Lippincott’s Illustrated Reviews: Biochemistr, Sixth Edition (2014)
UNIT I: Protein Structure and Function
Chapter 3. Globular Proteins
I. OVERVIEW
The previous chapter described the types of secondary and tertiary structures that are the bricks and mortar of protein architecture. By arranging these fundamental structural elements in different combinations, widely diverse proteins can be constructed that are capable of various specialized functions. This chapter examines the relationship between structure and function for the clinically important globular hemeproteins. Fibrous structural proteins are discussed in Chapter 4.
II. GLOBULAR HEMEPROTEINS
Hemeproteins are a group of specialized proteins that contain heme as a tightly bound prosthetic group. (See p. 54 for a discussion of prosthetic groups.) The role of the heme group is dictated by the environment created by the three-dimensional structure of the protein. For example, the heme group of a cytochrome functions as an electron carrier that is alternately oxidized and reduced (see p. 76). In contrast, the heme group of the enzyme catalase is part of the active site of the enzyme that catalyzes the breakdown of hydrogen peroxide (see p. 148). In hemoglobin and myoglobin, the two most abundant hemeproteins in humans, the heme group serves to reversibly bind oxygen.
Figure 3.1 A. Hemeprotein (cytochrome c). B. Structure of heme.
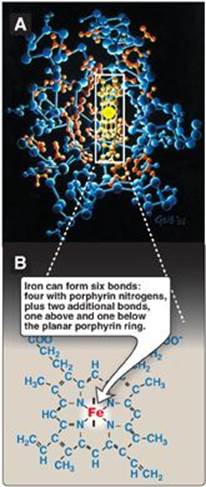
A. Structure of heme
Heme is a complex of protoporphyrin IX and ferrous iron (Fe2+) (Figure 3.1). The iron is held in the center of the heme molecule by bonds to the four nitrogens of the porphyrin ring. The heme Fe2+ can form two additional bonds, one on each side of the planar porphyrin ring. In myoglobin and hemoglobin, one of these positions is coordinated to the side chain of a histidine residue of the globin molecule, whereas the other position is available to bind oxygen (Figure 3.2). (See pp. 278 and 282 for a discussion of the synthesis and degradation of heme.)
Figure 3.2 A. Model of myoglobin showing helices A to H. B. Schematic diagram of the oxygen-binding site of myoglobin.
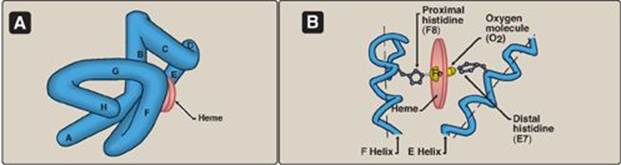
B. Structure and function of myoglobin
Myoglobin, a hemeprotein present in heart and skeletal muscle, functions both as a reservoir for oxygen and as an oxygen carrier that increases the rate of transport of oxygen within the muscle cell. [Note: Mouse myoglobin double knockouts (see p. 486) have, surprisingly, an apparently normal phenotype.] Myoglobin consists of a single polypeptide chain that is structurally similar to the individual polypeptide chains of the tetrameric hemoglobin molecule. This homology makes myoglobin a useful model for interpreting some of the more complex properties of hemoglobin.
1. α-Helical content: Myoglobin is a compact molecule, with approximately 80% of its polypeptide chain folded into eight stretches of α-helix. These α-helical regions, labeled A to H in Figure 3.2A, are terminated either by the presence of proline, whose five-membered ring cannot be accommodated in an α-helix (see p. 16) or by β-bends and loops stabilized by hydrogen bonds and ionic bonds (see p. 17). [Note: Ionic bonds are also termed electrostatic interactions or salt bridges.]
2. Location of polar and nonpolar amino acid residues: The interior of the myoglobin molecule is composed almost entirely of nonpolar amino acids. They are packed closely together, forming a structure stabilized by hydrophobic interactions between these clustered residues (see p. 19). In contrast, polar amino acids are located almost exclusively on the surface, where they can form hydrogen bonds, both with each other and with water.
3. Binding of the heme group: The heme group of the myoglobin molecule sits in a crevice, which is lined with nonpolar amino acids. Notable exceptions are two histidine residues (Figure 3.2B). One, the proximal histidine (F8), binds directly to the iron of heme. The second, or distal histidine (E7), does not directly interact with the heme group but helps stabilize the binding of oxygen to the ferrous iron. The protein, or globin, portion of myoglobin thus creates a special microenvironment for the heme that permits the reversible binding of one oxygen molecule (oxygenation). The simultaneous loss of electrons by the ferrous iron (oxidation to the ferric form) occurs only rarely.
Figure 3.3 A. Structure of hemoglobin showing the polypeptide backbone. B. Simplified drawing showing the helices.
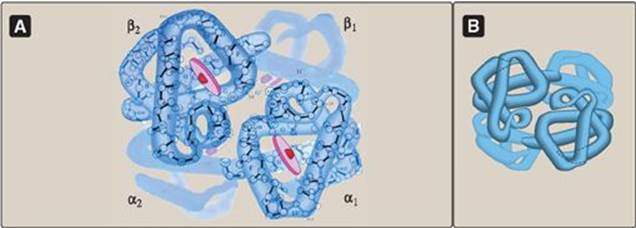
C. Structure and function of hemoglobin
Hemoglobin is found exclusively in red blood cells (RBC), where its main function is to transport oxygen (O2) from the lungs to the capillaries of the tissues. Hemoglobin A, the major hemoglobin in adults, is composed of four polypeptide chains (two α chains and two β chains) held together by noncovalent interactions (Figure 3.3). Each chain (subunit) has stretches of α-helical structure and a hydrophobic heme-binding pocket similar to that described for myoglobin. However, the tetrameric hemoglobin molecule is structurally and functionally more complex than myoglobin. For example, hemoglobin can transport H+ and CO2 from the tissues to the lungs and can carry four molecules of O2 from the lungs to the cells of the body. Furthermore, the oxygen-binding properties of hemoglobin are regulated by interaction with allosteric effectors (see p. 29).
Obtaining O2 from the atmosphere solely by diffusion greatly limits the size of organisms. Circulatory systems overcome this, but transport molecules such as hemoglobin are also required because O2 is only slightly soluble in aqueous solutions such as blood.
1. Quaternary structure of hemoglobin: The hemoglobin tetramer can be envisioned as being composed of two identical dimers, (αβ)1 and (αβ)2. The two polypeptide chains within each dimer are held tightly together primarily by hydrophobic interactions (Figure 3.4). [Note: In this instance, hydrophobic amino acid residues are localized not only in the interior of the molecule, but also in a region on the surface of each subunit. Multiple interchain hydrophobic interactions form strong associations between α-subunits and β-subunits in the dimers.] In contrast, the two dimers are held together primarily by polar bonds. The weaker interactions between the dimers allows them to move with respect to one other. This movement results in the two dimers occupying different relative positions in deoxyhemoglobin as compared with oxyhemoglobin (see Figure 3.4). [Note: The binding of O2 to the heme iron pulls the iron into the plane of the heme. Because the iron is also linked to the proximal histidine (F8), there is movement of the globin chains that alters the interface between the αβ dimers.]
Figure 3.4 Schematic diagram showing structural changes resulting from oxygenation and deoxygenation of hemoglobin.
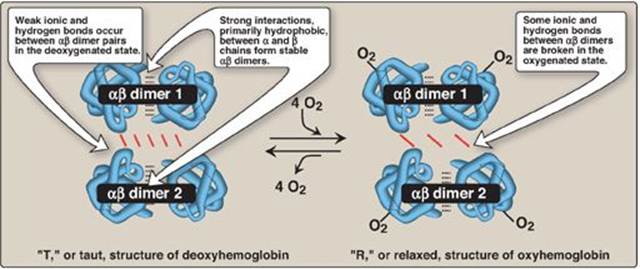
a. T form: The deoxy form of hemoglobin is called the “T,” or taut (tense) form. In the T form, the two αβ dimers interact through a network of ionic bonds and hydrogen bonds that constrain the movement of the polypeptide chains. The T conformation is the low-oxygen-affinity form of hemoglobin.
b. R form: The binding of O2 to hemoglobin causes the rupture of some of the polar bonds between the αβ dimers, allowing movement. This leads to a structure called the “R,” or relaxed form (see Figure 3.4). The R conformation is the high-oxygen-affinity form of hemoglobin.
D. Binding of oxygen to myoglobin and hemoglobin
Myoglobin can bind only one molecule of O2, because it contains only one heme group. In contrast, hemoglobin can bind four O2 molecules, one at each of its four heme groups. The degree of saturation (Y) of these oxygen-binding sites on all myoglobin or hemoglobin molecules can vary between zero (all sites are empty) and 100% (all sites are full), as shown in Figure 3.5. [Note: Pulse oximetry is a noninvasive, indirect method of measuring the O2 saturation of arterial blood based on differences in light absorption by oxyhemoglobin and deoxyhemoglobin.]
1. Oxygen-dissociation curve: A plot of Y measured at different partial pressures of oxygen (pO2) is called the oxygen-dissociation curve. [Note: pO2 may also be represented as PO2.] The curves for myoglobin and hemoglobin show important differences (see Figure 3.5). This graph illustrates that myoglobin has a higher oxygen affinity at all pO2 values than does hemoglobin. The partial pressure of oxygen needed to achieve half-saturation of the binding sites (P50) is approximately 1 mm Hg for myoglobin and 26 mm Hg for hemoglobin. The higher the oxygen affinity (that is, the more tightly oxygen binds), the lower the P50.
Figure 3.5 Oxygen-dissociation curves for myoglobin and hemoglobin (Hb).
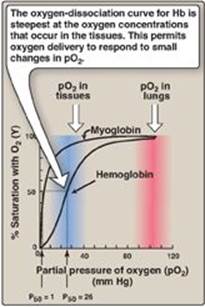
a. Myoglobin: The oxygen-dissociation curve for myoglobin has a hyperbolic shape (see Figure 3.5). This reflects the fact that myoglobin reversibly binds a single molecule of oxygen. Thus, oxygenated (MbO2) and deoxygenated (Mb) myoglobin exist in a simple equilibrium:
Mb + O2 ![]() MbO2
MbO2
The equilibrium is shifted to the right or to the left as oxygen is added to or removed from the system. [Note: Myoglobin is designed to bind oxygen released by hemoglobin at the low pO2 found in muscle. Myoglobin, in turn, releases oxygen within the muscle cell in response to oxygen demand.]
b. Hemoglobin: The oxygen-dissociation curve for hemoglobin is sigmoidal in shape (see Figure 3.5), indicating that the subunits cooperate in binding oxygen. Cooperative binding of oxygen by the four subunits of hemoglobin means that the binding of an oxygen molecule at one heme group increases the oxygen affinity of the remaining heme groups in the same hemoglobin tetramer (Figure 3.6). This effect is referred to as heme–heme interaction (see below). Although it is more difficult for the first oxygen molecule to bind to hemoglobin, the subsequent binding of oxygen occurs with high affinity, as shown by the steep upward curve in the region near 20–30 mm Hg (see Figure 3.5).
E. Allosteric effects
The ability of hemoglobin to reversibly bind oxygen is affected by the pO2 (through heme–heme interactions as described above), the pH of the environment, the partial pressure of carbon dioxide (pCO2) and the availability of 2,3-bisphosphoglycerate. These are collectively called allosteric (“other site”) effectors, because their interaction at one site on the hemoglobin molecule affects the binding of oxygen to heme groups at other sites on the molecule. [Note: The binding of oxygen to monomeric myoglobin is not influenced by allosteric effectors.]
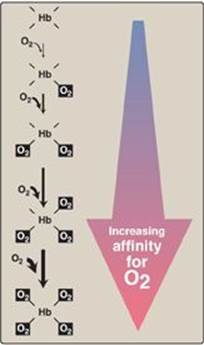
1. Heme–heme interactions: The sigmoidal oxygen-dissociation curve reflects specific structural changes that are initiated at one heme group and transmitted to other heme groups in the hemoglobin tetramer. The net effect is that the affinity of hemoglobin for the last oxygen bound is approximately 300 times greater than its affinity for the first oxygen bound.
Figure 3.6 Hemoglobin (Hb) binds successive molecules of oxygen with increasing affinity.
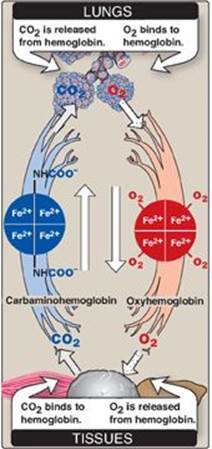
a. Loading and unloading oxygen: The cooperative binding of oxygen allows hemoglobin to deliver more oxygen to the tissues in response to relatively small changes in the partial pressure of oxygen. This can be seen in Figure 3.5, which indicates pO2 in the alveoli of the lung and the capillaries of the tissues. For example, in the lung, the concentration of oxygen is high, and hemoglobin becomes virtually saturated (or “loaded”) with oxygen. In contrast, in the peripheral tissues, oxyhemoglobin releases (or “unloads”) much of its oxygen for use in the oxidative metabolism of the tissues (Figure 3.7).
b. Significance of the sigmoidal oxygen-dissociation curve: The steep slope of the oxygen-dissociation curve over the range of oxygen concentrations that occur between the lungs and the tissues permits hemoglobin to carry and deliver oxygen efficiently from sites of high to sites of low pO2. A molecule with a hyperbolic oxygen-dissociation curve, such as myoglobin, could not achieve the same degree of oxygen release within this range of partial pressures of oxygen. Instead, it would have maximum affinity for oxygen throughout this oxygen pressure range and, therefore, would deliver no oxygen to the tissues.
Figure 3.7 Transport of oxygen and carbon dioxide by hemoglobin. Fe = iron.
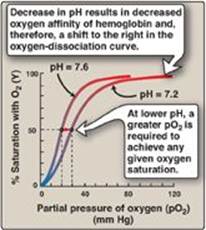
2. Bohr effect: The release of oxygen from hemoglobin is enhanced when the pH is lowered or when the hemoglobin is in the presence of an increased pCO2. Both result in a decreased oxygen affinity of hemoglobin and, therefore, a shift to the right in the oxygen-dissociation curve (Figure 3.8), and both, then, stabilize the T (deoxy) form. This change in oxygen binding is called the Bohr effect. Conversely, raising the pH or lowering the concentration of CO2 results in a greater affinity for oxygen, a shift to the left in the oxygen-dissociation curve, and stabilization of the R (oxy) form.
a. Source of the protons that lower the pH: The concentration of both H+ and CO2 in the capillaries of metabolically active tissues is higher than that observed in alveolar capillaries of the lungs, where CO2 is released into the expired air. In the tissues, CO2 is converted by carbonic anhydrase to carbonic acid:
CO2 + H2O ![]() H2CO3
H2CO3
which spontaneously loses a proton, becoming bicarbonate (the major blood buffer):
H2CO3 ![]() HCO3– + H+
HCO3– + H+
The H+ produced by this pair of reactions contributes to the lowering of pH. This differential pH gradient (that is, lungs having a higher pH and tissues a lower pH) favors the unloading of oxygen in the peripheral tissues and the loading of oxygen in the lung. Thus, the oxygen affinity of the hemoglobin molecule responds to small shifts in pH between the lungs and oxygen-consuming tissues, making hemoglobin a more efficient transporter of oxygen.
Figure 3.8 Effect of pH on the oxygen affinity of hemoglobin. Protons are allosteric effectors of hemoglobin.
b. Mechanism of the Bohr effect: The Bohr effect reflects the fact that the deoxy form of hemoglobin has a greater affinity for protons than does oxyhemoglobin. This effect is caused by ionizable groups such as specific histidine side chains that have a higher pKa in deoxyhemoglobin than in oxyhemoglobin. Therefore, an increase in the concentration of protons (resulting in a decrease in pH) causes these groups to become protonated (charged) and able to form ionic bonds (salt bridges). These bonds preferentially stabilize the deoxy form of hemoglobin, producing a decrease in oxygen affinity. [Note: Hemoglobin, then, is an important blood buffer.]
The Bohr effect can be represented schematically as:
![]()
where an increase in protons (or a lower pO2) shifts the equilibrium to the right (favoring deoxyhemoglobin), whereas an increase in pO2 (or a decrease in protons) shifts the equilibrium to the left.
Figure 3.9 Synthesis of 2,3-bisphosphoglycerate. [Note: ![]() is a phosphoryl group, PO32–.] In older literature, 2, 3-bisphosphoglycerate (2,3-BPG) may be referred to as 2,3-diphosphoglycerate (2,3-DPG).
is a phosphoryl group, PO32–.] In older literature, 2, 3-bisphosphoglycerate (2,3-BPG) may be referred to as 2,3-diphosphoglycerate (2,3-DPG).
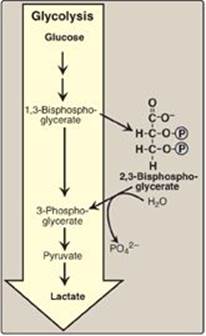
3. Effect of 2,3-bisphosphoglycerate on oxygen affinity: 2,3-Bisphosphoglycerate (2,3-BPG) is an important regulator of the binding of oxygen to hemoglobin. It is the most abundant organic phosphate in the RBC, where its concentration is approximately that of hemoglobin. 2,3-BPG is synthesized from an intermediate of the glycolytic pathway (Figure 3.9; see p. 101 for a discussion of 2,3-BPG synthesis in glycolysis).
a. Binding of 2,3-BPG to deoxyhemoglobin: 2,3-BPG decreases the O2 affinity of hemoglobin by binding to deoxyhemoglobin but not to oxyhemoglobin. This preferential binding stabilizes the T conformation of deoxyhemoglobin. The effect of binding 2,3-BPG can be represented schematically as:
![]()
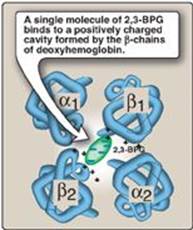
b. Binding site of 2,3-BPG: One molecule of 2,3-BPG binds to a pocket, formed by the two β-globin chains, in the center of the deoxyhemoglobin tetramer (Figure 3.10). This pocket contains several positively charged amino acids that form ionic bonds with the negatively charged phosphate groups of 2,3-BPG. [Note: Replacement of one of these amino acids can result in hemoglobin variants with abnormally high oxygen affinity that may be compensated for by increased RBC production (erythrocytosis).] 2,3-BPG is expelled with oxygenation of the hemoglobin.
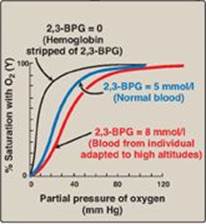
c. Shift of the oxygen-dissociation curve: Hemoglobin from which 2,3-BPG has been removed has a high affinity for oxygen. However, as seen in the RBC, the presence of 2,3-BPG significantly reduces the affinity of hemoglobin for oxygen, shifting the oxygen-dissociation curve to the right (Figure 3.11). This reduced affinity enables hemoglobin to release oxygen efficiently at the partial pressures found in the tissues.
Figure 3.10 Binding of 2,3-bisphosphoglycerate (2,3-BPG) by deoxyhemoglobin.
d. Response of 2,3-BPG levels to chronic hypoxia or anemia: The concentration of 2,3-BPG in the RBC increases in response to chronic hypoxia, such as that observed in chronic obstructive pulmonary disease (COPD) like emphysema, or at high altitudes, where circulating hemoglobin may have difficulty receiving sufficient oxygen. Intracellular levels of 2,3-BPG are also elevated in chronic anemia, in which fewer than normal RBCs are available to supply the body’s oxygen needs. Elevated 2,3-BPG levels lower the oxygen affinity of hemoglobin, permitting greater unloading of oxygen in the capillaries of the tissues (see Figure 3.11).
Figure 3.11 Allosteric effect of 2,3-bisphosphoglycerate (2,3-BPG) on the oxygen affinity of hemoglobin.
e. Role of 2,3-BPG in transfused blood: 2,3-BPG is essential for the normal oxygen transport function of hemoglobin. However, storing blood in the currently available media results in a decrease in 2,3-BPG. Stored blood displays an abnormally high oxygen affinity and fails to unload its bound oxygen properly in the tissues. Hemoglobin deficient in 2,3-BPG thus acts as an oxygen “trap” rather than as an oxygen transport system. Transfused RBC are able to restore their depleted supplies of 2,3-BPG in 6–24 hours. However, severely ill patients may be compromised if transfused with large quantities of such 2,3-BPG–“stripped” blood. [Note: The maximum storage time for RBC has been doubled (21 to 42 days, with median time of 15 days) by changes in H+, phosphate, and hexose sugar concentration and by the addition of adenine (see p. 291). Although the content of 2,3-BPG was not greatly improved in the long-term by these changes, adenosine triphosphate production was increased and improved RBC survival.]
4. Binding of CO2: Most of the CO2 produced in metabolism is hydrated and transported as bicarbonate ion (see p. 9). However, some CO2 is carried as carbamate bound to the N-terminal amino groups of hemoglobin (forming carbaminohemoglobin as shown in Figure 3.7), which can be represented schematically as follows:
Hb – NH2 + CO2 ![]() Hb – NH – COO- + H+
Hb – NH – COO- + H+
The binding of CO2 stabilizes the T or deoxy form of hemoglobin, resulting in a decrease in its affinity for oxygen (see p. 28) and a right shift in the oxygen-dissociation curve. In the lungs, CO2 dissociates from the hemoglobin and is released in the breath.
5. Binding of CO: Carbon monoxide (CO) binds tightly (but reversibly) to the hemoglobin iron, forming carboxyhemoglobin. When CO binds to one or more of the four heme sites, hemoglobin shifts to the R conformation, causing the remaining heme sites to bind oxygen with high affinity. This shifts the oxygen-dissociation curve to the left and changes the normal sigmoidal shape toward a hyperbola. As a result, the affected hemoglobin is unable to release oxygen to the tissues (Figure 3.12). [Note: The affinity of hemoglobin for CO is 220 times greater than for oxygen. Consequently, even minute concentrations of CO in the environment can produce toxic concentrations of carboxyhemoglobin in the blood. For example, increased levels of CO are found in the blood of tobacco smokers. CO toxicity appears to result from a combination of tissue hypoxia and direct CO-mediated damage at the cellular level.] CO poisoning is treated with 100% oxygen at high pressure (hyperbaric oxygen therapy), which facilitates the dissociation of CO from the hemoglobin. [Note: CO inhibits Complex IV of the electron transport chain (see p. 76).] In addition to O2, CO2, and CO, nitric oxide gas (NO) also is carried by hemoglobin. NO is a potent vasodilator (see p. 151). It can be taken up (salvaged) or released from RBC, thus modulating NO availability and influencing vessel diameter.
Figure 3.12 Effect of carbon monoxide (CO) on the oxygen affinity of hemoglobin. CO-Hb = carboxyhemoglobin (carbon monoxyhemoglobin).
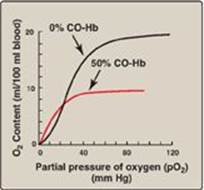
Figure 3.13 Normal adult human hemoglobins. [Note: The α-chains in these hemoglobins are identical.] Hb = hemoglobin.
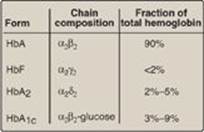
F. Minor hemoglobins
It is important to remember that human hemoglobin A (HbA) is just one member of a functionally and structurally related family of proteins, the hemoglobins (Figure 3.13). Each of these oxygen-carrying proteins is a tetramer, composed of two α-globin (or α-like) polypeptides and two β-globin (or β-like) polypeptides. Certain hemoglobins, such as HbF, are normally synthesized only during fetal development, whereas others, such as HbA2, are synthesized in the adult, although at low levels compared with HbA. HbA can also become modified by the covalent addition of a hexose (see p. 34).
1. Fetal hemoglobin: HbF is a tetramer consisting of two α chains identical to those found in HbA, plus two γ chains (α2γ2; see Figure 3.13). The γ chains are members of the β-globin gene family (see p. 35).
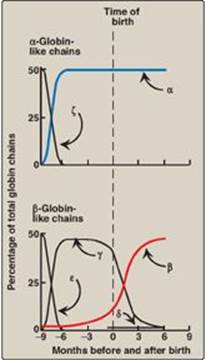
a. HbF synthesis during development: In the first month after conception, embryonic hemoglobins such as Hb Gower 1, composed of two α-like zeta (ζ) chains and two β-like epsilon (ε) chains (ζ2ε2), are synthesized by the embryonic yolk sac. In the fifth week of gestation, the site of globin synthesis shifts, first to the liver and then to the marrow, and the primary product is HbF. HbF is the major hemoglobin found in the fetus and newborn, accounting for about 60% of the total hemoglobin in the RBC during the last months of fetal life (Figure 3.14). HbA synthesis starts in the bone marrow at about the eighth month of pregnancy and gradually replaces HbF. (Figure 3.14 shows the relative production of each type of hemoglobin chain during fetal and postnatal life.) [Note: HbF represents less than 1% of the hemoglobin in most adults and is concentrated in RBC known as F cells.]
b. Binding of 2,3-BPG to HbF: Under physiologic conditions, HbF has a higher affinity for oxygen than does HbA as a result of HbF only weakly binding 2,3-BPG. [Note: The γ-globin chains of HbF lack some of the positively charged amino acids that are responsible for binding 2,3-BPG in the β-globin chains.] Because 2,3-BPG serves to reduce the affinity of hemoglobin for oxygen, the weaker interaction between 2,3-BPG and HbF results in a higher oxygen affinity for HbF relative to HbA. In contrast, if both HbA and HbF are stripped of their 2,3-BPG, they then have a similar affinity for oxygen. The higher oxygen affinity of HbF facilitates the transfer of oxygen from the maternal circulation across the placenta to the RBC of the fetus.
Figure 3.14 Developmental changes in hemoglobin.
2. Hemoglobin A2: HbA2 is a minor component of normal adult hemoglobin, first appearing shortly before birth and, ultimately, constituting about 2% of the total hemoglobin. It is composed of two α-globin chains and two δ-globin chains (α2δ2; see Figure 3.13).
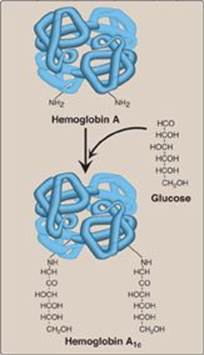
3. Hemoglobin A1c: Under physiologic conditions, HbA is slowly and nonenzymically glycosylated (glycated), the extent of glycosylation being dependent on the plasma concentration of a particular hexose. The most abundant form of glycosylated hemoglobin is HbA1c. It has glucose residues attached predominantly to the NH2 groups of the N-terminal valines of the β-globin chains (Figure 3.15). Increased amounts of HbA1c are found in RBC of patients with diabetes mellitus, because their HbA has contact with higher glucose concentrations during the 120-day lifetime of these cells. (See p. 340 for a discussion of the use of HbA1c levels in assessing average blood glucose levels in patients with diabetes.)
Figure 3.15 Nonenzymic addition of glucose to hemoglobin. The nonenzymic addition of a sugar to a protein is referred to as glycation.
III. ORGANIZATION OF THE GLOBIN GENES
To understand diseases resulting from genetic alterations in the structure or synthesis of hemoglobins, it is necessary to grasp how the hemoglobin genes, which direct the synthesis of the different globin chains, are structurally organized into gene families and also how they are expressed.
A. α-Gene family
The genes coding for the α-globin and β-globin subunits of the hemoglobin chains occur in two separate gene clusters (or families) located on two different chromosomes (Figure 3.16). The α-gene cluster on chromosome 16 contains two genes for the α-globin chains. It also contains the ζ gene that is expressed early in development as an α-globin-like component of embryonic hemoblobin. [Note: Globin gene famillies also contain globin-like genes that are not expressed, that is, their genetic information is not used to produce globin chains. These are called pseudogenes.]
Figure 3.16 Organization of the globin gene families. Hb = hemoglobin.
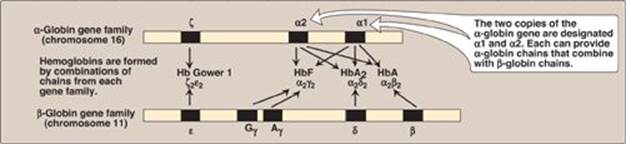
B. β-Gene family
A single gene for the β-globin chain is located on chromosome 11 (see Figure 3.16). There are an additional four β-globin-like genes: the ε gene (which, like the ζ gene, is expressed early in embryonic development), two γ genes (Gγ and Aγ that are expressed in HbF), and the δ gene that codes for the globin chain found in the minor adult hemoglobin HbA2.
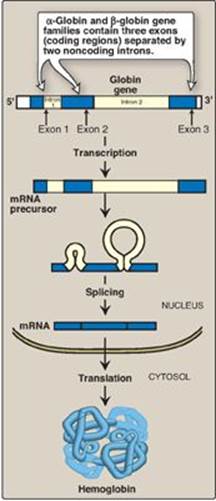
C. Steps in globin chain synthesis
Expression of a globin gene begins in the nucleus of RBC precursors, where the DNA sequence encoding the gene is transcribed. The RNA produced by transcription is actually a precursor of the messenger RNA (mRNA) that is used as a template for the synthesis of a globin chain. Before it can serve this function, two noncoding stretches of RNA (introns) must be removed from the mRNA precursor sequence and the remaining three fragments (exons) joined in a linear manner. The resulting mature mRNA enters the cytosol, where its genetic information is translated, producing a globin chain. (A summary of this process is shown in Figure 3.17. A more detailed description of gene expresion is presented in Unit VI, p. 395.)
Figure 3.17 Synthesis of globin chains. mRNA = messenger RNA.
IV. HEMOGLOBINOPATHIES
Hemoglobinopathies are defined as a group of genetic disorders caused by production of a structurally abnormal hemoglobin molecule; synthesis of insufficient quantities of normal hemoglobin; or, rarely, both. Sickle cell anemia (HbS), hemoglobin C disease (HbC), hemoglobin SC disease (HbS + HbC = HbSC), and the thalassemias are representative hemoglobinopathies that can have severe clinical consequences. The first three conditions result from production of hemoglobin with an altered amino acid sequence (qualitative hemoglobinopathy), whereas the thalassemias are caused by decreased production of normal hemoglobin (quantitative hemoglobinopathy).
A. Sickle cell anemia (hemoglobin S disease)
Sickle cell anemia, the most common of the RBC sickling diseases, is a genetic disorder of the blood caused by a single nucleotide substitution (a point mutation, see p. 433) in the gene for β-globin. It is the most common inherited blood disorder in the United States, affecting 50,000 Americans. It occurs primarily in the African American population, affecting one of 500 newborn African American infants in the United States. Sickle cell anemia is an autosomal recessive disorder. It occurs in individuals who have inherited two mutant genes (one from each parent) that code for synthesis of the β chains of the globin molecules. [Note: The mutant β-globin chain is designated βS, and the resulting hemoglobin, α2βS2, is referred to as HbS.] An infant does not begin showing symptoms of the disease until sufficient HbF has been replaced by HbS so that sickling can occur (see below). Sickle cell anemia is characterized by lifelong episodes of pain (“crises”); chronic hemolytic anemia with associated hyperbilirubinemia (see p. 284); and increased susceptibility to infections, usually beginning in infancy. [Note: The lifetime of a RBC in sickle cell anemia is less than 20 days, compared with 120 days for normal RBC, hence, the anemia.] Other symptoms include acute chest syndrome, stroke, splenic and renal dysfunction, and bone changes due to marrow hyperplasia. Heterozygotes, representing 1 in 12 African Americans, have one normal and one sickle cell gene. The blood cells of such heterozygotes contain both HbS and HbA. These individuals have sickle cell trait. They usually do not show clinical symptoms (but may under conditions of extreme physical exertion with dehydration) and can have a normal life span.
Figure 3.18 Amino acid substitutions in hemoglobin S (HbS) and hemoglobin C (HbC).
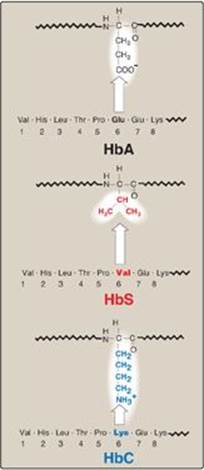
1. Amino acid substitution in HbS β chains: A molecule of HbS contains two normal α-globin chains and two mutant β-globin chains (βS), in which glutamate at position six has been replaced with valine (Figure 3.18). Therefore, during electrophoresis at alkaline pH, HbS migrates more slowly toward the anode (positive electrode) than does HbA (Figure 3.19). This altered mobility of HbS is a result of the absence of the negatively charged glutamate residues in the two β chains, thereby rendering HbS less negative than HbA. [Note: Electrophoresis of hemoglobin obtained from lysed RBC is routinely used in the diagnosis of sickle cell trait and sickle cell disease. DNA analysis also is used (see p. 472).]
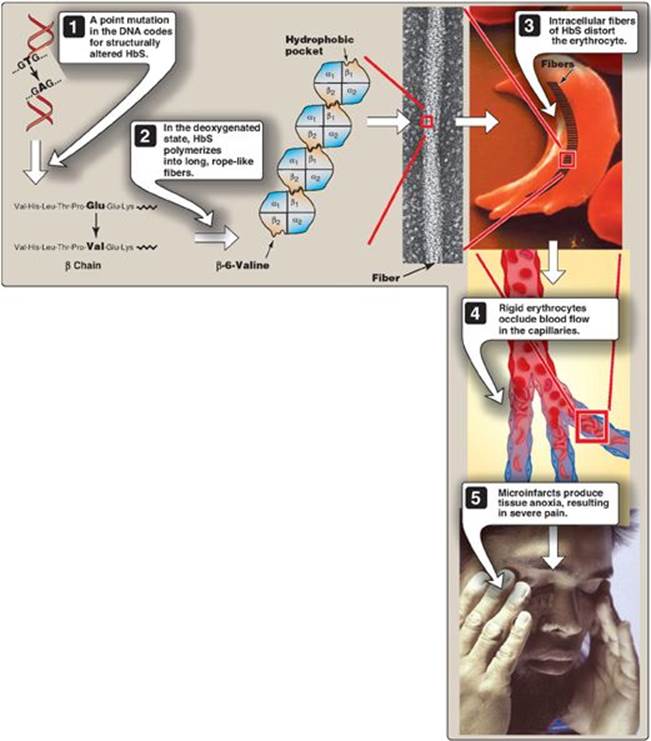
2. Sickling and tissue anoxia: The replacement of the charged glutamate with the nonpolar valine forms a protrusion on the β chain that fits into a complementary site on the β chain of another hemoglobin molecule in the cell (Figure 3.20). At low oxygen tension, deoxyhemoglobin S polymerizes inside the RBC, forming a network of insoluble fibrous polymers that stiffen and distort the cell, producing rigid, misshapen RBC. Such sickled cells frequently block the flow of blood in the narrow capillaries. This interruption in the supply of oxygen leads to localized anoxia (oxygen deprivation) in the tissue, causing pain and eventually death (infarction) of cells in the vicinity of the blockage. The anoxia also leads to an increase in deoxygenated HbS. [Note: The mean diameter of RBC is 7.5 µm, whereas that of the microvasculature is 3–4 µm. Compared to normal RBC, sickled cells have a decreased ability to deform and an increased tendency to adhere to vessel walls and so have difficulty moving through small vessels, thereby causing microvascular occlusion.]
3. Variables that increase sickling: The extent of sickling and, therefore, the severity of disease is enhanced by any variable that increases the proportion of HbS in the deoxy state (that is, reduces the affinity of HbS for O2). These variables include decreased pO2, increased pCO2, decreased pH, dehydration, and an increased concentration of 2,3-BPG in RBC.
4. Treatment: Therapy involves adequate hydration, analgesics, aggressive antibiotic therapy if infection is present, and transfusions in patients at high risk for fatal occlusion of blood vessels. Intermittent transfusions with packed RBC reduce the risk of stroke, but the benefits must be weighed against the complications of transfusion, which include iron overload (hemosiderosis), bloodborne infections, and immunologic complications. Hydroxyurea (hydroxycarbamide), an antitumor drug, is therapeutically useful because it increases circulating levels of HbF, which decreases RBC sickling. This leads to decreased frequency of painful crises and reduces mortality. [Note: The morbidity and mortality associated with sickle cell anemia has led to its inclusion in newborn screening panels to allow prophylactic antibiotic therapy to begin soon after the birth of an affected child.]
Figure 3.19 Diagram of hemoglobins (HbA), (HbS), and (HbC) after electrophoresis.
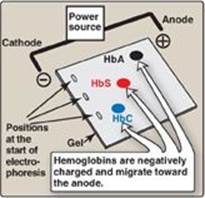
Figure 3.20 Molecular and cellular events leading to sickle cell crisis. HbS = hemoglobin S.
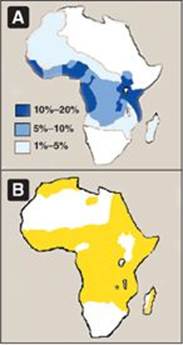
5. Possible selective advantage of the heterozygous state: The high frequency of the bS mutation among black Africans, despite its damaging effects in the homozygous state, suggests that a selective advantage exists for heterozygous individuals. For example, heterozygotes for the sickle cell gene are less susceptible to the severe malaria caused by the parasite Plasmodium falciparum. This organism spends an obligatory part of its life cycle in the RBC. One theory is that because these cells in individuals heterozygous for HbS, like those in homozygotes, have a shorter life span than normal, the parasite cannot complete the intracellular stage of its development. This fact may provide a selective advantage to heterozygotes living in regions where malaria is a major cause of death. Figure 3.21 illustrates that in Africa, the geographic distribution of sickle cell anemia is similar to that of malaria.
B. Hemoglobin C disease
Like HbS, HbC is a hemoglobin variant that has a single amino acid substitution in the sixth position of the β-globin chain (see Figure 3.18). In HbC, however, a lysine is substituted for the glutamate (as compared with a valine substitution in HbS). [Note: This substitution causes HbC to move more slowly toward the anode than HbA or HbS does (see Figure 3.19).] Rare patients homozygous for HbC generally have a relatively mild, chronic hemolytic anemia. These patients do not suffer from infarctive crises, and no specific therapy is required.
C. Hemoglobin SC disease
HbSC disease is another of the RBC sickling diseases. In this disease, some β-globin chains have the sickle cell mutation, whereas other β-globin chains carry the mutation found in HbC disease. [Note: Patients with HbSC disease are doubly heterozygous. They are called compound heterozygotes because both of their β-globin genes are abnormal, although different from each other.] Hemoglobin levels tend to be higher in HbSC disease than in sickle cell anemia and may even be at the low end of the normal range. The clinical course of adults with HbSC anemia differs from that of sickle cell anemia in that symptoms such as painful crises are less frequent and less severe. However, there is significant clinical variability.
D. Methemoglobinemias
Oxidation of the heme iron in hemoglobin to the ferric (Fe3+) state forms methemoglobin, which cannot bind O2. This oxidation may be caused by the action of certain drugs, such as nitrates, or endogenous products such as reactive oxygen species (see p. 148). The oxidation may also result from inherited defects, for example, certain mutations in the α- or β-globin chain promote the formation of methemoglobin (HbM). Additionally, a deficiency of NADH-cytochrome b5 reductase (also called NADH-methemoglobin reductase), the enzyme responsible for the conversion of methemoglobin (Fe3+) to hemoglobin (Fe2+), leads to the accumulation of HbM. [Note: The RBC of newborns have approximately half the capacity of those of adults to reduce HbM. They are, therefore, particularly susceptible to the effects of HbM-producing compounds.] The methemoglobinemias are characterized by “chocolate cyanosis” (a brownish blue coloration of the skin and mucous membranes and brown-colored blood) as a result of the dark-colored HbM. Symptoms are related to the degree of tissue hypoxia and include anxiety, headache, and dyspnea. In rare cases, coma and death can occur. Treatment is with methylene blue, which is oxidized as Fe+3 is reduced.
Figure 3.21 A. Distribution of sickle cell in Africa expressed as a percentage of the population with disease. B. Distribution of malaria in Africa.
E. Thalassemias
The thalassemias are hereditary hemolytic diseases in which an imbalance occurs in the synthesis of globin chains. As a group, they are the most common single gene disorders in humans. Normally, synthesis of the α- and β-globin chains is coordinated, so that each α-globin chain has a β-globin chain partner. This leads to the formation of α2β2 (HbA). In the thalassemias, the synthesis of either the α- or the β-globin chain is defective. A thalassemia can be caused by a variety of mutations, including entire gene deletions, or substitutions or deletions of one to many nucleotides in the DNA. [Note: Each thalassemia can be classified as either a disorder in which no globin chains are produced (αo- or βo-thalassemia), or one in which some chains are synthesized but at a reduced level (α+- or β+-thalassemia).]
Figure 3.22 A. β-Globin gene mutations in the β-thalassemias. B. Hemoglobin (Hb) tetramers formed in β-thalassemias.
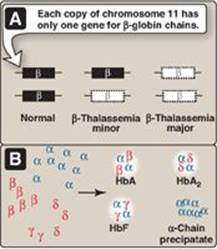
1. β-Thalassemias: In these disorders, synthesis of β-globin chains is decreased or absent, typically as a result of point mutations that affect the production of functional mRNA. However, α-globin chain synthesis is normal. Excess α-globin chains cannot form stable tetramers and so precipitate, causing the premature death of cells initially destined to become mature RBC. Increase in α2δ2 (HbA2) and α2γ2 (HbF) also occurs. There are only two copies of the β-globin gene in each cell (one on each chromosome 11). Therefore, individuals with β-globin gene defects have either β-thalassemia trait (β-thalassemia minor) if they have only one defective β-globin gene or β-thalassemia major (Cooley anemia) if both genes are defective (Figure 3.22). Because the β-globin gene is not expressed until late in fetal gestation, the physical manifestations of β-thalassemias appear only several months after birth. Those individuals with β-thalassemia minor make some β chains, and usually do not require specific treatment. However, those infants born with β-thalassemia major are seemingly healthy at birth but become severely anemic, usually during the first or second year of life due to ineffective erythropoiesis. Skeletal changes as a result of extramedullary hematopoiesis also are seen. These patients require regular transfusions of blood. [Note: Although this treatment is lifesaving, the cumulative effect of the transfusions is iron overload (a syndrome known as hemosiderosis). Use of iron chelation therapy has improved morbidity and mortality.] The only curative option available is hematopoietic stem cell transplantation.
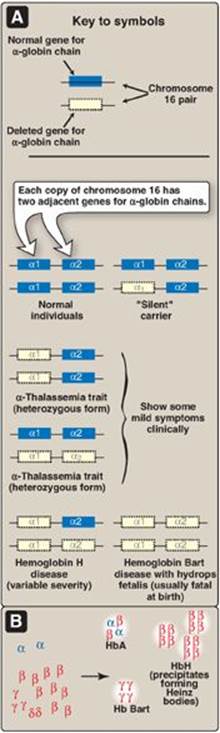
2. α-Thalassemias: In these disorders, synthesis of α-globin chains is decreased or absent, typically as a result of deletional mutations. Because each individual’s genome contains four copies of the α-globin gene (two on each chromosome 16), there are several levels of α-globin chain deficiencies (Figure 3.23). If one of the four genes is defective, the individual is termed a silent carrier of α-thalassemia, because no physical manifestations of the disease occur. If two α-globin genes are defective, the individual is designated as having α-thalassemia trait. If three α-globin genes are defective, the individual has hemoglobin H (β4) disease, a hemolytic anemia of variable severity. If all four α-globin genes are defective, hemoglobin Bart (γ4) disease with hydrops fetalis and fetal death results, because α-globin chains are required for the synthesis of HbF.
Figure 3.23 A. α-Globin gene deletions in the α-thalassemias. B. Hemoglobin (Hb) tetramers formed in α-thalassemias.
V. CHAPTER SUMMARY
Hemoglobin A (HbA), the major hemoglobin (Hb) in adults, is composed of four polypeptide chains (two α chains and two β chains, α2β2) held together by noncovalent interactions (Figure 3.24). The subunits occupy different relative positions in deoxyhemoglobin compared with oxyhemoglobin. The deoxy form of Hb is called the “T,” or taut (tense) conformation. It has a constrained structure that limits the movement of the polypeptide chains. The T form is the low-oxygen-affinity form of Hb. The binding of O2 to Hb causes rupture of some of the ionic and hydrogen bonds, and movement of the dimers. This leads to a structure called the “R,” or relaxed conformation. The R form is the high-oxygen-affinity form of Hb. The oxygen-dissociation curve for Hb is sigmoidal in shape (in contrast to that of myoglobin, which is hyperbolic), indicating that the subunits cooperate in binding O2. Cooperative binding of O2 by the four subunits of Hb means that the binding of an O2 molecule at one heme group increases the oxygen affinity of the remaining heme groups in the same Hb molecule. Hb’s ability to bind O2 reversibly is affected by the partial pressure of O2 (pO2) (through heme-heme interactions), the pH of the environment, the partial pressure of CO2 (pCO2), and the availability of 2,3-bisphosphoglycerate(2,3-BPG). For example, the release of O2 from Hb is enhanced when the pH is lowered or the pCO2 is increased (the Bohr effect), such as in exercising muscle, and the oxygen-dissociation curve of Hb is shifted to the right. To cope long-term with the effects of chronic hypoxia or anemia, the concentration of 2,3-BPG in red blood cells increases. 2,3-BPG binds to the Hb and decreases its oxygen affinity. It therefore also shifts the oxygen-dissociation curve to the right. Carbon monoxide (CO) binds tightly (but reversibly) to the Hb iron, forming carboxyhemoglobin. Hemoglobinopathies are disorders caused either by production of a structurally abnormalHb molecule; synthesis of insufficient quantities of normal Hb subunits, or, rarely, both (Figure 3.25). The sickling diseases sickle cell anemia (hemoglobin S disease) and hemoglobin SC disease as well as hemoglobin C disease and the thalassemias are representative hemoglobinopathies that can have severe clinical consequences.
Study Questions
Choose the ONE best answer.
3.1 Which one of the following statements concerning the hemoglobins is correct?
A. HbA is the most abundant hemoglobin in normal adults.
B. Fetal blood has a lower affinity for oxygen than does adult blood because HbF has an increased affinity for 2,3-bisphosphoglycerate.
C. The globin chain composition of HbF is α2δ2.
D. HbA1c differs from HbA by a single, genetically determined amino acid substitution.
E. HbA2 appears early in fetal life.
Correct answer = A. HbA accounts for over 90% of the hemoglobin in a normal adult. If HbA1c is included, the percentage rises to approximately 97%. Because 2,3-bisphosphoglycerate (2,3-BPG) reduces the affinity of hemoglobin for oxygen, the weaker interaction between 2,3-BPG and HbF results in a higher oxygen affinity for HbF relative to HbA. HbF consists of α2γ2. HbA1c is a glycosylated form of HbA, formed nonenzymically in red cells. HbA2 is a minor component of normal adult hemoglobin, first appearing shortly before birth and rising to adult levels (about 2% of the total hemoglobin) by age 6 months.
3.2 Which one of the following statements concerning the ability of acidosis to precipitate a crisis in sickle cell anemia is correct?
A. Acidosis decreases the solubility of HbS.
B. Acidosis increases the affinity of hemoglobin for O2.
C. Acidosis favors the conversion of hemoglobin from the taut to the relaxed conformation.
D. Acidosis shifts the oxygen-dissociation curve to the left.
E. Acidosis decreases the ability of 2,3-bisphosphoglycerate to bind to hemoglobin.
Correct answer = A. HbS is significantly less soluble in the deoxygenated form, compared with oxyhemoglobin S. A decrease in pH (acidosis) causes the oxygen-dissociation curve to shift to the right, indicating a decreased affinity for oxygen. This favors the formation of the deoxy, or taut, form of hemoglobin, and can precipitate a sickle cell crisis. The binding of 2,3-bisphosphoglycerate is increased, because it binds only to the deoxy form of hemoglobins.
3.3 Which one of the following statements concerning the binding of oxygen by hemoglobin is correct?
A. The Bohr effect results in a lower affinity for oxygen at higher pH values.
B. Carbon dioxide increases the oxygen affinity of hemoglobin by binding to the C-terminal groups of the polypeptide chains.
C. The oxygen affinity of hemoglobin increases as the percentage saturation increases.
D. The hemoglobin tetramer binds four molecules of 2,3-bisphosphoglycerate.
E. Oxyhemoglobin and deoxyhemoglobin have the same affinity for protons.
Correct answer = C. The binding of oxygen at one heme group increases the oxygen affinity of the remaining heme groups in the same molecule. A rise in pH results in increased affinity for oxygen. Carbon dioxide decreases oxygen affinity because it lowers the pH; moreover, binding of carbon dioxide to the N-termini stabilizes the taut, deoxy form. Hemoglobin binds one molecule of 2,3-bisphosphoglycerate. Deoxyhemoglobin has a greater affinity for protons and, therefore, is a weaker acid.
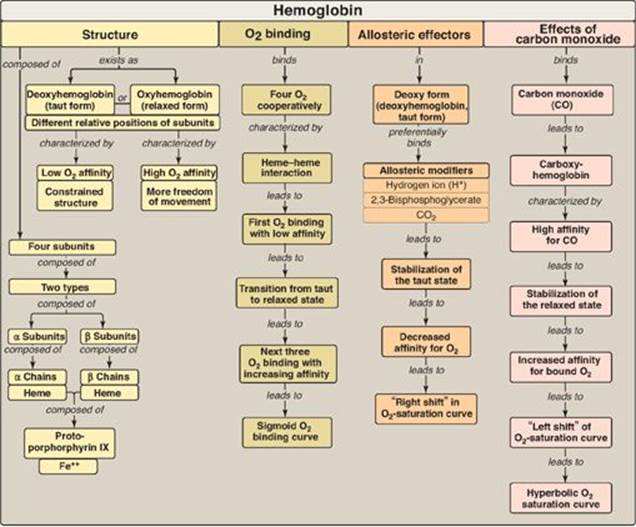
Figure 3.24 Key concept map for hemoglobin structure and function.
3.4 β-Lysine 82 in HbA is important for the binding of 2,3-bisphosphoglycerate. In Hb Helsinki, this amino acid has been replaced by methionine. Which of the following should be true concerning Hb Helsinki?
A. It should be stabilized in the taut, rather than the relaxed, form.
B. It should have increased O2 affinity and, consequently, decreased delivery of O2 to tissues.
C. Its O2-dissociation curve should be shifted to the right relative to HbA.
D. It results in anemia.
Correct answer = B. Substitution of lysine by methionine decreases the ability of negatively charged phosphate groups in 2,3-bisphosphoglycerate (2,3-BPG) to bind the b subunits of hemoglobin. Because 2,3-BPG decreases the O2 affinity of hemoglobin, a reduction in 2,3-BPG should result in increased O2 affinity and decreased delivery of O2 to tissues. The relaxed form is the high-oxygen-affinity form of hemoglobin. Increased O2 affinity (decreased delivery) results in a left shift in the O2-dissociation curve. Decreased O2 delivery is compensated for by increased RBC production.
3.5 Why is hemoglobin C disease a nonsickling disease?
In HbC, the polar glutamate is replaced by polar lysine rather than by nonpolar valine as in HbS.
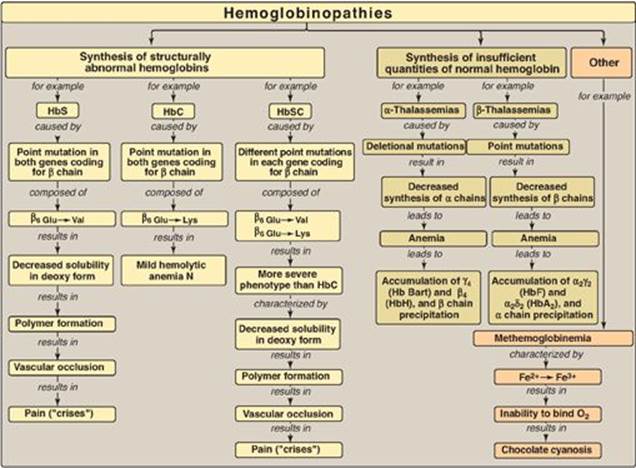
Figure 3.25 Key concept map for hemoglobinopathies. Hb = hemoglobin.
3.6 A 67-year-old man presented to the emergency department with a 1-week history of angina and shortness of breath. He complained that his face and extremities had a “blue color.” His medical history included chronic stable angina treated with isosorbide dinitrate and nitroglycerin. Blood obtained for analysis was brown colored. Which one of the following is the most likely diagnosis?
A. Carboxyhemoglobinemia
B. Hemoglobin SC disease
C. Methemoglobinemia
D. Sickle cell anemia
E. β-Thalassemia
Correct answer = C. Oxidation of the heme component of hemoglobin to the ferric (Fe3+) state forms methemoglobin. This may be caused by the action of certain drugs such as nitrates. The methemoglobinemias are characterized by chocolate cyanosis (a brownish blue coloration of the skin and mucous membranes and chocolate-colored blood) as a result of the dark-colored methemoglobin. Symptoms are related to tissue hypoxia and include anxiety, headache, and dyspnea. In rare cases, coma and death can occur. [Note: Benzocaine, an aromatic amine used as a topical anesthetic, is a cause of acquired methemoglobinemia.]
3.7 What would be true about the extent of red blood cell sickling in individuals with HbS and hereditary persistence of HbF?
Decreased. HbF reduces HbS concentration. It also inhibits polymerization of deoxy HbS.