Lippincott’s Illustrated Reviews: Biochemistr, Sixth Edition (2014)
UNIT VI: Storage and Expression of Genetic Information
Chapter 29. DNA Structure, Replication, and Repair
I. OVERVIEW
Nucleic acids are required for the storage and expression of genetic information. There are two chemically distinct types of nucleic acids: deoxyribonucleic acid (DNA) and ribonucleic acid ([RNA], see Chapter 30). DNA, the repository of genetic information, is present not only in chromosomes in the nucleus of eukaryotic organisms, but also in mitochondria and the chloroplasts of plants. Prokaryotic cells, which lack nuclei, have a single chromosome but may also contain nonchromosomal DNA in the form of plasmids. The genetic information found in DNA is copied and transmitted to daughter cells through DNA replication. The DNA contained in a fertilized egg encodes the information that directs the development of an organism. This development may involve the production of billions of cells. Each cell is specialized, expressing only those functions that are required for it to perform its role in maintaining the organism. Therefore, DNA must be able to not only replicate precisely each time a cell divides, but also to have the information that it contains be selectively expressed. Transcription (RNA synthesis) is the first stage in the expression of genetic information (see Chapter 30). Next, the code contained in the nucleotide sequence of messenger RNA molecules is translated (protein synthesis; see Chapter 31), thus completing gene expression. The regulation of gene expression is discussed in Chapter 32.
Figure 29.1 The “central dogma” of molecular biology.
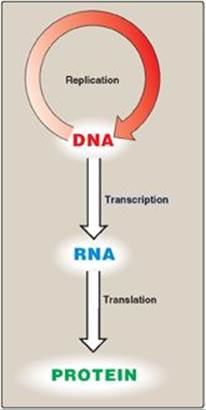
The flow of information from DNA to RNA to protein is termed the “central dogma” of molecular biology (Figure 29.1) and is descriptive of all organisms, with the exception of some viruses that have RNA as the repository of their genetic information.
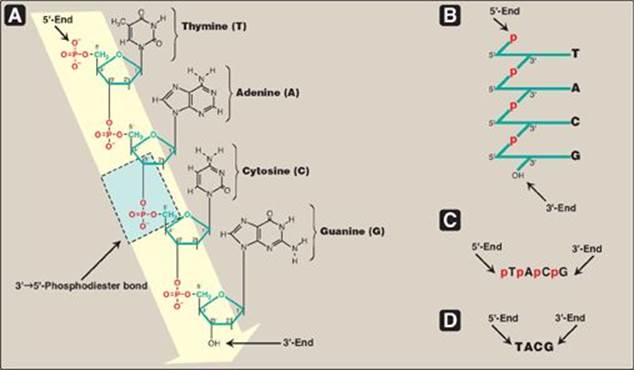
Figure 29.2 A. DNA chain with the nucleotide sequence shown written in the 5ʹ→3ʹ direction. A 3ʹ→5ʹ-phosphodiester bond is shown highlighted in the blue box, and the deoxyribose-phosphate backbone is shaded in yellow. B. The DNA chain written in a more stylized form, emphasizing the deoxyribose-phosphate backbone. C. A simpler representation of the nucleotide sequence. D. The simplest (and most common) representation, with the abbreviations for the bases written in the conventional 5ʹ→3ʹ direction.
II. STRUCTURE OF DNA
DNA is a polymer of deoxyribonucleoside monophosphates (dNMPs) covalently linked by 3→5–phosphodiester bonds. With the exception of a few viruses that contain single-stranded (ss) DNA, DNA exists as a double-stranded (ds) molecule, in which the two strands wind around each other, forming a double helix. [Note: The sequence of the linked dNMPs is primary structure, whereas the double helix is secondary structure.] In eukaryotic cells, DNA is found associated with various types of proteins (known collectively as nucleoprotein) present in the nucleus, whereas in prokaryotes, the protein–DNA complex is present in a nonmembrane-bound region known as the nucleoid.
A. 3ʹ→5ʹ-Phosphodiester bonds
Phosphodiester bonds join the 3-hydroxyl group of the deoxypentose of one nucleotide to the 5-hydroxyl group of the deoxypentose of an adjacent nucleotide through a phosphoryl group (Figure 29.2). The resulting long, unbranched chain has polarity, with both a 5-end (the end with the free phosphate) and a 3-end (the end with the free hydroxyl) that are not attached to other nucleotides. The bases located along the resulting deoxyribose–phosphate backbone are, by convention, always written in sequence from the 5-end of the chain to the 3-end. For example, the sequence of bases in the DNA shown in Figure 29.2D (5ʹ-TACG-3ʹ) is read “thymine, adenine, cytosine, guanine.” Phosphodiester linkages between nucleotides can be hydrolyzed enzymatically by a family of nucleases: deoxyribonucleases for DNA and ribonucleases for RNA, or cleaved hydrolytically by chemicals. [Note: Only RNA is cleaved by alkali.]
Figure 29.3 DNA double helix, illustrating some of its major structural features.
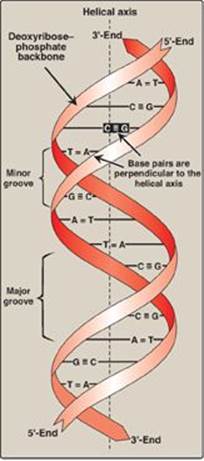
B. Double helix
In the double helix, the two chains are coiled around a common axis called the helical axis. The chains are paired in an antiparallel manner (that is, the 5ʹ-end of one strand is paired with the 3ʹ-end of the other strand) as shown in Figure 29.3. In the DNA helix, the hydrophilic deoxyribose–phosphate backbone of each chain is on the outside of the molecule, whereas the hydrophobic bases are stacked inside. The overall structure resembles a twisted ladder. The spatial relationship between the two strands in the helix creates a major (wide) groove and a minor (narrow) groove. These grooves provide access for the binding of regulatory proteins to their specific recognition sequences along the DNA chain. [Note: Certain anticancer drugs, such as dactinomycin (actinomycin D), exert their cytotoxic effect by intercalating into the narrow groove of the DNA double helix, thereby interfering with DNA (and RNA) synthesis.]
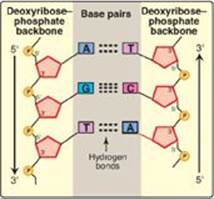
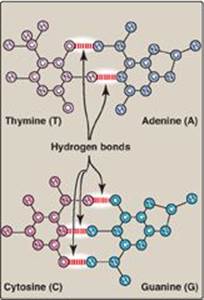
1. Base-pairing: The bases of one strand of DNA are paired with the bases of the second strand, so that an adenine (A) is always paired with a thymine (T) and a cytosine (C) is always paired with a guanine (G). [Note: The base pairs are perpendicular to the helical axis (see Figure 29.3).] Therefore, one polynucleotide chain of the DNA double helix is always the complement of the other. Given the sequence of bases on one chain, the sequence of bases on the complementary chain can be determined (Figure 29.4). [Note: The specific base-pairing in DNA leads to the Chargaff rule, which states that in any sample of dsDNA, the amount of A equals the amount of T, the amount of G equals the amount of C, and the total amount of purines equals the total amount of pyrimidines.] The base pairs are held together by hydrogen bonds: two between A and T and three between G and C (Figure 29.5). These hydrogen bonds, plus the hydrophobic interactions between the stacked bases, stabilize the structure of the double helix.
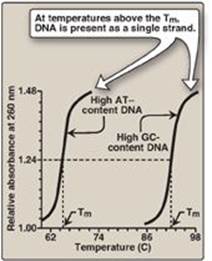
2. Separation of the two DNA strands in the double helix: The two strands of the double helix separate when hydrogen bonds between the paired bases are disrupted. Disruption can occur in the laboratory if the pH of the DNA solution is altered so that the nucleotide bases ionize, or if the solution is heated. [Note: Phosphodiester bonds are not broken by such treatment.] When DNA is heated, the temperature at which one half of the helical structure is lost is defined as the melting temperature (Tm). The loss of helical structure in DNA, called denaturation, can be monitored by measuring its absorbance at 260 nm. [Note: ssDNA has a higher relative absorbance at this wavelength than does dsDNA.] Because there are three hydrogen bonds between G and C but only two between A and T, DNA that contains high concentrations of A and T denatures at a lower temperature than G- and C-rich DNA (Figure 29.6). Under appropriate conditions, complementary DNA strands can reform the double helix by the process called renaturation (or reannealing). [Note: Separation of the two strands over short regions occurs during both DNA and RNA synthesis.]
Figure 29.4 Two complementary DNA sequences. T= thymine; A = adenine; C = cytosine; G = guanine.
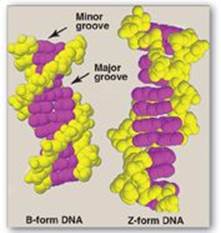
3. Structural forms of the double helix: There are three major structural forms of DNA: the B form (described by Watson and Crick in 1953), the A form, and the Z form. The B form is a right-handed helix with 10 base pairs per 360° turn (or twist) of the helix, and with the planes of the bases perpendicular to the helical axis. Chromosomal DNA is thought to consist primarily of B-DNA (Figure 29.7 shows a space-filling model of B-DNA). The A form is produced by moderately dehydrating the B form. It is also a right-handed helix, but there are 11 base pairs per turn, and the planes of the base pairs are tilted 20° away from the perpendicular to the helical axis. The conformation found in DNA–RNA hybrids or RNA–RNA double-stranded regions is probably very close to the A form. Z-DNA is a left-handed helix that contains 12 base pairs per turn (see Figure 29.7). [Note: The deoxyribose–phosphate backbone “zigzags,” hence, the name “Z”-DNA.] Stretches of Z-DNA can occur naturally in regions of DNA that have a sequence of alternating purines and pyrimidines (for example, poly GC). Transitions between the B and Z helical forms of DNA may play a role in regulating gene expression.
Figure 29.5 Hydrogen bonds between complementary bases.
C. Linear and circular DNA molecules
Each chromosome in the nucleus of a eukaryote contains one long, linear molecule of dsDNA, which is bound to a complex mixture of proteins (histone and nonhistone, see p. 409) to form chromatin. Eukaryotes have closed, circular, dsDNA molecules in their mitochondria, as do plant chloroplasts. A prokaryotic organism typically contains a single, circular, dsDNA molecule. [Note: Circular DNA is “supercoiled”, that is, the double helix crosses over on itself one or more times. Supercoiling can result in overwinding (positive supercoiling) or underwinding (negative supercoiling) of DNA. Supercoiling, a type of tertiary structure, compacts DNA.] Each prokaryotic chromosome is associated with nonhistone proteins that help compact the DNA to form a nucleoid. In addition, most species of bacteria also contain small, circular, extrachromosomal DNA molecules called plasmids. Plasmid DNA carries genetic information, and undergoes replication that may or may not be synchronized to chromosomal division. [Note: The use of plasmids as vectors in recombinant DNA technology is described in Chapter 33.]
Plasmids may carry genes that convey antibiotic resistance to the host bacterium and may facilitate the transfer of genetic information from one bacterium to another.
Figure 29.6 Melting temperatures (Tm) of DNA molecules with different nucleotide compositions.
III. STEPS IN PROKARYOTIC DNA SYNTHESIS
When the two strands of the DNA double helix are separated, each can serve as a template for the replication of a new complementary strand. This produces two daughter molecules, each of which contains two DNA strands with an antiparallel orientation (see Figure 29.3). This process is called semiconservative replication because, although the parental duplex is separated into two halves (and, therefore, is not “conserved” as an entity), each of the individual parental strands remains intact in one of the two new duplexes (Figure 29.8). The enzymes involved in the DNA replication process are template-directed polymerases that can synthesize the complementary sequence of each strand with extraordinary fidelity. The reactions described in this section were first known from studies of the bacterium Escherichia coli (E. coli), and the description given below refers to the process in prokaryotes. DNA synthesis in higher organisms is more complex but involves the same types of mechanisms. In either case, initiation of DNA replication commits the cell to continue the process until the entire genome has been replicated.
Figure 29.7 Structures of B-DNA and Z-DNA.
A. Separation of the two complementary DNA strands
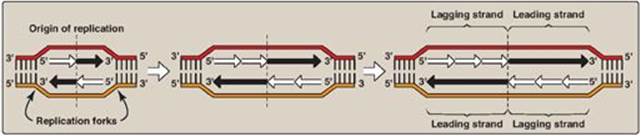
In order for the two strands of the parental dsDNA to be replicated, they must first separate (or “melt”) over a small region, because the polymerases use only ssDNA as a template. In prokaryotic organisms, DNA replication begins at a single, unique nucleotide sequence, a site called the origin of replication, or ori (Figure 29.9A). [Note: This sequence is referred to as a consensus sequence, because the order of nucleotides is essentially the same at each site.] The ori includes short, AT-rich segments that facilitate melting. In eukaryotes, replication begins at multiple sites along the DNA helix (Figure 29.9B). Having multiple origins of replication provides a mechanism for rapidly replicating the great length of eukaryotic DNA molecules.
B. Formation of the replication fork
As the two strands unwind and separate, synthesis occurs at two replication forks that move away from the origin in opposite directions (bidirectionally), generating a replication bubble (see Figure 29.9). [Note: The term “replication fork” derives from the Y-shaped structure in which the tines of the fork represent the separated strands (Figure 29.10).]
1. Proteins required for DNA strand separation: Initiation of DNA replication requires the recognition of the origin by a group of proteins that form the prepriming complex. These proteins are responsible for maintaining the separation of the parental strands, and for unwinding the double helix ahead of the advancing replication fork. These proteins include the following.
Figure 29.8 Semiconservative replication of DNA. T= thymine; A = adenine; C = cytosine; G = guanine.
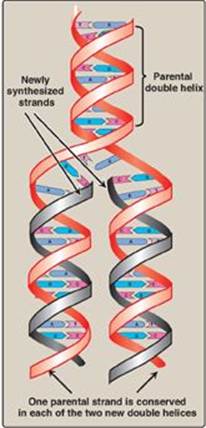
Figure 29.9 Replication of DNA: origins and replication forks. A. Small prokaryotic circular DNA. B. Very long eukaryotic DNA.
a. DnaA protein: DnaA protein binds to specific nucleotide sequences (DnaA boxes) within the origin of replication, causing the short, tandemly arranged (one after the other) AT-rich regions in the origin to melt. Melting is adenosine triphosphate (ATP) dependent, and results in strand separation with the formation of localized regions of ssDNA.
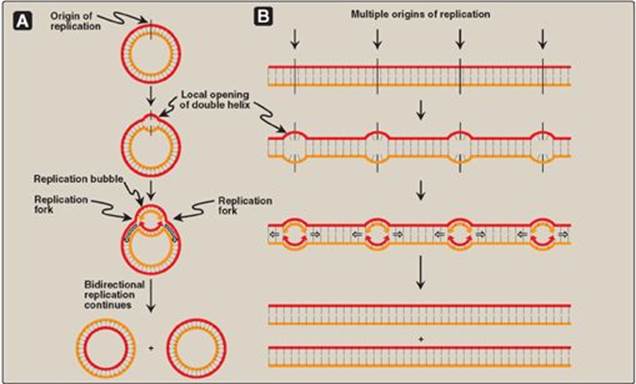
b. DNA helicases: These enzymes bind to ssDNA near the replication fork and then move into the neighboring double-stranded region, forcing the strands apart (in effect, unwinding the double helix). Helicases require energy provided by ATP (see Figure 29.10). Unwinding at the replication fork causes supercoiling in other regions of the DNA molecule. [Note: DnaB is the principal helicase of replication in E. coli. Its binding to DNA requires DnaC.]
c. Single-stranded DNA-binding protein: This protein binds to the ssDNA generated by helicases (see Figure 29.10). Binding is cooperative (that is, the binding of one molecule of single-stranded binding [SSB] protein makes it easier for additional molecules of SSB protein to bind tightly to the DNA strand). The SSB proteins are not enzymes, but rather serve to shift the equilibrium between dsDNA and ssDNA in the direction of the single-stranded forms. These proteins not only keep the two strands of DNA separated in the area of the replication origin, thus providing the single-stranded template required by polymerases, but also protect the DNA from nucleases that degrade ssDNA.
Figure 29.10 Proteins responsible for maintaining the separation of the parental strands and unwinding the double helix ahead of the advancing replication fork (![]() ). ADP = adenosine diphosphate; Pi = inorganic phosphate.
). ADP = adenosine diphosphate; Pi = inorganic phosphate.
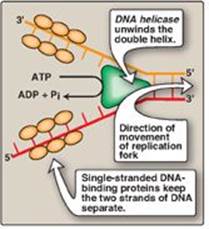
2. Solving the problem of supercoils: As the two strands of the double helix are separated, a problem is encountered, namely, the appearance of positive supercoils in the region of DNA ahead of the replication fork as a result of overwinding (Figure 29.11), and negative supercoils in the region behind the fork. The accumulating positive supercoils interfere with further unwinding of the double helix. [Note: Supercoiling can be demonstrated by tightly grasping one end of a helical telephone cord while twisting the other end. If the cord is twisted in the direction of tightening the coils, the cord will wrap around itself in space to form positive supercoils. If the cord is twisted in the direction of loosening the coils, the cord will wrap around itself in the opposite direction to form negative supercoils.] To solve this problem, there is a group of enzymes called DNA topoisomerases, which are responsible for removing supercoils in the helix by transiently cleaving one or both of the DNA strands.
Figure 29.11 Positive supercoiling resulting from DNA strand separation.
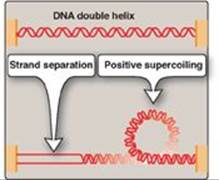
a. Type I DNA topoisomerases: These enzymes reversibly cleave one strand of the double helix. They have both strand-cutting and strand-resealing activities. They do not require ATP, but rather appear to store the energy from the phosphodiester bond they cleave, reusing the energy to reseal the strand (Figure 29.12). Each time a transient “nick” is created in one DNA strand, the intact DNA strand is passed through the break before it is resealed, thus relieving (“relaxing”) accumulated supercoils. Type I topoisomerases relax negative supercoils (that is, those that contain fewer turns of the helix than relaxed DNA) in E. coli, and both negative and positive supercoils (that is, those that contain fewer or more turns of the helix than relaxed DNA) in many prokaryotic cells (but not E. coli) and in eukaryotic cells.
Figure 29.12 Action of type I DNA topoisomerases.
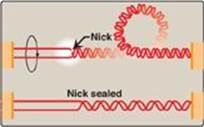
b. Type II DNA topoisomerases: These enzymes bind tightly to the DNA double helix and make transient breaks in both strands. The enzyme then causes a second stretch of the DNA double helix to pass through the break and, finally, reseals the break (Figure 29.13). As a result, both negative and positive supercoils can be relieved by this ATP-requiring process. DNA gyrase, a type II topoisomerase found in bacteria and plants, has the unusual property of being able to introduce negative supercoils into circular DNA using energy from the hydrolysis of ATP. This facilitates the replication of DNA because the negative supercoils neutralize the positive supercoils introduced during opening of the double helix. It also aids in the transient strand separation required during transcription (see p. 420).
Anticancer agents, such as the camptothecins, target human type I topoisomerases, whereas etoposide targets human type II topoisomerases. Bacterial DNA gyrase is a unique target of a group of antimicrobial agents called fluoroquinolones (for example, ciprofloxacin).
Figure 29.13 Action of type II DNA topoisomerase.
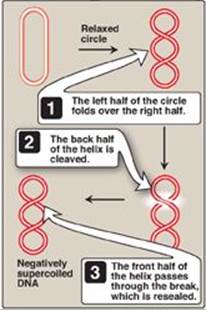
Figure 29.14 Discontinuous synthesis of DNA.
C. Direction of DNA replication
The DNA polymerases responsible for copying the DNA templates are only able to “read” the parental nucleotide sequences in the 3ʹ→5ʹ direction, and they synthesize the new DNA strands only in the 5ʹ→3ʹ (antiparallel) direction. Therefore, beginning with one parental double helix, the two newly synthesized stretches of nucleotide chains must grow in opposite directions, one in the 5ʹ→3ʹ direction toward the replication fork and one in the 5ʹ→3ʹ direction away from the replication fork (Figure 29.14). This feat is accomplished by a slightly different mechanism on each strand.
1. Leading strand: The strand that is being copied in the direction of the advancing replication fork is called the leading strand and is synthesized continuously.
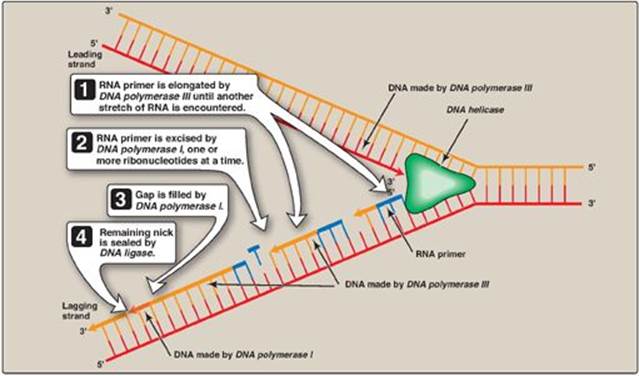
2. Lagging strand: The strand that is being copied in the direction away from the replication fork is synthesized discontinuously, with small fragments of DNA being copied near the replication fork. These short stretches of discontinuous DNA, termed Okazaki fragments, are eventually joined (ligated) to become a single, continuous strand. The new strand of DNA produced by this mechanism is termed the lagging strand.
Figure 29.15 Use of an RNA primer to initiate DNA synthesis. P = phosphate.
D. RNA primer
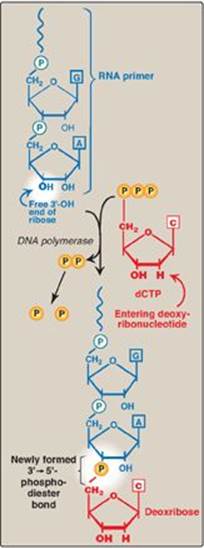
DNA polymerases cannot initiate synthesis of a complementary strand of DNA on a totally single-stranded template. Rather, they require an RNA primer, which is a short, double-stranded region consisting of RNA base-paired to the DNA template, with a free hydroxyl group on the 3ʹ-end of the RNA strand (Figure 29.15). This hydroxyl group serves as the first acceptor of a deoxynucleotide by action of a DNA polymerase. [Note: Recall that glycogen synthase also requires a primer (see p. 126).]
1. Primase: A specific RNA polymerase, called primase (DnaG), synthesizes the short stretches of RNA (approximately ten nucleotides long) that are complementary and antiparallel to the DNA template. In the resulting hybrid duplex, the U (uracil) in RNA pairs with A in DNA. As shown in Figure 29.16, these short RNA sequences are constantly being synthesized at the replication fork on the lagging strand, but only one RNA sequence at the origin of replication is required on the leading strand. The substrates for this process are 5ʹ-ribonucleoside triphosphates, and pyrophosphate is released as each ribonucleoside monophosphate is added through formation of a 3ʹ→5ʹ phosphodiester bond. [Note: The RNA primer is later removed as described on p. 405.]
Figure 29.16 Elongation of the leading and lagging strands. [Note: The DNA sliding clamp is not shown.]
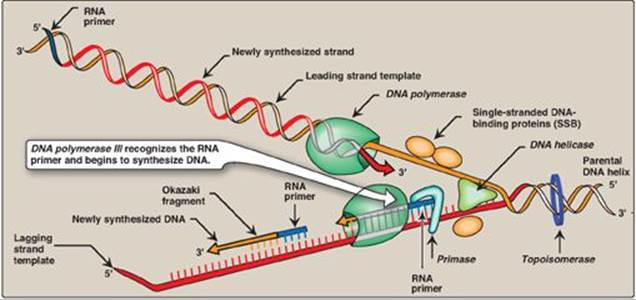
2. Primosome: The addition of primase converts the prepriming complex of proteins required for DNA strand separation (see p. 399) to a primosome. The primosome makes the RNA primer required for leading strand synthesis and initiates Okazaki fragment formation in lagging strand synthesis. As with DNA synthesis, the direction of synthesis of the primer is 5ʹ→3ʹ.
E. Chain elongation
Prokaryotic (and eukaryotic) DNA polymerases (DNA pols) elongate a new DNA strand by adding deoxyribonucleotides, one at a time, to the 3ʹ-end of the growing chain (see Figure 29.16). The sequence of nucleotides that are added is dictated by the base sequence of the template strand with which the incoming nucleotides are paired.
1. DNA polymerase III: DNA chain elongation is catalyzed by the multisubunit enzyme, DNA polymerase III (DNA pol III). Using the 3ʹ-hydroxyl group of the RNA primer as the acceptor of the first deoxyribonucleotide, DNA pol III begins to add nucleotides along the single-stranded template that specifies the sequence of bases in the newly synthesized chain. DNA pol III is a highly “processive” enzyme (that is, it remains bound to the template strand as it moves along and does not diffuse away and then rebind before adding each new nucleotide). The processivity of DNA pol III is the result of its β subunit forming a ring that encircles and moves along the template strand of the DNA, thus serving as a sliding DNA clamp. [Note: Clamp formation is facilitated by a protein complex, the clamp loader, and ATP hydrolysis.] The new strand grows in the 5ʹ→3ʹ direction, antiparallel to the parental strand (see Figure 29.16). The nucleotide substrates are 5ʹ-deoxyribonucleoside triphosphates. Pyrophosphate (PPi) is released when each new deoxynucleoside monophosphate is added to the growing chain (see Figure 29.15). Hydrolysis of PPi to 2Pi means that a total of two high-energy bonds are used to drive the addition of each deoxynucleotide.
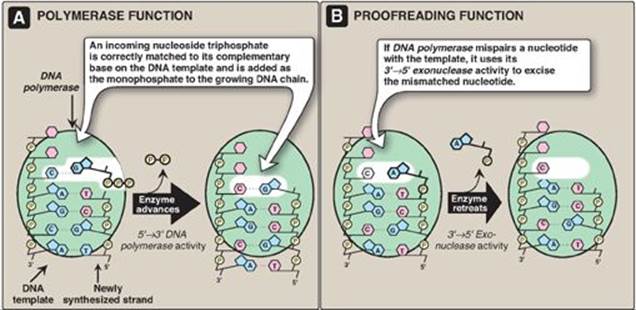
Figure 29.17 3ʹ→5ʹ Exonuclease activity enables DNA polymerase III to “proofread” the newly synthesized DNA strand.
The production of PPi with subsequent hydrolysis to 2Pi, as seen in DNA replication, is a common theme in biochemistry. Removal of the PPi product drives a reaction in the forward direction, making it essentially irreversible.
All four substrates (deoxyadenosine triphosphate [dATP], deoxythymidine triphosphate [dTTP], deoxycytidine triphosphate [dCTP], and deoxyguanosine triphosphate [dGTP]) must be present for DNA elongation to occur. If one of the four is in short supply, DNA synthesis stops when that nucleotide is depleted.
2. Proofreading of newly synthesized DNA: It is highly important for the survival of an organism that the nucleotide sequence of DNA be replicated with as few errors as possible. Misreading of the template sequence could result in deleterious, perhaps lethal, mutations. To ensure replication fidelity, DNA pol III has a “proofreading” activity (3ʹ→5ʹ exonuclease, Figure 29.17) in addition to its 5ʹ→3ʹ polymerase activity. As each nucleotide is added to the chain, DNA pol III checks to make certain the added nucleotide is, in fact, correctly matched to its complementary base on the template. If it is not, the 3ʹ→5ʹ exonuclease activity removes the error. [Note: The enzyme requires an improperly base-paired 3ʹ-hydroxy terminus and, therefore, does not degrade correctly paired nucleotide sequences.] For example, if the template base is C and the enzyme mistakenly inserts an A instead of a G into the new chain, the 3ʹ→5ʹ exonuclease activity hydrolytically removes the misplaced nucleotide. The 5ʹ→3ʹ polymerase activity then replaces it with the correct nucleotide containing G (see Figure 29.17). [Note: The proofreading exonuclease activity requires movement in the 3ʹ→5ʹ direction, not 5ʹ→3ʹ like the polymerase activity. This is because the excision must be done in the reverse direction from that of synthesis.]
F. Excision of RNA primers and their replacement by DNA
DNA pol III continues to synthesize DNA on the lagging strand until it is blocked by proximity to an RNA primer. When this occurs, the RNA is excised and the gap filled by DNA pol I.
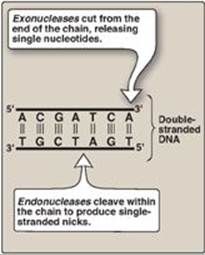
1. 5ʹ→3ʹ Exonuclease activity: In addition to having the 5ʹ→3ʹ polymerase activity that synthesizes DNA and the 3ʹ→5ʹ exonuclease activity that proofreads the newly synthesized DNA chain like DNA pol III, DNA pol I also has a 5ʹ→3ʹ exonuclease activity that is able to hydrolytically remove the RNA primer. [Note: These activities are exonucleases because they remove nucleotides from the end of the DNA chain, rather than cleaving the chain internally as do the endonucleases (Figure 29.18).] First, DNA pol I locates the space (nick) between the 3ʹ-end of the DNA newly synthesized by DNA pol III and the 5ʹ-end of the adjacent RNA primer. Next, DNA pol Ihydrolytically removes the RNA nucleotides “ahead” of itself, moving in the 5ʹ→3ʹ direction (5ʹ→3ʹ exonuclease activity). As it removes the RNA, DNA pol I replaces it with deoxyribonucleotides, synthesizing DNA in the 5ʹ→3ʹ direction (5ʹ→3ʹ polymerase activity). As it synthesizes the DNA, it also “proofreads” the new chain using its 3ʹ→5ʹ exonuclease activity to remove errors. This removal/synthesis/proofreading continues, one nucleotide at a time, until the RNA primer is totally degraded, and the gap is filled with DNA (Figure 29.19). [Note: DNA pol I uses its 5ʹ→3ʹ polymerase activity to fill in gaps generated during DNA repair (see p.410).]
Figure 29.18 Endonuclease versus exonuclease activity. [Note: Restriction endonucleases (see p. 465) cleave both strands.] T= thymine; A = adenine; C = cytosine; G = guanine.
2. Comparison of 5ʹ→3ʹ and 3ʹ→5ʹ exonucleases: The 5ʹ→3ʹ exonuclease activity of DNA pol I allows the polymerase, moving 5ʹ→3ʹ, to hydrolytically remove one or more nucleotides at a time from the 5ʹ end of the 20 to 30 nucleotide–long RNA primer. In contrast, the 3ʹ→5ʹ exonuclease activity of DNA pol I, as well as DNA pol II and III, allows these polymerases, moving 3ʹ→5ʹ, to hydrolytically remove one misplaced nucleotide at a time from the 3ʹ end of a growing DNA strand, increasing the fidelity of replication such that newly replicated DNA has one error per 107 nucleotides.
Figure 29.19 Removal of RNA primer and filling of the resulting “gaps” by DNA polymerase I.
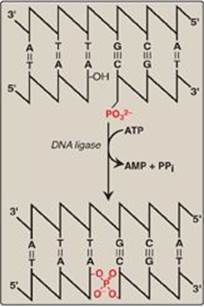
G. DNA ligase
The final phosphodiester linkage between the 5ʹ-phosphate group on the DNA chain synthesized by DNA pol III and the 3ʹ-hydroxyl group on the chain made by DNA pol I is catalyzed by DNA ligase (Figure 29.20). The joining of these two stretches of DNA requires energy, which in most organisms is provided by the cleavage of ATP to AMP + PPi.
H. Termination
Replication termination in E. coli is mediated by sequence-specific binding of the protein, Tus (terminus utilization substance) to replication termination sites (ter sites) on the DNA, stopping the movement of DNA polymerase.
Figure 29.20 Formation of a phosphodiester bond by DNA ligase. [Note: AMP is first linked to ligase, then to the 5ʹ phosphate, and then released.]
IV. EUKARYOTIC DNA REPLICATION
The process of eukaryotic DNA replication closely follows that of prokaryotic DNA synthesis. Some differences, such as the multiple origins of replication in eukaryotic cells versus single origins of replication in prokaryotes, have already been noted. Eukaryotic single-stranded DNA-binding proteins and ATP-dependent DNA helicases have been identified, whose functions are analogous to those of the prokaryotic enzymes previously discussed. In contrast, RNA primers are removed by RNase H and flap endonuclease-1 (FEN1) rather than by a DNA polymerase (Figure 29.21).
A. Eukaryotic cell cycle
The events surrounding eukaryotic DNA replication and cell division (mitosis) are coordinated to produce the cell cycle (Figure 29.22). The period preceding replication is called the G1 phase (Gap 1). DNA replication occurs during the S (synthesis) phase. Following DNA synthesis, there is another period (G2 phase, or Gap 2) before mitosis (M). Cells that have stopped dividing, such as mature T lymphocytes, are said to have gone out of the cell cycle into the G0 phase. Such quiescent cells can be stimulated to reenter the G1 phase to resume division. [Note: The cell cycle is controlled at a series of “checkpoints” that prevent entry into the next phase of the cycle until the preceding phase has been completed. Two key classes of proteins that control the progress of a cell through the cell cycle are the cyclins and cyclin-dependent kinases (Cdks).]
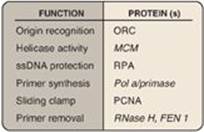
Figure 29.21 Proteins and their function in eukaryotic replication. ORC = origin recognition complex, MCM = minichromosome maintenance (complex), RPA = replication protein A, PCNA = proliferating cell nuclear antigen.
B. Eukaryotic DNA polymerases
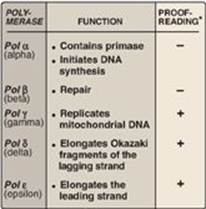
At least five high-fidelity eukaryotic DNA polymerases (pol) have been identified and categorized on the basis of molecular weight, cellular location, sensitivity to inhibitors, and the templates or substrates on which they act. They are designated by Greek letters rather than by Roman numerals (Figure 29.23).
1. Pol α: Pol α is a multisubunit enzyme. One subunit has primase activity, which initiates strand synthesis on the leading strand and at the beginning of each Okazaki fragment on the lagging strand. The primase subunit synthesizes a short RNA primer that is extended by the 5ʹ→3ʹ polymerase activity of pol α, generating a short piece of DNA. [Note: Pol α is also referred to as pol α/primase.]
2. Pol ε and pol d: Pol ε is recruited to complete DNA synthesis on the leading strand, whereas pol d elongates the Okazaki fragments of the lagging strand, each using 3ʹ→5ʹ exonuclease activity to proofread the newly synthesized DNA. [Note: DNA pol ε associates with proliferating cell nuclear antigen (PCNA), a protein that serves as a sliding DNA clamp in much the same way the β subunit of DNA pol III does in E. coli, thus ensuring high processivity.]
3. Pol β and pol γ: Pol β is involved in “gap filling” in DNA repair (see below). Pol γ replicates mitochondrial DNA.
Figure 29.22 The eukaryotic cell cycle. [Note: Cells can leave the cell cycle and enter a reversible quiescent state called G0.]
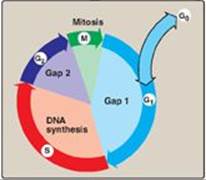
Figure 29.23 Activities of eukaryotic DNA polymerase (pol) *3ʹ→5ʹ exonuclease activity.
C. Telomeres
Telomeres are complexes of noncoding DNA plus proteins (collectively known as shelterin) located at the ends of linear chromosomes. They maintain the structural integrity of the chromosome, preventing attack by nucleases, and allow repair systems to distinguish a true end from a break in dsDNA. In humans, telomeric DNA consists of several thousand tandem repeats of a noncoding hexameric sequence, AGGGTT, base-paired to a complementary region of Cs and As. The GT-rich strand is longer than its CA complement, leaving ssDNA a few hundred nucleotides in length at the 3ʹ-end. The single-stranded region is thought to fold back on itself, forming a loop structure that is stabilized by protein.
1. Telomere shortening: Eukaryotic cells face a special problem in replicating the ends of their linear DNA molecules. Following removal of the RNA primer from the extreme 5ʹ-end of the lagging strand, there is no way to fill in the remaining gap with DNA. Consequently, in most normal human somatic cells, telomeres shorten with each successive cell division. Once telomeres are shortened beyond some critical length, the cell is no longer able to divide and is said to be senescent. In germ cells and other stem cells, as well as in cancer cells, telomeres do not shorten and the cells do not senesce. This is a result of the presence of a ribonucleoprotein, telomerase, which maintains telomeric length in these cells.
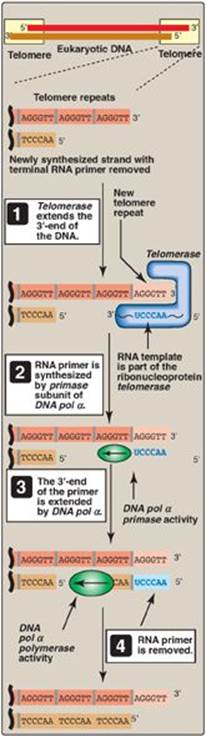
2. Telomerase: This complex contains a protein (Tert) that acts as a reverse transcriptase and a short piece of RNA (Terc) that acts as a template. The CA-rich RNA template base-pairs with the GT-rich, single-stranded 3ʹ-end of telomeric DNA (Figure 29.24). The reverse transcriptase uses the RNA template to synthesize DNA in the usual 5ʹ→3ʹ direction, extending the already longer 3ʹ-end. Telomerase then translocates to the newly synthesized end, and the process is repeated. Once the GT-rich strand has been lengthened, primase activity of DNA pol α can use it as a template to synthesize an RNA primer. The RNA primer is extended by DNA pol α, and then removed.
Telomeres may be viewed as mitotic clocks in that their length in most cells is inversely related to the number of times the cells have divided. The study of telomeres provides insight into the biology of aging and cancer.
Figure 29.24 Mechanism of action of telomerase. T= thymine; A = adenine; C = cytosine; G = guanine; pol = polymerase.
D. Reverse transcriptases
As seen with telomerase, reverse transcriptases are RNA-directed DNA polymerases. A reverse transcriptase is involved in the replication of retroviruses, such as human immunodeficiency virus (HIV). These viruses carry their genome in the form of ssRNA molecules. Following infection of a host cell, the viral enzyme reverse transcriptase uses the viral RNA as a template for the 5ʹ→3ʹ synthesis of viral DNA, which then becomes integrated into host chromosomes. Reverse transcriptase activity is also seen with transposons, DNA elements that can move about the genome (see p. 461). In eukaryotes, such elements are transcribed to RNA, the RNA is used as a template for DNA synthesis by a reverse transcriptase encoded by the transposon, and the DNA is randomly inserted into the genome. [Note: Transposons that involve an RNA intermediate are called retrotransposons or retroposons.]
E. Inhibition of DNA synthesis by nucleoside analogs
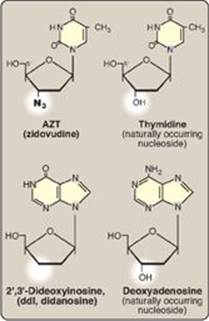
DNA chain growth can be blocked by the incorporation of certain nucleoside analogs that have been modified on the sugar portion (Figure 29.25). For example, removal of the hydroxyl group from the 3ʹ-carbon of the deoxyribose ring as in 2ʹ,3ʹ-dideoxyinosine ([ddI] also known as didanosine), or conversion of the deoxyribose to another sugar, such as arabinose, prevents further chain elongation. By blocking DNA replication, these compounds slow the division of rapidly growing cells and viruses. Cytosine arabinoside (cytarabine, or araC) has been used in anticancer chemotherapy, whereas adenine arabinoside (vidarabine, or araA) is an antiviral agent. Substitution on the sugar moiety, as seen in zidovudine (AZT, ZDV), also terminates DNA chain elongation. [Note: These drugs are generally supplied as nucleosides, which are then converted to the active nucleotides by cellular kinases.
Figure 29.25 Examples of nucleoside analogs that lack a 3ʹ-hydroxyl group. [Note: ddI is converted to its active form (ddATP).]
V. ORGANIZATION OF EUKARYOTIC DNA
A typical (diploid) human cell contains 46 chromosomes, whose total DNA is approximately 2 m long! It is difficult to imagine how such a large amount of genetic material can be effectively packaged into a volume the size of a cell nucleus so that it can be efficiently replicated, and its genetic information expressed. To do so requires the interaction of DNA with a large number of proteins, each of which performs a specific function in the ordered packaging of these long molecules of DNA. Eukaryotic DNA is associated with tightly bound basic proteins, called histones. These serve to order the DNA into fundamental structural units, called nucleosomes, which resemble beads on a string. Nucleosomes are further arranged into increasingly more complex structures that organize and condense the long DNA molecules into chromosomes that can be segregated during cell division. [Note: The complex of DNA and protein found inside the nuclei of eukaryotic cells is called chromatin.]
A. Histones and the formation of nucleosomes
There are five classes of histones, designated H1, H2A, H2B, H3, and H4. These small, evolutionally conserved proteins are positively charged at physiologic pH as a result of their high content of lysine and arginine. Because of their positive charge, they form ionic bonds with negatively charged DNA. Histones, along with ions such as Mg2+, help neutralize the negatively charged DNA phosphate groups.
1. Nucleosomes: Two molecules each of H2A, H2B, H3, and H4 form the octameric core of the individual nucleosome “beads.” Around this structural core, a segment of dsDNA is wound nearly twice, causing supercoiling (Figure 29.26). [Note: The N-terminal ends of these histones can be acetylated, methylated, or phosphorylated. These reversible covalent modifications influence how tightly the histones bind to the DNA, thereby affecting the expression of specific genes (see p. 422). Histone modification is an example of “epigenetics” or heritable changes in gene expression without alteration of the nucleotide sequence.] Neighboring nucleosomes are joined by “linker” DNA approximately 50 base pairs long. H1, of which there are several related species, is not found in the nucleosome core, but instead binds to the linker DNA chain between the nucleosome beads. H1 is the most tissue specific and species specific of the histones. It facilitates the packing of nucleosomes into more compact structures.
Figure 29.26 Organization of human DNA, illustrating the structure of nucleosomes. H = histone.
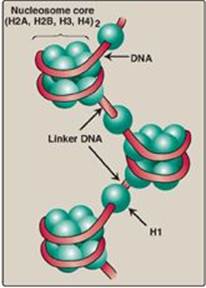
Figure 29.27 Structural organization of eukaryotic DNA. [Note: A 104 compaction is seen from ![]() to
to ![]() .] H = histone.
.] H = histone.
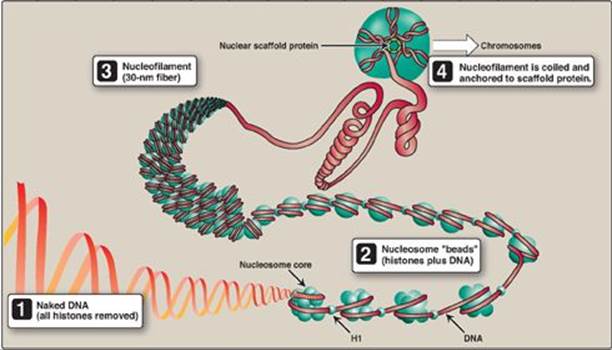
2. Higher levels of organization: Nucleosomes can be packed more tightly to form a polynucleosome (also called a nucleofilament). This structure assumes the shape of a coil, often referred to as a 30-nm fiber. The fiber is organized into loops that are anchored by a nuclear scaffold containing several proteins. Additional levels of organization lead to the final chromosomal structure (Figure 29.27).
B. Fate of nucleosomes during DNA replication
Parental nucleosomes are disassembled to allow access to DNA during replication. Once DNA is synthesized, nucleosomes form rapidly. Their histone proteins come both from new synthesis and from the transfer of intact parental histone octamers.
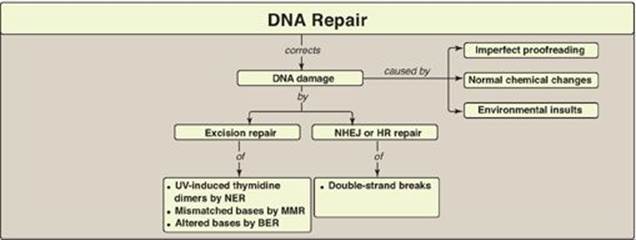
VI. DNA REPAIR
Despite the elaborate proofreading system employed during DNA synthesis, errors (including incorrect base-pairing or insertion of one to a few extra nucleotides) can occur. In addition, DNA is constantly being subjected to environmental insults that cause the alteration or removal of nucleotide bases. The damaging agents can be either chemicals (for example, nitrous acid, which can deaminate bases), or radiation (for example, non-ionizing ultraviolet light, which can fuse two pyrimidines adjacent to each other in the DNA, and high-energy ionizing radiation, which can cause double-strand breaks). Bases are also altered or lost spontaneously from mammalian DNA at a rate of many thousands per cell per day. If the damage is not repaired, a permanent change (mutation) is introduced that can result in any of a number of deleterious effects, including loss of control over the proliferation of the mutated cell, leading to cancer. Luckily, cells are remarkably efficient at repairing damage done to their DNA. Most of the repair systems involve recognition of the damage (lesion) on the DNA, removal or excision of the damage, replacement or filling the gap left by excision using the sister strand as a template for DNA synthesis, and ligation. These excision repair systems remove one to tens of nucleotides. [Note: Repair synthesis of DNA can occur outside of the S phase.]
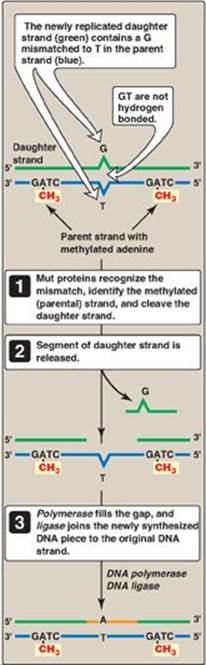
Figure 29.28 Methyl-directed mismatch repair in E. coli. [Note: The Mut proteins, S and L, recognize the mismatch and identify the parental (methylated) strand, and Mut H cleaves the daughter strand.] A = adenine; C= cytosine; G = guanine; T = thymine.
A. Repair of mismatched bases (mismatch repair)
Sometimes replication errors escape the proofreading function during DNA synthesis, causing a mismatch of one to several bases. In E. coli, mismatch repair (MMR) is mediated by a group of proteins known as the Mut proteins (Figure 29.28). Homologous proteins are present in humans. [Note: MMR reduces the error rate of replication from one in ten million to one in a billion.]
1. Identification of the mismatched strand: When a mismatch occurs, the Mut proteins that identify the mispaired nucleotide(s) must be able to discriminate between the correct strand and the strand with the mismatch. Discrimination is based on the degree of methylation. GATC sequences, which are found approximately once every thousand nucleotides, are methylated on the adenine (A) residue. This methylation is not done immediately after synthesis, so the newly synthesized DNA is hemimethylated (that is, the parental strand is methylated, but the daughter strand is not). The methylated parental strand is assumed to be correct, and it is the daughter strand that gets repaired. [Note: The exact mechanism by which the daughter strand is identified in eukaryotes is not yet known.]
2. Repair of damaged DNA: When the strand containing the mismatch is identified, an endonuclease nicks the strand, and the mismatched nucleotide(s) is/are removed by an exonuclease. Additional nucleotides at the 5ʹ- and 3ʹ-ends of the mismatch are also removed. The gap left by removal of the nucleotides is filled, using the sister strand as a template, by a DNA polymerase, typically DNA pol I. The 3ʹ-hydroxyl of the newly synthesized DNA is joined to the 5ʹ-phosphate of the remaining stretch of the original DNA strand by DNA ligase (see p. 406).
Mutation to the proteins involved in mismatch repair in humans is associated with hereditary nonpolyposis colorectal cancer (HNPCC), also known as Lynch syndrome. With HNPCC, there is an increased risk for developing colon cancer (as well as other cancers); however, only about 5% of all colon cancer is the result of mutations in mismatch repair.
B. Repair of damage caused by ultraviolet light (nucleotide excision repair)
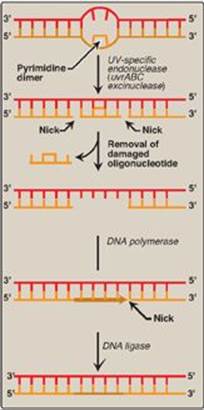
Exposure of a cell to ultrtaviolet (UV) light can result in the covalent joining of two adjacent pyrimidines (usually thymines), producing a dimer. These thymine dimers prevent DNA pol from replicating the DNA strand beyond the site of dimer formation. Thymine dimers are excised in bacteria by UvrABC proteins in a process known as nucleotide excision repair (NER) as illustrated in Figure 29.29. A related pathway is present in humans (see below).
1. Recognition and excision of dimers by UV-specific endonuclease: First, a UV-specific endonuclease (called uvrABC excinuclease) recognizes the dimer and cleaves the damaged strand on both the 5ʹ-side and 3ʹ-side of the dimer. A short oligonucleotide containing the dimer is released, leaving a gap in the DNA strand that formerly contained the dimer. This gap is filled in using a DNA polymerase and DNA ligase.
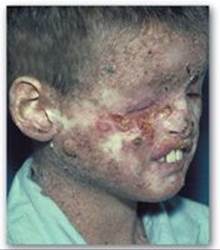
2. UV radiation and cancer: Pyrimidine dimers can be formed in the skin cells of humans exposed to unfiltered sunlight. In the rare genetic disease xeroderma pigmentosum (XP), the cells cannot repair the damaged DNA, resulting in extensive accumulation of mutations and, consequently, early and numerous skin cancers (Figure 29.30). XP can be caused by defects in any of the several genes that code for the XP proteins required for nucleotide excision repair of UV damage in humans.
Figure 29.29 Nucleotide excision repair of pyrimidine dimers in E. coli DNA. UV = ultraviolet.
C. Repair of base alterations (base excision repair)
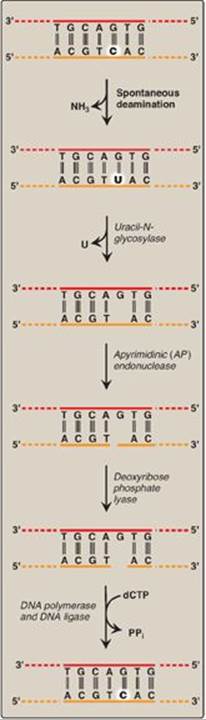
The bases of DNA can be altered, either spontaneously, as is the case with cytosine, which slowly undergoes deamination (the loss of its amino group) to form uracil, or by the action of deaminating or alkylating compounds. For example, nitrous acid, which is formed by the cell from precursors such as the nitrosamines, nitrites, and nitrates, is a potent compound that deaminates cytosine, adenine (to hypoxanthine), and guanine (to xanthine). Bases can also be lost spontaneously. For example, approximately 10,000 purine bases are lost this way per cell per day. Lesions involving base alterations or loss can be corrected by base excision repair ([BER] Figure 29.31).
1. Removal of abnormal bases: In BER, abnormal bases, such as uracil, which can occur in DNA either by deamination of cytosine or improper use of dUTP instead of dTTP during DNA synthesis, are recognized by specific glycosylases that hydrolytically cleave them from the deoxyribose–phosphate backbone of the strand. This leaves an apyrimidinic site (or apurinic, if a purine was removed), both referred to as AP sites.
2. Recognition and repair of an AP site: Specific AP-endonucleases recognize that a base is missing and initiate the process of excision and gap-filling by making an endonucleolytic cut just to the 5ʹ-side of the AP site. A deoxyribose phosphate lyase removes the single, base-free, sugar phosphate residue. DNA polymerase and DNA ligase complete the repair process.
Figure 29.30 Patient with xeroderma pigmentosum.
D. Repair of double-strand breaks
Ionizing radiation or oxidative free radicals (see p. 148) can cause double-strand breaks in DNA that are potentially lethal to the cell. Such breaks also occur naturally during gene rearrangements. dsDNA breaks cannot be corrected by the previously described strategy of excising the damage on one strand and using the remaining strand as a template for replacing the missing nucleotide(s). Instead, they are repaired by one of two systems. The first is nonhomologous end-joining (NHEJ), in which a group of proteins mediates the recognition, processing, and ligation of the ends of two DNA fragments. However, some DNA is lost in the process. Consequently, this mechanism of repair is error prone and mutagenic. Defects in this repair system are associated with a predisposition to cancer and immunodeficiency syndromes. The second repair system, homologous recombination (HR), uses the enzymes that normally perform genetic recombination between homologous chromosomes during meiosis. This system is much less error prone than NHEJ because any DNA that was lost is replaced using homologous DNA as a template. [Note: Mutations to the proteins, BRCA1 or 2 (breast cancer 1 or 2), which are involved in HR, increase the risk for developing breast cancer.]
Figure 29.31 Correction of base alterations by base excision repair. T= thymine; A = adenine; C = cytosine; G = guanine; PPi = pyrophosphate.
VII. CHAPTER SUMMARY
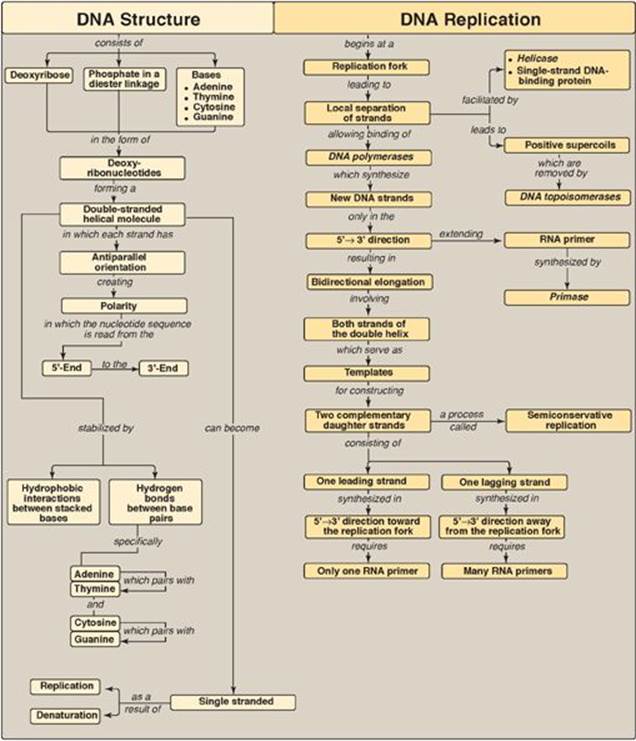
DNA is a polymer of deoxyribonucleoside monophosphates covalently linked by 3ʹ→5ʹ-phosphodiester bonds (Figure 29.32). The resulting long, unbranched chain has polarity, with both a 5ʹ-end and a 3ʹ-end. The sequence of nucleotides is read 5ʹ→3ʹ. DNA exists as a double-stranded molecule, in which the two chains are paired in an antiparallel manner, and wind around each other, forming a double helix. Adenine pairs with thymine, and cytosine pairs with guanine. Each strand of the double helix serves as a template for constructing a complementary daughter strand (semiconservative replication). DNA replication occurs in the S phase of the cell cycle and begins at the origin of replication. As the two strands unwind and separate, synthesis occurs at two replication forks that move away from the origin in opposite directions (bidirectionally). Helicase unwinds the double helix. As the two strands of the double helix are separated, positive supercoils are produced in the region of DNA ahead of the replication fork and negative supercoils behind the fork. DNA topoisomerases types I and IIremove supercoils. DNA polymerases (pol) synthesize new DNA strands only in the 5ʹ→3ʹ direction. Therefore, one of the newly synthesized stretches of nucleotide chains must grow in the 5ʹ→3ʹ direction toward the replication fork (leading strand) and one in the 5ʹ→3ʹ direction away from the replication fork (lagging strand). DNA pols require a primer. The primer for de novo DNA synthesis is a short stretch of RNA synthesized by primase. The leading strand only needs one RNA primer, whereas the lagging strand needs many. In E. coli, DNA chain elongation is catalyzed by DNA pol III, using 5ʹ-deoxyribonucleoside triphosphates as substrates. The enzyme “proofreads” the newly synthesized DNA, removing terminal mismatched nucleotides with its 3ʹ→5ʹ exonuclease activity. RNA primers are removed by DNA pol I, using its 5ʹ→3ʹ exonuclease activity. This enzyme fills the gaps with DNA, proofreading as it synthesizes. The final phosphodiester linkage is catalyzed by DNA ligase. There are at least five high-fidelity eukaryotic DNA polymerases. Pol α is a multisubunit enzyme, one subunit of which is a primase. Pol α 5ʹ→3ʹ polymerase activity adds a short piece of DNA to the RNA primer. Pol e completes DNA synthesis on the leading strand, whereas pol d elongates each lagging strand fragment. Pol βis involved with DNA repair, and pol γ replicates mitochondrial DNA. Pols e, d, and g use 3ʹ→5ʹ exonuclease activity to proofread. Nucleoside analogs containing modified sugars can be used to block DNA chain growth. They are useful in anticancer and antiviral chemotherapy. Telomeres are stretches of highly repetitive DNA complexed with protein that protect the ends of linear chromosomes. As most cells divide and age, these sequences are shortened, contributing to senescence. In cells that do not senesce (for example, germline and cancer cells), telomerase employs its enzyme component reverse transcriptase to extend the telomeres, using its RNA as a template. There are five classes of positively charged histone (H) proteins. Two each of histones H2A, H2B, H3, and H4 form an octameric structural core around which DNA is wrapped creating a nucleosome. The DNA connecting the nucleosomes, called linker DNA, is bound to H1. Nucleosomes can be packed more tightly to form a nucleofilament. Additional levels of organization create a chromosome. Most DNA damage can be corrected by excision repair involving recognition and removal of the damage by repair proteins, followed by replacement in E. coli by DNA pol and joining by ligase. Ultraviolet light can cause thymine dimers that are recognized and removed by uvrABC proteins of nucleotide excision repair. Defects in the XP proteins needed for thymine dimer repair in humans result in xeroderma pigmentosum. Mismatched bases are repaired by a similar process of recognition and removal by Mut proteins in E. coli. The extent of methylation is used for strand identification in prokaryotes. Defective mismatch repair by homologous proteins in humans is associated with hereditary nonpolyposis colorectal cancer. Abnormal bases (such as uracil) are removed by glycosylases in base excision repair, and the sugar phosphate at the apyrimidinic or apurinic (AP) site is cut out. Double-strand breaks in DNA are repaired by nonhomologous end-joining (error prone) and homologous recombination.
Figure 29.32 Key concept map for DNA structure, replication, and repair. Key concept map for DNA structure, replication, and repair. NHEJ = nonhomologous end-joining; HR = homologous recombination; NER = nucleotide excision repair; MMR = mismatch repair; BER = base excision repair; UV = ultraviolet light.
Study Questions
Choose the ONE best answer.
29.1 A 10-year-old girl is brought by her parents to the dermatologist. She has many freckles on her face, neck, arms, and hands, and the parents report that she is unusually sensitive to sunlight. Two basal cell carcinomas are identified on her face. Based on the clinical picture, which of the following processes is most likely to be defective in this patient?
A. Repair of double-strand breaks by error-prone homologous recombination
B. Removal of mismatched bases from the 3ʹ-end of Okazaki fragments by a methyl-directed process
C. Removal of pyrimidine dimers from DNA by nucleotide excision repair
D. Removal of uracil from DNA by base excision repair
Correct answer = C. The sensitivity to sunlight, extensive freckling on parts of the body exposed to the sun, and presence of skin cancer at a young age indicate that the patient most likely suffers from xeroderma pigmentosum (XP). These patients are deficient in any one of several XP proteins required for nucleotide excision repair of pyrimidine dimers in ultraviolet light–damaged DNA. Double-strand breaks are repaired by nonhomologous end-joining (error prone) or homologous recombination (error free). Methylation is not used for strand discrimination in eukaryotic mismatch repair. Uracil is removed from DNA molecules by a specific glycosylase in base excision repair, but a defect here does not cause XP.
29.2 Telomeres are complexes of DNA and protein that protect the ends of linear chromosomes. In most normal human somatic cells, telomeres shorten with each division. In stem cells and in cancer cells, however, telomeric length is maintained. In the synthesis of telomeres:
A. telomerase, a ribonucleoprotein, provides both the RNA and the protein needed for synthesis.
B. the RNA of telomerase serves as a primer.
C. the RNA of telomerase is a ribozyme.
D. the protein of telomerase is a DNA-directed DNA polymerase.
E. the shorter 3ʹ→5ʹ strand gets extended.
F. the direction of synthesis is 3ʹ→5ʹ.
Correct answer = A. Telomerase is a ribonucleoprotein particle required for telomere maintenance. Telomerase contains an RNA that serves as the template, not the primer, for the synthesis of telomeric DNA by the reverse transcriptase of telomerase. Telomeric RNA has no catalytic activity. As a reverse transcriptase, telomerase synthesizes DNA using its RNA template and so is an RNA-directed DNA polymerase. The direction of synthesis, as with all DNA synthesis, is 5ʹ→3ʹ, and it is the 3ʹ-end of the already longer 5ʹ→3ʹ strand that gets extended.
29.3 While studying the structure of a small gene that was sequenced during the Human Genome Project, an investigator notices that one strand of the DNA molecule contains 20 As, 25 Gs, 30 Cs, and 22 Ts. How many of each base is found in the complete double-stranded molecule?
A. A = 40, G = 50, C = 60, T = 44
B. A = 44, G = 60, C = 50, T = 40
C. A = 45, G = 45, C = 52, T = 52
D. A = 50, G = 47, C = 50, T = 47
E. A = 42, G = 55, C = 55, T = 42
Correct answer = E. The two DNA strands are complementary to each other, with A base-paired with T and G base-paired with C. So, for example, the 20 As on the first strand would be paired with 20 Ts on the second strand, the 25 Gs on the first strand would be paired with 25 Cs on the second strand, and so forth. When these are all added together, the correct numbers of each base are indicated in choice E. Notice that, in the correct answer, A = T and G = C.
29.4 List the order in which the following enzymes participate in prokaryotic replication.
A. Ligase
B. Polymerase I (3ʹ→5ʹ exonuclease activity)
C. Polymerase I (5ʹ→3ʹ exonuclease activity)
D. Polymerase I (5ʹ→3ʹ polymerase activity)
E. Polymerase III
F. Primase
Correct answer: F, E, C, D, B, A. Primase makes the RNA primer; Polymerase III extends the primer with DNA (and proofreads); polymerase I removes the primer with its 5ʹ→3ʹ exonuclease activity, fills in the gap with its 5ʹ→3ʹ polymerase activity, and removes errors with its 3ʹ→5ʹ exonuclease activity; and ligase makes the 5ʹ→3ʹ phosphodiester bond that links the DNA made by polymerase I and polymerase III.
29.5 Dideoxynucleotides lack a 3ʹ-hydroxyl group. Why would incorporation of a dideoxynucleotide into DNA stop replication?
The lack of the 3ʹ-OH group prevents formation of the 3ʹ-hydroxyl → 5ʹ-phosphate bond that links one nucleotide to the next in DNA.