. [Note: The salvage of pyrimidine nucleosides is the basis for using uridine in the treatment of hereditary orotic aciduria.]
VII. CHAPTER SUMMARY
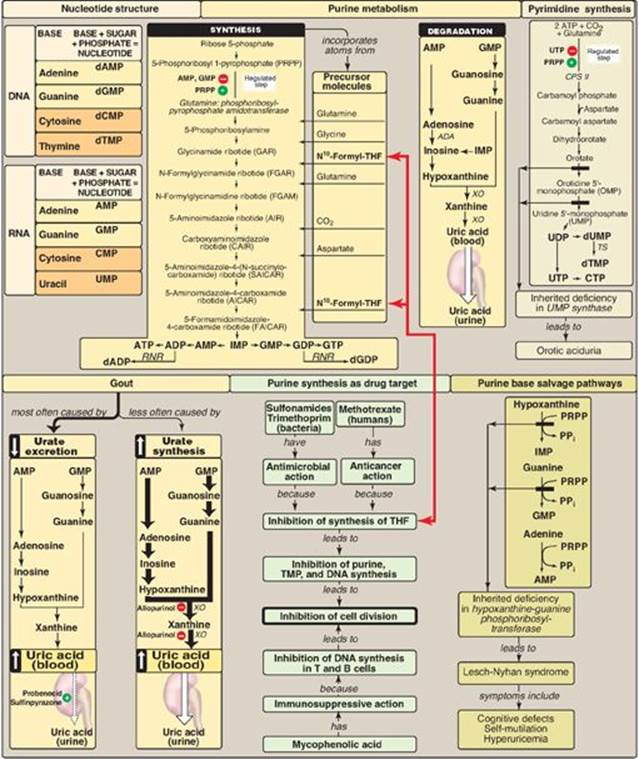
Nucleotides are composed of a nitrogenous base (adenine = A, guanine = G, cytosine = C, uracil = U, and thymine = T); a pentose; and one, two, or three phosphate groups (Figure 22.24). A and G are purines, and C, U, and T are pyrimidines. If the sugar is ribose, the nucleotide is a ribonucleoside phosphate (for example, adenosine monophosphate [AMP]), and it can have several functions in the cell, including being a component of RNA. If the sugar is deoxyribose, the nucleotide is a deoxyribonucleoside phosphate (for example, deoxyAMP), and will be found almost exclusively as a component of DNA. The committed step in purine synthesis uses 5-phosphoribosyl-1-pyrophosphate ([PRPP], an “activated pentose” that provides the ribose-phosphate group for de novo purine and pyrimidine synthesis and salvage) and nitrogen from glutamine to produce phosphoribosyl amine. The enzyme is glutamine:PRPP amidotransferase and is inhibited by AMP and guanosine monophosphate ([GMP] the end products of the pathway) and activated by PRPP. Purine nucleotides can also be produced from preformed purine bases by using salvage reactions catalyzed by adenine phosphoribosyltransferase (APRT) and hypoxanthine-guanine phosphoribosyltransferase (HGPRT). A near-total deficiency of HGPRT causes Lesch-Nyhan syndrome, a severe, heritable form of hyperuricemia accompanied by compulsive self-mutilation. All deoxyribonucleotides are synthesized from ribonucleotides by the enzyme ribonucleotide reductase. This enzyme is highly regulated (for example, it is strongly inhibited by deoxyadenosine triphosphate [dATP], a compound that is overproduced in bone marrow cells in individuals with adenosine deaminase deficiency). This syndrome causes severe combined immunodeficiency disease. The end product of purine degradation is uric acid, a compound whose overproduction or undersecretion causes hyperuricemia that, if accompanied by the deposition of monosodium urate crystals in joints and soft tissues and an inflammatory response to those crystals, results in gout. The first step in pyrimidine synthesis, the production of carbamoyl phosphate by carbamoyl phosphate synthetase II, is the regulated step in this pathway (it is inhibited by uridine triphosphate [UTP] and activated by PRPP). The UTP produced by this pathway can be converted to cytidine triphosphate. Deoxyuridine monophosphate can be converted to deoxythymidine monophosphate using thymidylate synthase, an enzyme targeted by anticancer drugs such as 5-fluorouracil. The regeneration of tetrahydrofolate from dihydrofolate produced in the thymidylate synthase reaction requires dihydrofolate reductase, an enzyme targeted by the drug, methotrexate. Pyrimidine degradation results in soluble products.
Figure 22.24 Key concept map for nucleotide metabolism. THF = tetrahydrofolate; ADA= adenosine deaminase; XO = xanthine oxidase; TS = thymidylate synthase; RNR = ribonucleotide reductase; CPS II = carbamyl phosphate synthetase II; AMP = adenosine monophosphate; GMP = guanosine monophosphate; CMP = cytidine monophosphate; TMP = thymidine monophosphate; IMP = inosine monophosphate; d = deoxy; PPi = pyrophosphate.
Study Questions
Choose the ONE correct answer.
22.1 Azaserine, a drug with research applications, inhibits glutamine-dependent enzymes. Incorporation of which of the ring nitrogens (N) in the generic purine structure shown would most likely be affected by azaserine?
A. 1
B. 3
C. 7
D. 9
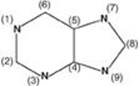
Correct answer = D. The nitrogen (N) at position 9 is supplied by glutamine in the first step of purine de novo synthesis, and its incorporation would be affected by azaserine. The N at position 1 is supplied by aspartate and at position 7 by glycine. The N at position 3 is also supplied by glutamine, but azaserine would have inhibited purine synthesis prior to this step.
22.2 A 42-year-old male patient undergoing radiation therapy for prostate cancer develops severe pain in the metatarsal phalangeal joint of his right big toe. Monosodium urate crystals are detected by polarized light microscopy in fluid obtained from this joint by arthrocentesis. This patient’s pain is directly caused by the overproduction of the end product of which of the following metabolic pathways?
A. De novo pyrimidine biosynthesis
B. Pyrimidine degradation
C. De novo purine biosynthesis
D. Purine salvage
E. Purine degradation
Correct answer = E. The patient’s pain is caused by gout, resulting from an inflammatory response to the crystallization of excess urate in his joints. Radiation therapy caused cell death, with degradation of nucleic acids and their constituent purines. Uric acid, the end product of purine degradation, is a relatively insoluble compound that can cause gout (and kidney stones). Pyrimidine metabolism is not associated with uric acid production. Overproduction of purines can indirectly result in hyperuricemia. Purine salvage decreases uric acid production.
22.3 Which one of the following enzymes of nucleotide metabolism is correctly paired with its pharmacologic inhibitor?
A. Dihydrofolate reductase—methotrexate
B. Inosine monophosphate dehydrogenase—hydroxyurea
C. Ribonucleotide reductase—5-fluorouracil
D. Thymidylate synthase—allopurinol
E. Xanthine oxidase—probenecid
Correct answer = A. Methotrexate interferes with folate metabolism by acting as a competitive inhibitor of the enzyme dihydrofolate reductase. This starves cells for tetrahydrofolate and makes them unable to synthesize purines and thymidine monophosphate. Inosine monophosphate dehydrogenase is inhibited by mycophenolic acid. Ribonucleotide reductase is inhibited by hydroxyurea. Thymidylate synthase is inhibited by 5-fluorouracil. Xanthine oxidase is inhibited by allopurinol. Probenecid increases renal excretion of urate but does not inhibit its production.
22.4 A 1-year-old female patient is lethargic, weak, and anemic. Her height and weight are low for her age. Her urine contains an elevated level of orotic acid. Activity of uridine monophosphate synthase is low. Administration of which of the following is most likely to alleviate her symptoms?
A. Adenine
B. Guanine
C. Hypoxanthine
D. Thymidine
E. Uridine
Correct answer = E. The elevated excretion of orotic acid and low activity of uridine monophosphate (UMP) synthase indicate that the patient has orotic aciduria, a rare genetic disorder affecting de novo pyrimidine synthesis. Deficiencies in one or both catalytic domains of UMP synthase leave the patient unable to synthesize pyrimidines. Uridine, a pyrimidine nucleoside, is a useful treatment because it bypasses the missing activities and can be salvaged to UMP, which can be converted to all the other pyrimidines. Although thymidine is a pyrimidine nucleoside, it cannot be converted to other pyrimidines. Hypoxanthine, guanine, and adenine are all purine bases and cannot be converted to pyrimidines.
22.5 What laboratory test would help in distinguishing an orotic aciduria caused by ornithine transcarbamylase deficiency from that caused by uridine monophosphate synthase deficiency?
Blood ammonia level would be expected to be elevated in ornithine transcarbamylase deficiency but not in uridine monophosphate synthase deficiency.