Lippincott’s Illustrated Reviews: Biochemistr, Sixth Edition (2014)
APPENDIX. Clinical Cases
I. INTEGRATIVE CASES
Metabolic pathways, initially presented in isolation, are, in fact, linked to form an interconnected network. The following four integrative case studies illustrate how a perturbation in one process can result in perturbations in other processes of the network.
CASE 1: CHEST PAIN
Patient Presentation: A 35-year-old man with severe substernal chest pain of about 2 hoursʹ duration is brought by ambulance to his local hospital at 5 a.m. The pain is accompanied by dyspnea (shortness of breath), diaphoresis (sweating), and nausea.
Focused History: The patient reports episodes of exertional chest pain in the last few months, but they were less severe and of short duration. He smokes (2–3 packs per day), drinks alcohol only rarely, eats a “typical” diet, and walks with his wife most weekends. His blood pressure has been normal. Family history reveals that his father and paternal aunt died of heart disease at age 45 years and 39 years, respectively. His mother and younger (age 31 years) brother are said to be in good health.
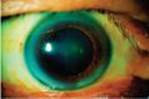
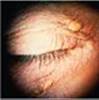
Physical Examination (Pertinent Findings): The patient is pale and clammy and is in distress due to chest pain. Blood pressure and respiratory rate are elevated. Lipid deposits are noted on the periphery of his corneas (corneal arcus; see left Image) and under the skin on and around his eyelids (xanthelasmas; see right Image). No deposits on his tendons (xanthomas) are detected.
Corneal arcus
Xanthelasmas
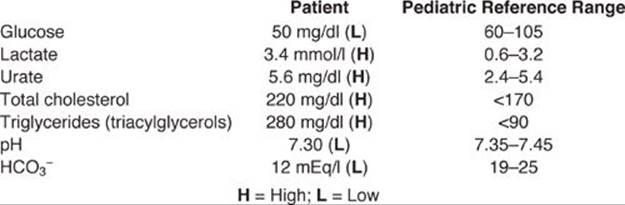
Pertinent Test Results: The patient’s electrocardiogram is consistent with a myocardial infarction (MI). Angiography reveals areas of severe stenosis (narrowing) of several coronary arteries. Initial results from the clinical laboratory include:

Diagnosis: MI, the irreversible necrosis (death) of heart muscle secondary to ischemia (decreased blood supply) is caused by the occlusion (blockage) of a blood vessel most commonly by a blood clot (thrombus). The patient subsequently is determined to have heterozygous familial hypercholesterolemia (FH), also known as type IIa hyperlipidemia.
Immediate Treatment: The patient is given O2, a vasodilator, pain medication, and drugs to dissolve blood clots (thrombolytics) and reduce clotting (antithrombotics).
Long-term Treatment: Lipid-lowering drugs (for example, high-potency statins, bile acid [BA] sequestrants, and niacin), daily aspirin; β-blockers; and counseling on nutrition, exercise, and smoking cessation would be part of the long-term treatment plan.
Prognosis: Patients with heterozygous FH have ~50% of the normal number of functional LDL receptors and a hypercholesterolemia (two to three times normal) that puts them at high risk (>50% risk) for premature coronary heart disease (CHD). However, less than 5% of patients with hypercholesterolemia have FH.
|
Nutrition Nugget: Dietary recommendations for individuals with heterozygous FH include limiting saturated fats to <7% of total calories and cholesterol to <200 mg/day; substituting unsaturated fats for saturated fats; and adding soluble fiber (10–20 g/day) and plant sterols (2 g/day) for their hypocholesterolemic effects. Fiber increases BA excretion. This results in increased hepatic uptake of cholesterol-rich LDLs to supply the substrate for BA synthesis. Plant sterols decrease cholesterol absorption in the intestine. |
Genetics Gem: FH is caused by hundreds of different mutations in the gene for the LDL receptor (on chromosome 19) that affect receptor amount and/or function. FH is an autosomal-dominant disease in which homozygotes are more seriously affected than heterozygotes. Heterozygous FH has an incidence of ~1:500 in the general population. It is associated with increased risk of cardiovascular disease. Genetic screening of the first-degree relatives of the patient would identify affected individuals to allow their treatment. |
REVIEW QUESTIONS: Choose the ONE best answer.
RQ1. Triacylglycerols are glycerol-based lipids. Which of the following is also a glycerol-based lipid?
A. Ganglioside GM2
B. Phosphatidylcholine
C. Prostaglandin PGI2
D. Sphingomyelin
E. Vitamin D
RQ2. Statins are of benefit to patients with hypercholesterolemia because they:
A. decrease a rate-limiting and regulated step of de novo cholesterol biosynthesis by inhibiting hydroxymethylglutaryl coenzyme A (HMG CoA) reductase.
B. decrease expression of the gene for the LDL receptor by preventing the movement of the sterol regulatory element–binding protein-2 (SREBP-2) in complex with SREBP cleavage–activating protein (SCAP) from the membrane of the endoplasmic reticulum to the membrane of the Golgi.
C. increase the oxidation of cholesterol to CO2 + H2O.
D. interfere with the absorption of bile salts in the enterohepatic circulation, thereby causing the liver to take up cholesterol from the blood for use in bile acid synthesis.
E. reduce cholesterol by increasing steroid hormone and vitamin D synthesis.
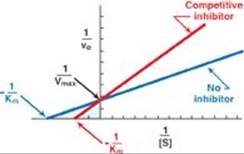
RQ3. Statins are competitive inhibitors of HMG CoA reductase. Which of the following statements about competitive inhibitors is correct?
A. Competitive inhibitors are examples of irreversible inhibitors.
B. Competitive inhibitors increase both the apparent Michaelis constant (Km) and the apparent maximal velocity (Vmax).
C. Competitive inhibitors increase the apparent Km and have no effect on the Vmax.
D. Competitive inhibitors decrease both the apparent Km and the apparent Vmax.
E. Competitive inhibitors have no effect on the Km and decrease the apparent Vmax.
RQ4. In a myocardial infarction, a blood clot forms as a result of injury to a blood vessel that leads to production of a platelet plug and a meshwork of thrombin. The clot occludes the blood vessel, preventing blood flow and, therefore, delivery of O2. Destruction of the clot (thrombolysis) restores blood flow. Which one of the following is an example of a thrombolytic agent?
A. Activated protein C complex
B. Antithrombin III
C. Aspirin
D. Factor XIII
E. Heparin
F. Tissue plasminogen activator
G. Vitamin K
H. Warfarin
RQ5. Decreased tissue perfusion results in hypoxia (decreased O2 availability). Relative to normoxia, in hypoxia the:
A. electron transport will be upregulated to provide protons for adenosine triphosphate synthesis.
B. ratio of the oxidized form of nicotinamide adenine dinucleotide (NAD+) to the reduced form (NADH) will increase.
C. pyruvate dehydrogenase complex will be active.
D. process of substrate-level phosphorylation will be increased in the cytosol.
E. tricarboxylic acid cycle will be upregulated to provide the reducing equivalents needed for oxidative phosphorylation to occur.
RQ6. Genetic screening of the patient’s first-degree relatives would be accomplished by mutation analysis via polymerase chain reaction–based amplification followed by automated sequencing of the promoter region and the 18 exons of the LDL receptor gene. This process would involve the:
A. generation and use of complementary DNA (cDNA).
B. initiation of DNA synthesis with dideoxynucleotides.
C. isolation of genomic DNA from germ cells.
D. use of fluorescently labeled nucleotides.
THOUGHT QUESTIONS
TQ1. Relative to an individual with familial defective LDL receptors, what would be the expected phenotype in an individual with familial defective apolipoprotein B-100? With apolipoprotein E-2, the isoform that only poorly binds its receptor?
TQ2. Why was aspirin prescribed? Hint: What pathway of lipid metabolism is affected by aspirin?
TQ3. Heart muscle normally uses aerobic metabolism to meet its energy needs. However, in hypoxia, anaerobic glycolysis is increased. What allosteric activator of glycolysis is responsible for this effect? With hypoxia, what will be the end product of glycolysis?
TQ4. One of the reasons for encouraging smoking cessation and exercise in the patient is that these changes raise the level of HDLs, and elevated HDL reduces the risk for CHD. How does a rise in HDL reduce the risk for CHD?
CASE 2: SEVERE FASTING HYPOGLYCEMIA
Patient Presentation: Jason S. is a 4-month-old boy whose mother is concerned about the “twitching” movements he makes just before feedings. She tells the pediatrician that the movements started about 1 week ago, are most apparent in the morning, and disappear shortly after eating.
Focused History: Jason is the product of a normal pregnancy and delivery. He appeared normal at birth. He has been at the 30th percentile for both weight and length since birth. His immunizations are up to date. Jason last ate a few hours ago.
Physical Examination (Pertinent Findings): Jason appears sleepy and feels clammy to the touch. His respiratory rate is elevated. His temperature is normal. Jason has a protuberant, firm abdomen that appears to be nontender. His liver is palpable 4 cm below the right costal margin and is smooth. His kidneys are enlarged and symmetrical.
Pertinent Test Results:
Jason is sent to the regional children’s hospital for further evaluation. Ultrasound studies confirm hepatomegaly and renomegaly and show no evidence of tumors. A liver biopsy is performed. The hepatocytes are distended. Staining reveals large amounts of lipid (primarily triacylglycerol) and carbohydrate. Liver glycogen is elevated in amount and normal in structure. Enzyme assay using liver homogenate treated with detergent reveals less than 10% of the normal activity of glucose 6-phosphatase, an enzyme of the endoplasmic reticulum (ER) membrane in the liver and the kidneys.
Diagnosis: Glucose 6-phosphatase deficiency (glycogen storage disease [GSD] type Ia, von Gierke disease)
Treatment (Immediate): Jason was given glucose intravenously, and his blood glucose level rose into the normal range. However, as the day progressed, it fell to well below normal. Administration of glucagon had no effect on blood glucose levels but increased blood lactate. Jason’s blood glucose levels were able to be maintained by constant infusion of glucose.
Prognosis: Individuals with glucose 6-phosphatase deficiency develop hepatic adenomas starting in the second decade of life and are at increased risk for hepatic carcinoma. Kidney glomerular function is impaired and can result in kidney failure. Patients are at increased risk for developing gout, but this rarely occurs before puberty.
|
Nutrition Nugget: Long-term medical nutrition therapy for Jason is designed to maintain his blood glucose levels in the normal range. Frequent (every 2–3 hours) daytime feedings rich in carbohydrate (provided by uncooked cornstarch that is slowly hydrolyzed) and nighttime nasogastric infusion (pump assisted) of glucose are advised. Avoidance of fructose and galactose is recommended because they are metabolized to glycolytic intermediates and lactate and can exacerbate the metabolic problems. Calcium and vitamin D supplements are prescribed. |
Genetics Gem: GSD Ia is an autosomal-recessive disorder caused by more than 100 known mutations to the gene for glucose 6-phosphatase located on chromosome 17. It has an incidence of 1:100,000 and accounts for approximately 25% of all cases of GSDs in the United States. It is one of the few genetic causes of hypoglycemia in newborns. GSD Ia is not routinely screened for in newborns. [Note: Deficiency of the translocase that moves glucose 6-phosphate out of the cytosol and into the ER is the cause of GSD Ib.] |
REVIEW QUESTIONS: Choose the ONE best answer.
RQ1. Jason is hypoglycemic because:
A. free (nonphosphorylated) glucose cannot be produced from either glycogenolysis or gluconeogenesis as a result of the deficiency in glucose 6-phosphatase.
B. glycogen phosphorylase is dephosphorylated and inactive, and glycogen cannot be degraded.
C. hormone-sensitive lipase is dephosphorylated and inactive, and fatty acid substrates for gluconeogenesis cannot be generated.
D. the decrease in the insulin/glucagon ratio upregulates glucose transporters in the liver and kidneys, resulting in increased uptake of blood glucose.
RQ2. Jason was prescribed calcium supplements because chronic acidosis can cause bone demineralization, resulting in osteopenia. Vitamin D was also prescribed because vitamin D:
A. binds Gq protein–coupled membrane receptors and causes a rise in inositol trisphosphate with release of calcium from intracellular stores.
B. cannot be synthesized by humans and, therefore, must be supplied in the diet.
C. is a fat-soluble vitamin that increases intestinal absorption of calcium.
D. is the coenzyme-prosthetic group for calbindin, a calcium transporter in the intestine.
RQ3. The hepatomegaly and renomegaly seen in Jason are primarily the result of an increase in the amount of glycogen stored in these organs. What is the basis for glycogen accumulation in these organs?
A. Glycolysis is downregulated, which pushes glucose to glycogenesis.
B. Increased oxidation of fatty acids spares glucose for glycogenesis.
C. Glucose 6-phosphate is an allosteric activator of glycogen synthase b.
D. The rise in the insulin/glucagon ratio favors glycogenesis.
RQ4. Glucose 6-phosphatase is an integral protein of the membrane of the endoplasmic reticulum. Which of the following statements about such proteins is correct?
A. If glycosylated, the carbohydrate is on the portion of the protein that extends into the cytosol.
B. They are synthesized on ribosomes that are free in the cytosol.
C. The membrane-spanning domain consists of hydrophilic amino acids.
D. The initial targeting signal is an amino terminal hydrophobic signal sequence.
THOUGHT QUESTIONS
TQ1. What is the likely reason for Jason’s twitching movements?
TQ2. Why was the liver homogenate treated with detergent? Hint: Think about where the enzyme is located.
TQ3. Why is Jason’s blood glucose level unaffected by glucagon? Hint: What is the role of glucagon in normal individuals who experience a drop in blood glucose?
TQ4. Why are urate and lactate elevated in a disorder of glycogen metabolism? Hint: It is the result of a decrease in inorganic phosphate (Pi), but why is Pi decreased?
TQ5. A. Why are triacylglycerols and cholesterol elevated? Hint: Glucose is the primary carbon source for their synthesis.
B. Why are ketone bodies not elevated?
CASE 3: HYPERGLYCEMIA AND HYPERKETONEMIA
Patient Presentation: A 40-year-old woman was brought to the hospital in a disoriented, confused state by her husband.
Focused History: As noted on her medical alert bracelet, the patient has had type 1 diabetes (T1D) for the last 24 years. Her husband reports that this is her first medical emergency in 2 years.
Physical Examination (Pertinent Findings): The patient displayed signs of dehydration (such as dry mucous membranes and skin, poor skin turgor, and low blood pressure) and acidosis (such as deep and rapid breathing [Kussmaul respiration]). Her breath had a faintly fruity odor. Her temperature was normal.
Pertinent Test Results: Rapid, bedside tests were strongly positive for glucose and acetoacetate and negative for protein. Results on blood tests performed by the clinical laboratory are shown below:
Microscopic examination of her urine revealed a urinary tract infection (UTI).
Diagnosis: The patient is in diabetic ketoacidosis that was precipitated by a UTI. [Note: Diabetes increases the risk for infections such as UTIs.]
Immediate Treatment: The patient was rehydrated with normal saline given intravenously (IV). She also was given insulin IV. Blood glucose, ketone bodies, and electrolytes were measured periodically. The patient was started on an antibiotic for her UTI.
Long-term Treatment: Diabetes increases the risk for macrovascular complications (such as coronary artery disease and stroke) and microvascular complications (such as retinopathy, nephropathy, and neuropathy). Ongoing monitoring for these complications will be continued.
Prognosis: Diabetes is the sixth leading cause of death by disease in the United States. Individuals with diabetes have a reduced life expectancy relative to those without diabetes.
|
Nutrition Nugget: Monitoring total intake of carbohydrate is primary in blood glucose control. Carbohydrate should come from whole grains, vegetables, legumes, and fruits. Low-fat dairy products and nuts and fish rich in ω-3 fatty acids are encouraged. Intake of saturated and trans fats should be minimized. |
Genetics Gem: Autoimmune destruction of pancreatic β cells is characteristic of T1D. Of the genetic loci that confer risk for T1D, the human-leukocyte antigen (HLA) region on chromosome 6 has the strongest association. The majority of genes in the HLA region are involved in the immune response. |
REVIEW QUESTIONS: Choose the ONE best answer.
RQ1. Which of the following statements concerning T1D is correct?
A. Diagnosis can be made by measuring the level of glucose or glycosylated hemoglobin (HbA1C) in the blood.
B. During periods of physiologic stress, the urine of an individual with T1D would likely test negative for reducing sugars.
C. T1D is associated with obesity and a sedentary lifestyle.
D. The characteristic metabolic abnormalities seen in T1D result from insensitivity to both insulin and glucagon.
E. Treatment with exogenous insulin allows normalization of blood glucose (euglycemia).
RQ2. Diabetic ketoacidosis occurs when the rate of ketone body production is greater than the rate of utilization. Which of the following statements concerning ketone body metabolism is correct? Ketone bodies:
A. are made in mitochondria from acetyl coenzyme A (CoA) produced by the oxidation of glucose.
B. are utilized by many tissues, particularly the liver, after conversion to acetyl CoA.
C. include acetoacetate, which can impart a fruity odor to the breath.
D. require albumin for transport through the blood.
E. utilized in energy metabolism are organic acids that can add to the proton load of the body.
RQ3. Adipose lipolysis followed by β-oxidation of the fatty acid products is required for the generation of ketone bodies. Which of the following statements concerning the generation and use of fatty acids is correct?
A. Mitochondrial β-oxidation of fatty acids is inhibited by malonyl CoA.
B. Production of fatty acids from adipose lipolysis is upregulated by insulin.
C. The acetyl CoA product of fatty acid β-oxidation favors the use of pyruvate for gluconeogenesis by activating the pyruvate dehydrogenase complex.
D. The β-oxidation of fatty acids utilizes reducing equivalents generated by gluconeogenesis.
E. The fatty acids produced by lipolysis are taken up by the brain and oxidized for energy.
THOUGHT QUESTIONS
TQI. At admission, the patient was hypoinsulinemic, and she was given insulin. Why did the patient’s hypoinsulinemia result in hyperglycemia? Hint: What is the role of insulin in glucose metabolism?
TQ2. Why is there glucose in the patient’s urine (glucosuria)? How is the glucosuria related to her dehydrated state?
TQ3. Why is the majority of the acetyl CoA from fatty acid β-oxidation being used for ketogenesis rather than being oxidized in the tricarboxylic acid cycle?
TQ4. Was the patient in positive or negative nitrogen balance when she was brought to the hospital?
TQ5. What response to the ketoacidosis is apparent in the patient? What response is likely occurring in the kidney? Hint: In addition to conversion to urea, how is toxic ammonia removed from the body?
TQ6. What would be true about the levels of ketone bodies and glucose during periods of physiologic stress in individuals with impaired fatty acid oxidation?
CASE 4: HYPOGLYCEMIA, HYPERKETONEMIA, AND LIVER DYSFUNCTION
Patient Presentation: AK, a 59-year-old male with slurred speech, ataxia (loss of skeletal muscle coordination), and abdominal pain, was dropped off at the Emergency Department (ED).
Focused History: AK is known to the ED staff from previous visits. He has a 6-year history of chronic, heavy alcohol consumption. He is not known to take illicit drugs. At this ED visit, AK reports that he has been drinking heavily in the past day or so. He cannot recall having eaten anything in that time. There is evidence of recent vomiting, but no blood is apparent.
Physical Examination (Pertinent Findings): The physical examination was remarkable for the emaciated appearance of the patient. (His body mass index was later determined to be 17.5, which put him in the underweight category.) The cheeks of his face were erythematous (red in color) due to dilated blood vessels in the skin (telangiectasia). Eye movement was normal. Neither icterus (jaundice) nor edema (swelling due to fluid retention) was seen. The liver was slightly enlarged. Bedside tests revealed hypoglycemia and hyperketonemia (as acetoacetate). Blood was drawn and sent to the clinical laboratory.
Pertinent Test Results:
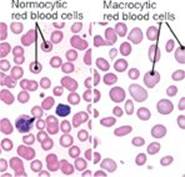
Additional Tests: Complete blood count (CBC) and blood smear revealed a macrocytic anemia (right Image). Folate and B12 levels were ordered.
Diagnosis: Alcoholism
Treatment (Immediate): Thiamine and glucose were given intravenously.
Prognosis: Alcoholism (alcohol dependence) is the third most common cause of preventable death in the United States. People with alcoholism are at increased risk for liver cirrhosis, pancreatitis, gastrointestinal bleeding, and some cancers.
|
Nutrition Nugget: Those with alcoholism are at risk for vitamin deficiencies as a result of decreased intake and absorption. Thiamine (vitamin B1) deficiency is common and can have serious consequences such as Wernicke-Korsakoff syndrome with its neurologic effects. Thiamine pyrophosphate (TPP), the coenzyme form, is required for the dehydrogenase-mediated oxidation of α-keto acids (such as pyruvate) as well as the transfer of two-carbon ketol groups by transketolase in the reversible sugar interconversions in the pentose phosphate pathway. |
Genetics Gem: Acetaldehyde, the product of ethanol oxidation by the hepatic, cytosolic, nicotinamide adenine dinucleotide (NAD+)-requiring enzyme alcohol dehydrogenase (ADH), is oxidized to acetate by the mitochondrial, NAD+-requiring aldehyde dehydrogenase (ALDH2). The majority of individuals of East Asian (but not European or African) heritage have a single nucleotide polymorphism (SNP) that renders ALDH2 essentially inactive. This results in aldehyde-induced facial flushing and mild to moderate intoxication after consumption of small amounts of ethanol. |
REVIEW QUESTIONS: Choose the ONE best answer.
RQ1. Many of the metabolic consequences of chronic excessive alcohol consumption seen in the patient are the result of an increase in the ratio of reduced nicotinamide adenine dinucleotide (NADH) to its oxidized form (NAD+) in both the cytoplasm and mitochondrion. Which of the following statements concerning the effects of the rise in mitochondrial NADH is correct?
A. Fatty acid oxidation is increased.
B. Gluconeogenesis is increased.
C. Lipolysis is inhibited.
D. The tricarboxylic acid cycle is inhibited.
E. The reduction of malate to oxaloacetate in the malate–aspartate shuttle is increased.
RQ2. Ethanol can also be oxidized by cytochrome P450 (CYP) enzymes, and CYP2E1 is an important example. CYP2E1, which is ethanol inducible, generates reactive oxygen species (ROS) in its metabolism of ethanol. Which of the following statements concerning the CYP proteins is correct?
A. CYP proteins are heme-containing dioxygenases.
B. CYP proteins of the inner mitochondrial membrane are involved in detoxification reactions.
C. CYP proteins of the smooth endoplasmic reticulum membrane are involved in the synthesis of steroid hormones, bile acids, and calcitriol.
D. ROS such as hydrogen peroxide generated by CYP2E1 can be oxidized by glutathione peroxidase.
E. The pentose phosphate pathway is an important source of the nicotinamide adenine dinucleotide phosphate (NADPH) that provides the reducing equivalents needed for activity of CYP proteins and the regeneration of functional glutathione.
RQ3. Alcohol is known to modulate the levels of serotonin in the central nervous system, where it functions as a neurotransmitter. Which of the following statements about serotonin is correct? Serotonin is:
A. associated with anxiety and depression.
B. degraded via methylation by monoamine oxidase, which also degrades the catecholamines.
C. released by activated platelets.
D. synthesized from tyrosine in a two-step process that utilizes a tetrahydrobiopterin-requiring hydroxylase and a pyridoxal phosphate-requiring carboxylase.
RQ4. Chronic, excessive consumption of alcohol is a leading cause of acute pancreatitis, a painful inflammatory condition that results from autodigestion of the gland by premature activation of pancreatic enzymes. Which of the following statements concerning the pancreas is correct?
A. Autodigestion of the pancreas would be expected to result in a decrease in pancreatic proteins in the blood.
B. In individuals who progress from acute to chronic pancreatitis, with the characteristic structural changes that result in decreased pancreatic function, diabetes and steatorrhea are expected findings.
C. In response to secretin, the exocrine pancreas secretes protons to lower the pH in the intestinal lumen.
D. Pancreatitis may also be seen in individuals with hypercholesterolemia.
THOUGHT QUESTIONS
TQ1. A. What effect does the rise in cytosolic NADH seen with ethanol metabolism have on glycolysis? Hint: What coenzyme is required in glycolysis?
B. How does this relate to the fatty liver (steatosis) commonly seen in alcohol-dependent individuals?
TQ2. Why might individuals with a history of gouty attacks be advised to reduce their consumption of ethanol?
TQ3. Why might prothrombin time be affected in alcohol-dependent individuals?
TQ4. Folate and vitamin B12 deficiencies cause a macrocytic anemia that may be seen in those with alcoholism. Why is it advisable to measure vitamin B12 levels before supplementing with folate in an individual with macrocytic anemia?
II. INTEGRATIVE CASE ANSWERS
CASE 1: Answers to Review Questions
RQ1. Answer = B. Phosphatidylcholine is a glycerol-based phospholipid derived from diacylglycerol phosphate (phosphatidic acid) and cytidine diphosphate-choline. Gangliosides are derived from ceramides, lipids with a sphingosine backbone. Prostaglandins of the 2 series are derived from the 20-carbon polyunsaturated fatty acid arachadonic acid. Sphingomyelin is a sphingophospholipid derived from ceramide. Vitamin D is derived from an intermediate in the biosynthetic pathway for the sterol cholesterol.
RQ2. Answer = A. Statins inhibit hydroxymethylglutaryl coenzyme A (HMG CoA) reductase, thereby preventing the reduction of HMG CoA to mevalonate and decreasing cholesterol biosynthesis (see Figure). The decrease in cholesterol content caused by statins results in movement of the sterol regulatory element–binding protein-2 (SREBP-2) in complex with SREBP cleavage–activating protein (SCAP) from the endoplasmic reticular membrane to the Golgi membrane, where SREBP-2 is cleaved, generating a transcription factor that moves to the nucleus and binds to the sterol regulatory element upstream of the genes for HMG CoA reductase and the low-density lipoprotein (LDL) receptor, increasing their expression. Humans are unable to degrade the steroid nucleus to CO2 + H2O. Bile acid sequestrants, such as cholestyramine, prevent the absorption of bile salts by the liver, thereby increasing their excretion. The liver then takes up cholesterol via the LDL receptor and uses it to make bile acids, thereby reducing blood cholesterol levels. Steroid hormones are synthesized from cholesterol, and vitamin D is synthesized from an intermediate (7-dehydrocholesterol) in the cholesterol biosynthetic pathway. Therefore, inhibition of cholesterol synthesis would be expected to decrease their production as well.
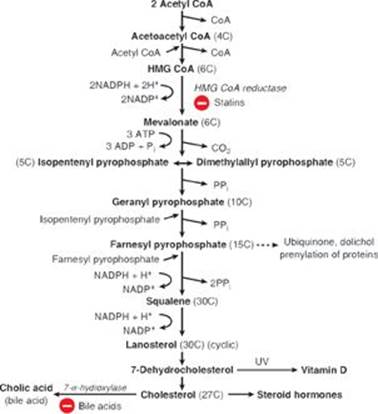
RQ3. Answer = C. Competitive inhibitors bind to the same site as the substrate. Once bound, they prevent the substrate from binding. This results in an increase in the apparent Km, that substrate concentration that gives one half of the maximal velocity (Vmax). However, because the inhibition can be reversed by adding additional substrate, the Vmax is unchanged (see Figure). It is noncompetitive inhibitors that decrease the apparent Vmax and have no effect on Km.
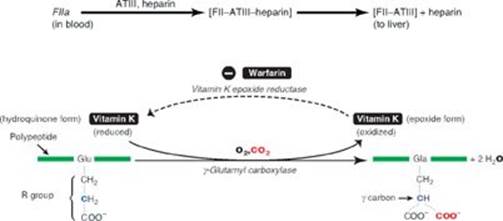
RQ4. Answer = F. Tissue plasminogen activator converts plasminogen to plasmin that degrades fibrin (fibrinolysis), thereby degrading the clot (thrombolysis). Aspirin, an inhibitor of cyclooxygenase, is an antiplatelet drug. Antithrombin III (ATIII) removes thrombin from the blood, and its action is potentiated by heparin. Activated protein C (APC) complex cleaves the accessory proteins factor (F)Va and FVIIIa. ATIII and APC are involved in anticoagulation. FXIII is a transglutaminase that cross-links the fibrin mesh. Vitamin K is a fat-soluble vitamin required for the γ-carboxylation of FII, FVII, FIX, and FX. Warfarin prevents regeneration of the functional, reduced form of vitamin K.
RQ5. Answer = D. In hypoxia, substrate level phosphorylation in glycolysis provides adenosine triphosphate (ATP). Oxidative phosphorylation is inhibited by the lack of O2. Because the rate of ATP synthesis by oxidative phosphorylation controls the rate of cellular respiration, electron transport is inhibited. The resulting rise in the ratio of the reduced form of nicotinamide adenine dinucleotide (NADH) to the oxidized form (NAD+) inhibits the tricarboxylic acid cycle and the pyruvate dehydrogenase complex.
RQ6. Answer = D. Fluorescently labeled nucleotides allow the base sequence of the DNA of interest to be detected. Complementary DNA (cDNA) is generated from processed mRNA and would not contain the promoter. Dideoxynucleotides lack the 3-OH needed to form the 3’→5’ phosphodiester bond that joins the nucleotides and, thus, will terminate DNA synthesis. Genomic DNA obtained from white cells isolated from a blood sample would be the source of the DNA.
CASE 1: Answers to Thought Questions
TQ1. The phenotype would be the same. In familial defective apolipoprotein B-100, LDL receptors are normal in number and function, but the ligand for the receptor is altered such that binding to the receptor is decreased. Decreased ligand–receptor binding results in increased levels of LDL in the blood with hypercholesterolemia. [Note: The phenotype would be the same in individuals with a gain-of-function mutation to PCSK9, the protease that decreases recycling of the LDL receptor, thereby increasing its degradation.] With the apolipoprotein E-2 isoform, chylomicron remnants and intermediate-density lipoproteins would accumulate in blood.
TQ2. Aspirin irreversibly inhibits cyclooxygenase (COX) and, therefore, the synthesis of prostaglandins (PGs), such as PGI2 in vascular endothelial cells, and thromboxanes (TXs), such as TXA2 in activated platelets. TXA2promotes vasoconstriction and formation of a platelet plug, whereas PGI2 inhibits these events. Because platelets are anucleate, they can’t overcome this inhibition by synthesizing more COX. However, endothelial cells have a nucleus. Aspirin, then, inhibits formation of blood clots by preventing production of TXA2 for the life of the platelet.
TQ3. The decrease in ATP (as the result of a decrease in O2 and, thus, a decrease in oxidative phosphorylation) causes an increase in adenosine monophosphate (AMP). AMP allosterically activates phosphofructokinase-1, the key regulated enzyme of glycolysis. The rise in glycolysis increases the production of ATP by substrate-level phosphorylation. It also increases the ratio of the reduced to oxidized forms of nicotinamide adenine dinucleotide (NAD). Under anaerobic conditions, pyruvate produced in glycolysis is reduced to lactate by lactate dehydrogenase as NADH is oxidized to NAD+. NAD+ is required for continued glycolysis. Because fewer ATP molecules are produced per molecule of substrate in substrate-level phosphorylation relative to oxidative phosphorylation, there is a compensatory increase in the rate of glycolysis under anaerobic conditions.
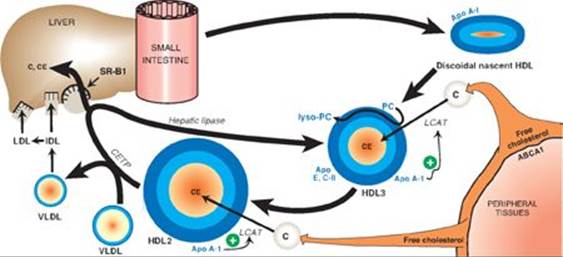
TQ4. High-density lipoprotein (HDL) functions in “reverse” cholesterol transport. It takes cholesterol from nonhepatic (peripheral) tissues (for example, the endothelial layer of arteries) and brings it to the liver (see Figure below). The ABCA1 transporter mediates the efflux of cholesterol to HDL. The cholesterol is esterified by lecithin-cholesterol acyl transferase (LCAT) that requires apo A-1 as a coenzyme. Some cholesteryl ester is transferred to very-low-density lipoproteins (VLDLs) by cholesteryl-ester transfer protein (CETP) in exchange for triacyl-glycerol. The remainder is taken up by a scavenger receptor (SR-B1) on the surface of hepatocytes. The liver can use the cholesterol from HDL in the synthesis of bile acids. Removal of cholesterol from endothelial cells prevents its accumulation (as cholesterol or cholesteryl ester), decreasing the risk of heart disease. [Note: In contrast, LDL carries cholesterol from the liver to peripheral tissues.]
CASE 2: Answers to Review Questions
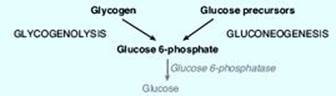
RQ1. Answer = A. Deficiency of glucose 6-phosphatase prevents the glucose 6-phosphate generated by glycogenolysis and gluconeogenesis from being dephosphorylated and released into the blood (see Figure below). Blood glucose levels fall, and a severe, fasting hypoglycemia results. [Note: Jason’s symptoms appeared only recently because, at age 4 months, he is going longer periods between feedings.] Hypoglycemia stimulates release of glucagon, which leads to phosphorylation and activation of glycogen phosphorylase kinase that phosphorylates and activates glycogen phosphorylase. Epinephrine is also released and leads to phosphorylation and activation of hormone-sensitive lipase. However, typical fatty acids cannot serve as substrates for gluconeogenesis. The glucose transporters in the liver and kidneys are insulin insensitive.
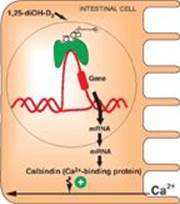
RQ2. Answer = C. Vitamin D is a fat-soluble vitamin that functions as a steroid hormone. In complex with its intracellular nuclear receptor, it increases transcription of the gene for calbindin, a calcium transporter protein in the intestine (see Figure). Vitamin D does not bind to a membrane receptor and does not produce second messengers. It can be synthesized in the skin by the action of ultraviolet light on an intermediate of cholesterol synthesis, 7-dehydrocholesterol. Of the fat-soluble vitamins (A, D, E, and K), only vitamin K functions as a coenzyme.
RQ3. Answer = C. Glucose 6-phosphate is a positive allosteric effector of the covalently inhibited (phosphorylated) glycogen synthase b. With the rise in glucose 6-phosphate, glycogen synthesis is activated and glycogen stores are increased in both the liver and kidneys. The increased availability of glucose 6-phosphate also drives glycolysis. The increase in glycolysis provides substrates for lipogenesis, thereby increasing synthesis of fatty acids and triacylglycerols (TAGs). In hypoglycemia, the insulin/glucagon ratio is low, not high.
RQ4. Answer = D. Membrane proteins are initially targeted to the endoplasmic reticulum (ER) by an amino terminal hydrophobic signal sequence. Glycosylation is the most common posttranslational modification found in proteins. The glycosylated portion of membrane proteins is found on the outside face of the membrane. The membrane-spanning domain consists of approximately 22 hydrophobic amino acids. Proteins destined for secretion or for membrane, the ER lumen, Golgi, or lysosomes are synthesized on ribosomes associated with the ER.
CASE 2: Answers to Thought Questions
TQ1. The twitching is the result of the adrenergic response to hypoglycemia and is mediated by the rise in epinephrine. The adrenergic response includes tremor and sweating. Neuroglycopenia (impaired delivery of glucose to the brain) results in impairment of brain function that can lead to seizures, coma, and death. Neuroglycopenic symptoms develop if the hyperglycemia persists.
TQ2. Detergents are amphipathic molecules (that is they have both hydrophilic [polar] and hydrophobic [nonpolar] regions). Detergents solubilize membranes, thereby disrupting membrane structure. If the problem were the translocase needed to move the glucose 6-phosphate substrate into the ER, rather than the phosphatase, disruption of the ER membrane would allow the substrate access to the phosphatase.
TQ3. Glucagon, a peptide hormone released in hypoglycemia, binds its plasma membrane G protein–coupled receptor on hepatocytes. The as subunit of the trimeric G protein is activated (guanosine diphosphate is replaced by guanosine triphosphate), separates from the β and γ subunits, and activates adenylyl cyclase that generates cyclic adenosine monophosphate (cAMP) from adenosine triphosphate (ATP). cAMP activates protein kinase A (PKA) that phosphorylates and activates glycogen phosphorylase kinase, which phosphorylates and activates glycogen phosphorylase. The phosphorylase degrades glycogen, generating glucose 1-phosphate that is converted to glucose 6-phosphate. With glucose 6-phosphatase deficiency, the degradative process stops here (see Figure). Consequently, administration of glucagon is unable to cause a rise in blood glucose. [Note: Epinephrine would be similarly ineffective.]
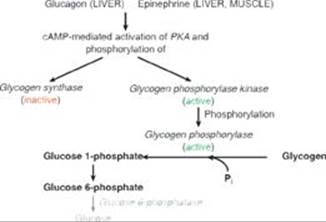
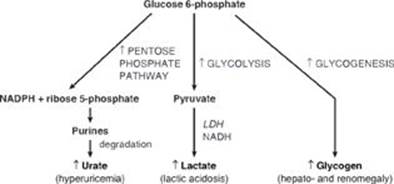
TQ4. The availability of inorganic phosphate (Pi) is decreased because it is trapped as phosphorylated glycolytic intermediates as a result of the upregulation of glycolysis by the rise in glucose 6-phosphate. Urate is elevated because the trapping of Pi decreases the ability to phosphorylate adenosine diphosphate (ADP) to ATP, and the fall in ATP causes a rise in adenosine monophosphate (AMP). The AMP is degraded to urate. Additionally, the availability of glucose 6-phosphate drives the pentose phosphate pathway, resulting in a rise in ribose 5-phosphate (from ribulose 5-phosphate) and, consequently, a rise in purine synthesis. Purines made beyond need are degraded to urate (see Figure below). [Note: The decrease in Pi reduces the activity of glycogen phosphorylase, resulting in increased storage of glycogen with a normal structure.] Lactate is elevated because the decrease in phosphorylation of ADP to ATP results in a decrease in cellular respiration (respiratory control) as a result of these processes being coupled. As a consequence, reduced nicotinamide adenine dinucleotide (NADH) from glycolysis cannot be oxidized by complex I of the electron transport chain. Instead, it is oxidized by cytosolic lactate dehydrogenase (LDH) with its coenzyme NADH as pyruvate is reduced to lactate. [Note: Pyruvate is increased as a result of the increase in glycolysis.] The lactate ionizes, releasing protons (H+) and leading to a metabolic acidosis (low pH caused here by increased production of acid). Respiratory compensation causes an increased respiratory rate.
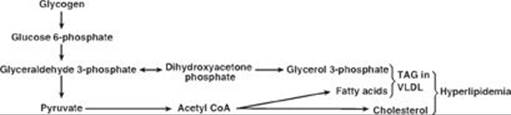
TQ5. Increased glycolysis results in increased availability of glycerol 3-phosphate for hepatic TAG synthesis. Additionally, some of the pyruvate generated in glycolysis will be oxidatively decarboxylated to acetyl coenzyme A (CoA). However, the tricarboxylic acid cycle is inhibited by the rise in NADH, and the acetyl CoA is transported to the cytosol as citrate. The rise of acetyl CoA in the cytosol results in increased fatty acid synthesis. Recall that citrate is an allosteric activator of acetyl CoA carboxylase (ACC). The malonyl product of ACC inhibits fatty acid oxidation at the carnitine palmitoyltransferase I step. Because mitochondrial fatty acid oxidation generates the acetyl CoA substrate for hepatic ketogenesis, ketone body levels do not rise. The fatty acids get esterified to the glycerol backbone, resulting in an increase in TAGs that get sent out of liver as components of very-low-density lipoproteins (VLDLs). [Note: The hypoglycemia results in release of epinephrine and the activation of TAG lipolysis with release of free fatty acids into the blood. The fatty acids are used in hepatic TAG synthesis.] The acetyl CoA is also a substrate for cholesterol synthesis. Thus, the increase in glycolysis results in the hyperlipidemia seen in the patient (see Figure).
CASE 3: Answers to Review Questions
RQ1. Correct answer = A. Diabetes is characterized by hyperglycemia. Chronic hyperglycemia can result in the nonenzymatic glycosylation (glycation) of hemoglobin (Hb) producing HbA1c. Therefore, measurement of glucose or HbA1c in the blood is used to diagnose diabetes. In response to physiologic stress (for example, a urinary tract infection), secretion of counterregulatory hormones (such as the catecholamines) results in a rise in blood glucose. Glucose is a reducing sugar. It is type 2 diabetes (T2D) that is associated with obesity and a sedentary lifestyle and is caused by insensitivity to insulin (insulin resistance). T1D is caused by lack of insulin as a result of the autoimmune destruction of pancreatic β cells. Even individuals on a program of tight glycemic control do not achieve euglycemia.
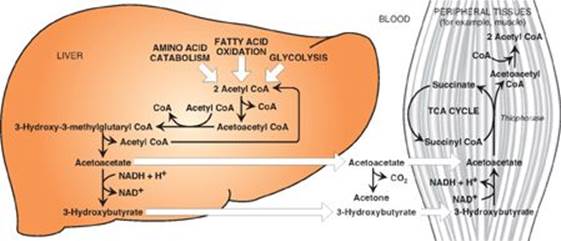
RQ2. Correct answer = E. 3-Hydroxybutyrate and acetoacetate are organic acids, and their ionization contributes to the proton load of the body. Ketone bodies are made in the mitochondria of liver cells using acetyl coenzyme A (CoA) generated primarily from the β-oxidation of fatty acids (see Figure below). Because they are water soluble, they do not require a transporter. Liver cannot use them because it lacks the enzyme thiophorase, which moves CoA from succinyl CoA to acetoacetate for conversion to two molecules of acetyl CoA. It is the acetone released in the breath that can impart a fruity odor.
RQ3. Correct answer = A. Malonyl CoA, an intermediate of fatty acid synthesis, inhibits fatty acid β-oxidation through inhibition of carnitine palmitoyltransferase I. Lipolysis occurs when the insulin-to-counterregulatory hormone ratio decreases. Acetyl CoA, the product of fatty acid β-oxidation, inhibits pyruvate dehydrogenase (PDH) through activation of PDH kinase and activates pyruvate carboxylase. Acetyl CoA then pushes pyruvate to gluconeogenesis. β-Oxidation generates reduced nicotinamide adenine dinucleotide (NADH), the reducing equivalent required for gluconeogenesis. The blood–brain barrier inhibits use of fatty acids by the brain.
CASE 3: Answers to Thought Questions
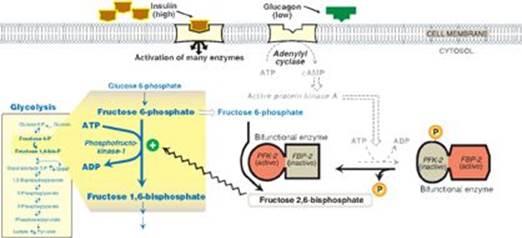
TQ1. Hypoinsulinemia results in hyperglycemia because insulin is required for the uptake of blood glucose by muscle and adipose tissue. Their glucose transporter (GLUT-4) is insulin dependent in that insulin is required for movement of the transporter to the cell surface from intracellular storage sites. Insulin is also required to suppress hepatic gluconeogenesis. Insulin suppresses the release of glucagon from pancreatic α cells. The resulting rise in the insulin-to-glucagon ratio results in the dephosphorylation and activation of the kinase domain of bifunctional phosphofructokinase-2 (PFK-2). The fructose 2,6-bisphosphate produced by PFK-2 inhibits fructose 1,6-bisphosphatase, thereby inhibiting gluconeogenesis, and activates phosphofructokinase-1 (PFK-1) of glycolysis (see Figure). With hypoinsulinemia, the failure to take up glucose from the blood while simultaneously sending it out into the blood results in hyperglycemia.
TQ2. The blood glucose level has exceeded the capacity of the kidney to reabsorb glucose (via a sodium-dependent glucose transporter [SGLT]). The high concentration of glucose in the urine osmotically draws water from the body. This causes increased urination (polyuria) with loss of water that results in dehydration.
TQ3. The NADH generated in fatty acid β-oxidation inhibits the tricarboxylic acid (TCA) cycle at the three NADH-producing dehydrogenase steps. This shifts acetyl CoA away from oxidation in the TCA cycle and toward use as a substrate in hepatic ketogenesis.
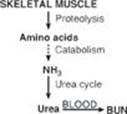
TQ4. The patient was in negative nitrogen balance: more nitrogen was going out than coming in. This is reflected in the elevated blood urea nitrogen (BUN) level seen in the patient (see Figure). [Note: The BUN value also reflects dehydration.] Muscle proteolysis and amino acid catabolism are occurring as a result of the fall in insulin. (Recall that skeletal muscle does not express the glucagon receptor.) Amino acid catabolism produces ammonia (NH3), which is converted to urea by the hepatic urea cycle and sent into the blood. [Note: Urea in the urine is reported as urinary urea nitrogen (UUN).]
TQ5. The Kussmaul respiration seen in the patient is a respiratory response to the metabolic acidosis. Hyperventilation blows off carbon dioxide (CO2) and water, reducing the concentration of protons (H+) and bicarbonate (HCO3−) as reflected in the following equation:
H+ + HCO3− ↔ H2CO3 (carbonic acid) ↔ CO2 + H2O.
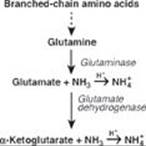
The renal response includes, in part, the excretion of H+ as ammonium (NH4+). Degradation of branched-chain amino acids in skeletal muscle results in the release of large amounts of glutamine into the blood. The kidneys take up and catabolize the glutamine generating NH3 in the process. The NH3 is converted to NH4+ by secreted H+ and is excreted (see Figure). [Note: When ketone bodies are plentiful, enterocytes shift to using them as a fuel instead of glutamine. This increases the amount of glutamine going to the kidney.]
TQ6. Because fatty acid β-oxidation supplies the acetyl CoA substrate for ketogenesis, impaired β-oxidation decreases the ability to make ketone bodies. Ketone bodies are an alternate to the use of glucose, and, thus, dependence on glucose increases. Because fatty acid β-oxidation supplies the NADH and the nucleoside triphosphates needed for gluconeogenesis, glucose production decreases. The result is a hypoketotic hypoglycemia. Recall that this was seen with medium-chain acyl CoA dehydrogenase (MCAD) deficiency.
CASE 4: Answers to Review Questions
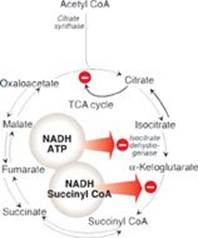
RQ1. Answer = D. The rise in reduced nicotinamide adenine dinucleotide (NADH) in the mitochondria decreases the tricarboxylic acid (TCA) cycle, fatty acid oxidation, and gluconeogenesis. NADH inhibits the isocitrate dehydrogenase reaction, the key regulated step of the TCA cycle, and the α-ketoglutarate dehydrogenase reaction (see Figure). It also favors the reduction of oxaloacetate to malate (not malate to oxaloacetate), decreasing the availability of oxaloacetate for condensation with acetyl coenzyme A (CoA) in the TCA cycle and for gluconeogenesis. Fatty acid oxidation requires NAD+ for the 3-hydroxyacyl CoA dehydrogenase step and, thus, is inhibited by the rise in NADH. The decrease in fatty acid oxidation decreases the production of adenosine triphosphate (ATP) and acetyl CoA (the allosteric activator of pyruvate carboxylase) needed for gluconeogenesis. Lipolysis is activated in fasting as a consequence of the fall in insulin and the rise in catecholamines that result in activation of hormone-sensitive lipase.
RQ2. Answer = E. The irreversible, oxidative portion of the pentose phosphate pathway provides the nicotinamide adenine dinucleotide phosphate (NADPH) that supplies the reducing equivalents needed for activity of cytochrome P450 (CYP) proteins and for the regeneration of functional (reduced) glutathione. It is also an important source of NADPH for reductive biosynthetic processes in the cytosol, such as fatty acid and cholesterol synthesis. CYP proteins are monooxygenases (mixed-function oxidases). They incorporate one O atom from O2 into the substrate as the other is reduced to water. It is the CYP proteins of the smooth endoplasmic reticulum membrane that are involved in detoxification reactions. Those of the inner mitochondrial membrane are involved in the synthesis of steroid hormones, bile acids, and calcitriol. Reactive oxygen species are reduced by glutathione peroxidase as glutathione is oxidized.
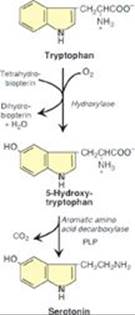
RQ3. Answer = C. Serotonin is released by activated platelets and causes vasoconstriction and platelet aggregation. [Note: Platelets do not synthesize serotonin, but they take up serotonin that was made in the intestine and secreted into the blood.] Serotonin is associated with a feeling of well-being. It is degraded by monoamine oxidase (MAO) that catalyzes oxidative deamination. It is catechol-O-methyl transferase (COMT) that catalyzes the methylation step in the degradation of the catecholamines. Serotonin is synthesized from tryptophan in a two-step process that utilizes tetrahydrobiopterin-requiring tryptophan hydroxylase and a pyridoxal phosphate (PLP)-requiring decarboxylase (see Figure).
RQ4. Answer = B. The exocrine pancreas secretes enzymes required for the digestion of dietary carbohydrate, protein, and fat. The endocrine pancreas secretes the peptide hormones insulin and glucagon. Damage that affects the functions of the pancreas would lead to diabetes (decreased insulin) and steatorrhea (fatty stool), with the latter the consequence of maldigestion of dietary fat. As was seen with the rise of troponins in a myocardial infarction and transaminases in liver damage, loss of cellular integrity (as would be seen in autodigestion of the pancreas) results in proteins that normally are intracellular being found in higher-than-normal concentrations in the blood. Secretin causes the pancreas to release bicarbonate to raise the pH of the chyme coming to the intestine from the stomach. Pancreatic enzymes work best at neutral or slightly alkaline pH. Pancreatitis is seen in individuals with hypertriglyceridemia as a result of a deficiency in lipoprotein lipase or its coenzyme, apolipoprotein C-II.
CASE 4: Answers to Thought Questions
TQ1. A. The rise in cytosolic NADH seen with ethanol metabolism inhibits glycolysis. The glyceraldehyde 3-phosphate dehydrogenase step requires NAD+, which gets reduced as glyceraldehyde 3-phosphate gets oxidized. With the rise in NADH, glyceraldehyde 3-phosphate accumulates.
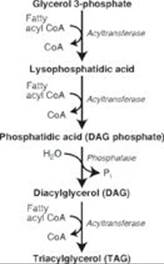
B. The glyceraldehyde 3-phosphate from glycolysis is converted to glycerol 3-phosphate, the initial acceptor of fatty acids in triacylglycerol (TAG) synthesis (see Figure). Fatty acids are available because of increased synthesis (from acetyl CoA, which is increased as a result of both increased production from the acetate product of acetaldehyde and decreased use in the TCA cycle), increased availability from lipolysis in adipose tissue, and decreased degradation. The TAGs produced in the liver accumulate (due, in part, to decreased production of very-low-density lipoproteins) and cause fatty liver (steatosis). Hepatic steatosis is an early (and reversible) stage in alcohol-induced liver disease. Subsequent stages are alcohol-related hepatitis (sometimes reversible) and cirrhosis (irreversible).
TQ2. The rise in NADH favors the reduction of pyruvate to lactate by lactate dehydrogenase. Lactate decreases the renal excretion of uric acid, thereby causing hyperuricemia, a necessary step in an acute gouty attack. [Note: The shift from pyruvate to lactate decreases the availability of pyruvate, a substrate for gluconeogenesis. This contributes to the hypoglycemia seen in the patient.]
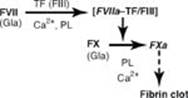
TQ3. Prothrombin time (PT) measures the time it takes for plasma to clot after the addition of tissue factor (FIII), thereby allowing evaluation of the extrinsic (and common) pathways of coagulation (see Figure). In the extrinsic pathway, FIII activates FVII. FVII, like most of the proteins of clotting, is made by the liver. Alcohol-induced liver damage can decrease its synthesis. Additionally, FVII has a short half-life and, as a g-carboxyglutamate (Gla)-containing protein, its synthesis requires vitamin K. Poor nutrition can result in decreased availability of vitamin K and, therefore, decreased ability to clot. [Note: Severe liver disease results in prolonged PT and activated partial thromboplasin time, or aPPt.]
TQ4. Administration of folate can mask a deficiency in vitamin B12 by reversing the hemotologic manifestations of the deficiency. However, folate has no effect on the neurologic damage caused by B12 deficiency. Over time, then, the neurologic effects can become severe and irreversible. Thus, folate can mask a deficiency of B12 and prevent treatment until the neuropathy is apparent.
III. FOCUSED CASES
CASE 1: MICROCYTIC ANEMIA
Patient Presentation: JS is a 24-year-old man who is being evaluated as a follow-up to a pre-placement medical evaluation he had prior to starting his new job.
Focused History: JS has no significant medical issues. His family history is unremarkable, but he knows little of the health status of those family members who remain in Greece.
Pertinent Findings: The physical examination was normal. Routine analysis of his blood included the following results:

Based on the data, hemoglobin (Hb) electrophoresis was performed. The results:

Diagnosis: JS has β-thalassemia trait (β-thalassemia minor) that is causing a microcytic anemia (see Image). Ethnicity (such as being of Mediterranean origin) influences the risk for thalassemia.
Treatment: None is required at this time. Patients are advised that iron supplements will not prevent their anemia.
Prognosis: β-thalassemia trait does not cause mortality or significant morbidity. Patients should be informed of the genetic nature of their autosomal recessive condition for family planning considerations because homozygous β-thalassemia (Cooley anemia) is a serious disorder.
CASE-RELATED QUESTIONS: Choose the ONE best answer.
Q1. Mutations to the gene for β globin that result in decreased production of the protein are the cause of β-thalassemia. The mutations primarily affect gene transcription or posttranscriptional processing of the messenger RNA (mRNA) product. Which of the following statements concerning mRNA is correct?
A. Eukaryotic mRNA is polycistronic.
B. mRNA synthesis involves trans-acting factors binding to cis-acting elements.
C. mRNA synthesis is terminated at the DNA base sequence thymine adenine guanine (TAG).
D. Polyadenylation of the 5ʹ end of eukaryotic mRNA requires a methyl donor.
E. Splicing of eukaryotic mRNA involves removal of exons and joining of introns by the proteasome.
Q2. Hemoglobin A (HbA), a tetramer of 2 α and 2 β globin chains, delivers O2 from the lungs to the tissues and protons and CO2 from the tissues to the lungs. Increased concentration of which of the following will result in decreased O2 delivery by HbA?
A. 2,3-Bisphosphoglycerate
B. Carbon dioxide
C. Carbon monoxide
D. Protons
Q3. What is the basis for the increase in HbA2 and HbF in the β-thalassemias?
Q4. Why is the allele-specific oligonucleotide (ASO) hybridization technique useful in the diagnosis of all cases of sickle cell anemia but not all cases of β-thalassemia?
CASE 2: SKIN RASH
Patient Presentation: KL is a 34-year-old woman who presents with a red, nonitchy rash on her left thigh and flulike symptoms.
Focused History: KL reports that the rash first appeared a little over 2 weeks ago. It started out small but has gotten larger. She also thinks she is getting the flu because her muscles and joints ache (myalgia and arthralgia, respectively), and she has had a headache for the last few days. Upon questioning, KL tells you she and her husband took a camping trip through New England last month.
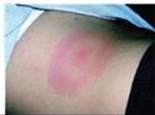
Pertinent Findings: The physical examination is remarkable for the presence of a red, circular, flat lesion about 11 cm in size that resembled a bullseye (erythema migrans) (see image). KL also has a low-grade fever.
Diagnosis: KL has Lyme disease caused by the bacterium Borrelia burgdorferi, which is transmitted by the bite of a tick in the genus Ixodes. Infected ticks are endemic in the Northeast region of the United States.
Treatment: The patient is prescribed doxycycline, an antibiotic in the tetracycline family. Monitoring of KL will continue until all symptoms have completely resolved. Blood is drawn for clinical laboratory tests.
Prognosis: Patients treated with the appropriate antibiotic in the early stages of Lyme disease typically recover quickly and completely.
CASE-RELATED QUESTIONS: Choose the ONE best answer.
Q1. Antibiotics in the tetracycline class inhibit protein synthesis (translation) at the initiation step. Which of the following statements about translation is correct?
A. In eukaryotic translation, the initiating amino acid is formylated methionine.
B. Only the charged initiating transfer RNA goes directly to the ribosomal A site.
C. Peptidyl transferase is a ribozyme that forms the peptide bond between two amino acids.
D. Prokaryotic translation can be inhibited by the phosphorylation of initiation factor 2.
E. Termination of translation is independent of guanosine triphosphate hydrolysis.
F. The Shine-Dalgarno sequence facilitates the binding of the large ribosomal subunit to eukaryotic messenger RNA (mRNA).
Q2. The Centers for Disease Control and Prevention recommends a two-tier testing procedure for Lyme disease that involves a screening enzyme-linked immunosorbent assay (ELISA) follow by a confirmatory Western blot analysis on any sample with a positive or equivocal ELISA result. Which of the following statements about these testing procedures is correct?
A. Both techniques are used to detect specific mRNAs.
B. Both techniques involve the use of antibodies.
C. ELISAs require the use of electrophoresis.
D. Western blots require use of the polymerase chain reaction.
Q3. Why are eukaryotic cells unaffected by antibiotics in the tetracycline class?
CASE 3: BLOOD ON THE TOOTHBRUSH
Patient Presentation: LT is an 84-year-old man whose gums have been bleeding for several months.
Focused History: LT is a widower and lives alone in a suburban community on the East Coast. He no longer drives. His two children live on the West Coast and come east infrequently. Since the death of his wife 11 months ago, he has been isolated and finds it hard to get out of the house. His appetite has changed, and he is content with cereal, coffee, and packaged snacks. Chewing is difficult.
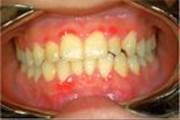
Pertinent Findings: The physical examination was remarkable for the presence of swollen dark-colored gums (see Image). Several of the patient’s teeth were loose, including one that anchors his dental bridge. Several black and blue marks (ecchymoses) were noted on the legs, and an unhealed sore was present on the right wrist. Inspection of his scalp revealed tiny red spots (petechia) around some of the hair follicles. Blood was drawn for testing.
Results of tests on the patient’s blood:

The test for blood in his stool (occult blood test) was negative.
Results of follow-up tests (obtained several days after the appointment) included:
![]()
Diagnosis: Vitamin C deficiency with a microcytic, hypochromic anemia secondary to the deficiency
Treatment: LT was prescribed Vitamin C (as oral ascorbic acid) and iron (as oral ferrous sulfate) supplements. He will also be referred to social services.
Prognosis: The prognosis for recovery is excellent.
CASE-RELATED QUESTIONS: Choose the ONE best answer.
Q1. Which of the following statements about vitamin C is correct? Vitamin C is:
A. a competitor of iron absorption in the intestine.
B. a fat-soluble vitamin with a 3-month supply typically stored in adipose tissue.
C. a coenzyme in several enzymic reactions such as the hydroxylation of proline.
D. required for the cross-linking of collagen.
Q2. In contrast to the microcytic anemia characteristic of iron deficiency (common in older adults), a macrocytic anemia is seen with deficiencies of vitamin B12 and/or folic acid. These vitamin deficiencies are also common in older adults. Which of the following statements concerning these vitamins is correct?
A. An inability to absorb B12 results in pernicious anemia.
B. Both vitamins cause changes in gene expression.
C. Folic acid plays a key role in energy metabolism in most cells.
D. Treatment with methotrexate can result in toxic levels of the coenzyme form of folic acid.
E. Vitamin B12 is the coenzyme for enzymes catalyzing amino acid deaminations, decarboxylations, and transaminations.
Q3. How do hemolytic anemias differ from nutritional anemias?
CASE 4: RAPID HEART RATE, HEADACHE, AND SWEATING
Patient Presentation: BE is a 45-year-old woman who presents with concerns about sudden (paroxysmal), intense, brief episodes of headache, sweating (diaphoresis), and a racing heart (palpitations).
Focused History: BE reports that the attacks started about 3 weeks ago. They last from 2 to 10 minutes, during which time she feels quite anxious. During the attacks it feels as though her heart is skipping beats (arrhythmia). At first she thought the attacks were related to recent stress at work and maybe even menopause. The last time it happened, she was in the drug store and had her blood pressure taken. She was told it was 165/110 mm Hg. BE notes that she has lost weight (about 8 lbs) in this period even though her appetite has been good.
Pertinent Findings: The physical examination was remarkable for the patient’s thin, pale appearance. Blood pressure was elevated (150/100 mm Hg), as was the heart rate (110–120 beats/minute). Based on the patient’s history, blood levels of normetanephrine and metanephrine were ordered. They were found to be elevated.
Diagnosis: Pheochromocytoma, a rare catecholamine-secreting tumor of the adrenal medulla
Treatment: Imaging studies of the abdomen were performed to locate the tumor. Surgical resection of the tumor was performed. The tumor was found to be nonmalignant. Follow-up measurement of plasma metanephrines was performed 2 weeks later and was in the normal range.
Prognosis: The 5-year survival rate for nonmalignant pheochromocytomas is over 95%.
CASE-RELATED QUESTIONS: Choose the ONE best answer.
Q1. Pheochromocytomas secrete norepinephrine and epinephrine. Which of the following statements concerning the synthesis and degradation of these two biogenic amines is correct?
A. The substrate for their synthesis is tryptophan, which is hydroxylated to 3,4-dihydroxyphenylalanine (DOPA) by tetrahydrobiopterin-requiring tryptophan hydroxylase.
B. The conversion of DOPA to dopamine utilizes a pyridoxal phosphate–requiring carboxylase.
C. The conversion of norepinephrine to epinephrine requires vitamin C.
D. Degradation involves methylation by catechol-O-methytransferase and produces normetanephrine from norepinephrine and metanephrine from epinephrine.
E. Normetanephrine and metanephrine are oxidatively deaminated to homovanillic acid by monoamine oxidase.
Q2. Which of the following statements concerning the actions of epinephrine and/or norepinephrine are correct?
A. Norepinephrine functions as a neurotransmitter and a hormone.
B. They are initiated by autophosphorylation of select tyrosine residues in their receptors.
C. They are mediated by binding to adrenergic receptors, which are a class of nuclear receptors.
D. They result in the activation of glycogen and triacylglycerol synthesis.
Q3. Norepinephrine bound to certain receptors causes vasoconstriction and an increase in blood pressure. Why might norepinephrine be used clinically in the treatment of septic shock?
CASE 5: SUN SENSITIVITY
Patient Presentation: AZ is a 6-year-old boy who is being evaluated for freckle-like areas of hyperpigmentation on his face, neck, forearms, and lower legs.
Focused History: AZ’s father reports that the boy has always been quite sensitive to the sun. His skin turns red (erythema) and his eyes hurt (photophobia) if he is exposed to the sun for any period of time.
Pertinent Findings: The physical examination was remarkable for the presence of thickened, scaly areas (actinic keratosis) and hyperpigmented areas on skin exposed to ultraviolet (UV) radiation from the sun. Small dilated blood vessels (telangiectasia) were also seen. Tissue from several sites on his face was biopsied, and two were later determined to be squamous cell carcinomas.
Diagnosis: Xeroderma pigmentosum, a rare defect in nucleotide excision repair of DNA
Treatment: Protection from sunlight through use of sunscreens such as protective clothing that reflect UV radiation and chemicals that absorb it is essential. Frequent skin and eye examinations are recommended.
Prognosis: Most patients with xeroderma pigmentosum die at an early age from skin cancers. However, survival beyond middle age is possible.
CASE-RELATED QUESTIONS: Choose the ONE best answer.
Q1. Which of the following statements about DNA repair mechanisms is correct? DNA repair:
A. is performed only by eukaryotes.
B. of double-strand breaks is error free.
C. of mismatched bases involves repair of the parental strand.
D. of ultraviolet radiation–induced pyrimidine dimers involves removal of a short oligonucleotide containing the dimer.
E. of uracil produced by the deamination of cytosine requires the actions of endo- and exonucleases to remove the uracil base.
Q2. Which one of the following statements about DNA synthesis (replication) is correct? Replication:
A. in both eukaryotes and prokaryotes requires an RNA primer.
B. in eukaryotes requires condensation of chromatin.
C. in prokaryotes is accomplished by a single DNA polymerase.
D. is initiated at random sites in the genome.
E. produces a polymer of deoxyribonucleoside monophosphates linked by 5ʹ→3ʹ phosphodiester bonds.
Q3. What is the difference between DNA proofreading and repair?
CASE 6: DARK URINE AND YELLOW SCLERAE
Patient Presentation: JF is a 13-year-old girl who presents with fatigue and yellow sclerae.
Focused History: JF began treatment about 4 days ago with a sulfonamide antibiotic and a urinary analgesic for a urinary tract infection. She had been told that her urine would change color (become reddish) with the analgesic, but she reports that it has gotten darker (more brownish) over the last 2 days. Last night her mother noticed that her eyes had a yellow tint. JF says she feels as though she has no energy.
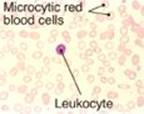
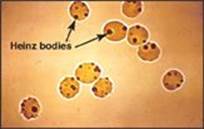
Pertinent Findings: The physical examination was remarkable for the pale appearance of the patient, mild scleral icterus (jaundice), mild splenomegaly, and an increased heart rate (tachycardia). JF’s urine tested positive for hemoglobin (hemoglobinuria). A peripheral blood smear reveals a lower-than-normal number of red blood cells (RBCs), with some containing precipitated hemoglobin (Heinz bodies; see Image), and a higher-than-normal number of reticulocytes (immature RBCs). Results of the complete blood count (CBC) and blood chemistry tests are pending.
Diagnosis: Glucose 6-phosphate dehydrogenase (G6PD) deficiency, an X-linked disorder that causes hemolysis (RBC lysis)
Treatment: G6PD deficiency can result in a hemolytic anemia in affected individuals exposed to oxidative agents. JF will be switched to a different antibiotic. She will be advised that she is susceptible to certain drugs (for example, sulfa drugs), foods (fava or broad beans), and certain chemicals (for example, naphthalene), and must avoid exposure to them.
Prognosis: In the absence of exposure to oxidative agents, G6PD deficiency does not cause mortality or significant morbidity.
CASE-RELATED QUESTIONS: Choose the ONE best answer.
Q1. Glucose 6-phosphate dehydrogenase (G6PD) catalyzes the regulated step in the pentose phosphate pathway. Which of the following statements concerning G6PD and the pentose phosphate pathway is correct?
A. Deficiency of G6PD occurs only in red blood cells.
B. Deficiency of G6PD results in an inability to keep glutathione in its functional, reduced form.
C. The pentose phosphate pathway includes one reversible reductive reaction followed by a series of phosphorylated sugar interconversions.
D. The reduced nicotinamide adenine dinucleotide phosphate (NADPH) product of the pentose phosphate pathway is utilized in processes such as fatty acid oxidation.
Q2. When received, the results of the blood count were consistent with a hemolytic anemia. Blood chemistry tests revealed an elevation in the bilirubin level. Which of the following statements concerning bilirubin is correct?
A. Hyperbilirubinemia can cause deposition of bilirubin in the skin and sclerae resulting in jaundice.
B. The solubility of bilirubin is increased by conjugating it with two molecules of ascorbic acid in the liver.
C. The conjugated form of bilirubin increases in the blood with a hemolytic anemia.
D. Phototherapy can increase the solubility of the excess bilirubin generated in the porphyrias.
Q3. Why is urinary urobilinogen increased relative to normal in hemolytic jaundice and absent in obstructive jaundice?
CASE 7: JOINT PAIN
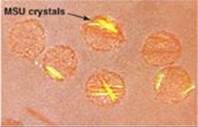
Patient Presentation: BJ is a 22-year-old male who presents for follow-up 10 days after having been treated in the Emergency Department (ED) for severe inflammation at the base of his thumb.
Focused History: This was BJ’s first occurrence of severe joint pain. In the ED, he was given an anti-inflammatory medication. Fluid aspirated from the carpometacarpal joint of the thumb was negative for organisms but positive for needle-shaped monosodium urate (MSU) crystals (see Image). The inflammatory symptoms have since resolved. BJ reports he is in good health otherwise, with no significant past medical history. His body mass index (BMI) is 31. No tophi (deposits of MSU crystals under the skin) were detected in the physical examination.
Pertinent Findings: Results on a 24-hour urine specimen and blood tests requested in advance of this visit reveal that BJ is not an undersecretor of uric acid. His blood urate was 8.5 mg/dl (reference = 2.5–8.0). The unusually young age of presentation is suggestive of an enzymopathy of purine metabolism, and additional blood tests are ordered.
Diagnosis: Gout (MSU crystal deposition disease), a type of inflammatory arthritis
Treatment: BJ was given prescriptions for allopurinol and colchicine. The treatment goals are to reduce his blood urate levels to <6.0 mg/dl and prevent additional attacks. He was advised to lose weight because being overweight or obese is a risk factor for gout. His BMI of 31 puts him in the obese category. He was also given written information on the association between diet and gout.
Prognosis: Gout increases the risk of developing renal stones. It is also associated with hypertension, diabetes, and heart disease.
CASE-RELATED QUESTIONS: Choose the ONE best answer.
Q1. Allopurinol is converted in the body to oxypurinol, which functions as a noncompetitive inhibitor of an enzyme in purine metabolism. Which of the following statements concerning purine metabolism and its regulation is correct?
A. As a noncompetitive inhibitor, oxypurinol increases the apparent Michaelis constant (Km) of the target enzyme.
B. Colchicine inhibits xanthine oxidase, an enzyme of purine degradation.
C. Glutamate provides two of the nitrogen atoms of the purine ring.
D. In purine nucleotide synthesis, the ring system is first constructed and then attached to ribose 5-phosphate.
E. Oxypurinol inhibits the amidotransferase that initiates degradation of the purine ring system.
F. Partial or complete enzymic deficiencies in the salvage of purine bases are characterized by hyperuricemia.
Q2. Purines are one type of nitrogenous base found in nucleotides. Pyrimidines are the other. Which of the following statements is true of the pyrimidines?
A. Carbamoyl phosphate synthetase I is the regulated enzymic activity in pyrimidine ring synthesis.
B. Methotrexate decreases synthesis of the pyrimidine nucleotide thymidine monophosphate.
C. Orotic aciduria is a pathology of pyrimidine degradation.
D. Pyrimidine nucleotide synthesis is independent of 5-phosphoribosyl-1-pyrophosphate (PRPP).
Q3. BJ is subsequently shown to have a form of PRPP synthetase that shows increased enzymic activity. Why does this result in hyperuricemia?
CASE 8: NO BOWEL MOVEMENT
Patient Presentation: MW is a 48-hour-old female who has not yet had a bowel movement.
Focused History: MW is the full-term product of a normal pregnancy and delivery. She appeared normal at birth. MW is the first child of parents of Northern European ethnicity. The parents are both in good health, and their family histories are unremarkable.
Pertinent Findings: MW has a distended abdomen. She recently vomited small amounts of bilious (green-colored) material.
Diagnosis: Meconium ileus (obstruction of the ileum by meconium, the first stool produced by newborns) was confirmed by abdominal x-rays. About 98% of full-term newborns with meconium ileus have cystic fibrosis (CF). Diagnosis of CF was subsequently confirmed with a chloride sweat test.
Treatment: The ileus was successfully treated nonsurgically. For management of the CF, the family was referred to the CF center at the regional children’s hospital.
Prognosis: CF is the most common life-limiting autosomal-recessive disease in Caucasians.
CASE-RELATED QUESTIONS: Choose the ONE best answer.
Q1. CF is the result of mutations to the gene that codes for the cystic fibrosis transmembrane conductance regulator (CFTR) protein that functions as a chloride channel in the apical membrane of epithelial cells on a mucosal surface. Which of the following statements concerning CF is correct?
A. Clinical manifestations of CF are the consequence of chloride retention with increased water reabsorption that causes mucous on the epithelial surface to be excessively thick and sticky.
B. Excessive pancreatic secretion of insulin commonly results in hypoglycemia.
C. Genetic testing for CF may involve the use of a set of probes for the most common mutations, a technique known as restriction fragment length polymorphism analysis.
D. Some mutations result in premature degradation of the CFTR protein through tagging with ubiquinone followed by proteasome-mediated proteolysis.
E. The most common mutation, ΔF508, results in the loss of a codon for phenylalanine and is classified as a frameshift mutation.
Q2. The CFTR protein is an intrinsic plasma membrane glycoprotein. Targeting of proteins destined to function as components of membranes:
A. includes transport to and through the Golgi.
B. involves an amino-terminal signal sequence that is retained in the functional protein.
C. occurs after the protein has been completely synthesized (that is, posttranslationally).
D. requires the presence of mannose 6-phosphate residues on the protein.
Q3. Why might steatorrhea be seen with CF?
CASE 9: ELEVATED AMMONIA
Patient Presentation: RL is a 40-hour-old male with signs of cerebral edema.
Focused History: RL is the full-term product of a normal pregnancy and delivery. He appeared normal at birth. At age 36 hours he became irritable, lethargic, and hypothermic. He fed only poorly and vomited. He also displayed tachypneic (rapid) breathing and neurologic posturing. At age 38 hours he had a seizure.
Pertinent Findings: Respiratory alkalosis (increased pH, decreased CO2 [hypocapnia]), increased ammonia level, and decreased blood urea nitrogen level were found. An amino acid screen revealed that argininosuccinate was increased more than 60-fold over baseline, and citrulline was increased 4-fold. Glutamine was elevated, and arginine was decreased relative to normal.
Diagnosis: Urea cycle enzyme defect with neonatal onset
Treatment: Hemodialysis was performed to remove ammonia. Sodium phenylacetate and sodium benzoate were administered to aid in excretion of waste nitrogen, as was arginine. Long-term treatment will include lifelong limitation of dietary protein; supplementation with essential amino acids; and administration of arginine, sodium phenylacetate, and sodium phenylbutyrate.
Prognosis: Survival into adulthood is possible. The degree of neurologic impairment is related to the degree and extent of the hyperammonemia.
CASE-RELATED QUESTIONS: Choose the ONE best answer.
Q1. Based on the findings, which enzyme of the urea cycle is most likely to be deficient in this patient?
A. Arginase
B. Argininosuccinate lyase
C. Argininosuccinate synthetase
D. Carbamoyl phosphate synthetase I
E. Ornithine transcarbamoylase
Q2. Why is arginine supplementation helpful in this case?
Q3. In individuals with partial (milder) deficiency of urea cycle enzymes, the level of which one of the following would be expected to be decreased during periods of physiologic stress?
A. Alanine
B. Ammonia
C. Glutamine
D. Insulin
E. pH
CASE 10: CALF PAIN
Patient Presentation: CR is a 19-year-old female who is being evaluated for pain and swelling in her right calf.
Focused History: Ten days ago, CR had her spleen removed following a bicycle accident in which she fractured her tibial eminence, necessitating immobilization of the right knee. She has had a good recovery from the surgery. CR is no longer taking pain medication but has continued her oral contraceptives.
Pertinent Findings: CR’s right calf is reddish in color (erythematous) and warm to the touch. It is visibly swollen. The left calf is normal in appearance and is without pain. An ultrasound is ordered.
Diagnosis: CR has a deep venous thrombosis (DVT). Oral contraceptives are a risk factor for DVT, as are surgery and immobilization.
Treatment (Immediate): Heparin and warfarin are administered.
Prognosis: In the 10 years following a DVT, about one third of individuals have a recurrence.
CASE-RELATED QUESTIONS: Choose the ONE best answer.
Q1. A deep venous thrombosis is a blood clot that occludes the lumen of a deep vein, most commonly in the leg. Which of the following statements about the clotting cascade is correct?
A. A deficiency in factor (F)IX of the intrinsic pathway results in hemophilia A.
B. FIII of the extrinsic pathway is a serine protease.
C. Formation of the fibrin meshwork is referred to as primary hemostasis.
D. Thrombin proteolytically activates components of the extrinsic, intrinsic, and common pathways.
E. Vitamin K is required for the activation of fibrinogen.
Q2. Which one of the following would increase the risk of thrombosis?
A. Excess production of antithrombin
B. Excess production of protein S
C. Expression of FV Leiden
D. Hypoprothrobinemia
E. von Willebrand disease
Q3. Compare and contrast the actions of heparin and warfarin.
IV. FOCUSED CASES: ANSWERS TO CASE-BASED QUESTIONS
CASE 1: Anemia with β-Thalassemia Minor
Q1. Answer = B. Transcription (synthesis of single-stranded RNA from the template strand of double-stranded DNA) requires the binding of proteins (trans-acting factors) to sequences on the DNA (cis-acting elements). Eukaryotic messenger RNA (mRNA) is monocistronic because it contains information from just one gene (cistron). The base sequence TAG (thymine adenine guanine) in the coding strand of DNA is U(uracil)AG in the mRNA. UAG is a signal that terminates translation (protein synthesis), not transcription. It is formation of the 5ʹ cap of eukaryotic mRNA that requires methylation (using S-adenosylmethionine), not 3ʹ polyadenylation. Splicing is the spliceosome-mediated process by which introns are removed from eukaryotic mRNA and exons joined.
Q2. Answer = C. Carbon monoxide (CO) increases the affinity of hemoglobin (Hb)A for O2, thereby decreasing the ability of HbA to offload O2 in the tissues. CO stabilizes the R (relaxed) or oxygenated form and shifts the O2dissociation curve to the left (see Figure). The other choices decrease the affinity for O2, stabilize the T (tense) or deoxygenated form, and cause a right shift in the curve.
Q3. HbA2 and HbF do not contain β globin. As β globin production decreases, synthesis of HbA2 (α2δ2) and HbF (α2γ2) increases.
Q4. Sickle cell anemia is caused by a single point mutation (A→T) in the gene for β globin that results in the replacement of glutamate by valine at the sixth amino acid position in the protein. Mutational analysis using allele-specific oligonucleotide (ASO) probes for that mutation (βS) and for the normal sequence (βA) is used in diagnosis (see Figure). β-Thalassemia, in contrast, is caused by hundreds of different mutations. Mutational analysis using ASO probes can assess common mutations, including point mutations, in at-risk populations (for example, those of Greek ancestry). However, less common mutations are often not included in the panel and can be detected only by DNA sequencing.
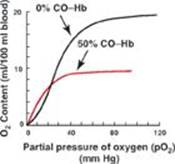
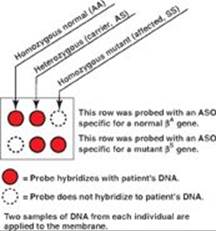
CASE 2: Skin Rash with Lyme Disease
Q1. Answer = C. Peptide bond formation between the amino acid in the A site of the ribosome and the amino acid last added to the growing peptide in the P site is catalyzed by an RNA of the large ribosomal subunit. Any RNA with catalytic activity is referred to as a ribozyme (see Figure below). Formylated methionine is used to initiate prokaryotic translation. The charged initiating transfer RNA (tRNA) is the only tRNA that goes directly to the P site, leaving the A site available for the tRNA carrying the next amino acid of the protein being made. Eukaryotic translation is inhibited by the phosphorylation of initiation factor 2 (eIF-2). The Shine-Dalgarno (SD) sequence is found in prokaryotic messenger RNA (mRNA) and facilitates the interaction of the mRNA with the small ribosomal subunit. In eukaryotes, the cap-binding proteins perform that task.
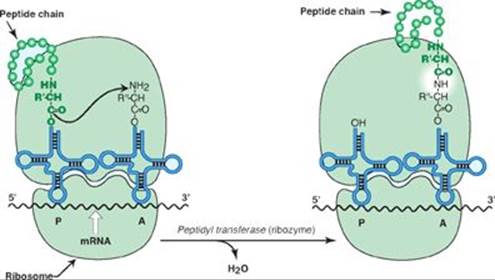
Q2. Answer = B. The enzyme-linked immunosorbent assay (ELISA) and Western blot are used to analyze proteins. Each makes use of antibodies to detect and quantify the protein of interest. It is Western blots that utilize electrophoresis. The polymerase chain reaction (PCR) is used to amplify DNA.
Q3. Antibiotics in the tetracycline family inhibit protein synthesis by binding to and blocking the A site of the small ribosomal subunit (30S) in prokaryotes. Tetracycline specifically interacts with the 16S ribosomal RNA (rRNA) component of the 30S subunit, inhibiting translation initiation. Eukaryotes do not contain 16S rRNA (see Figure). Their small (40S) subunit contains 18S rRNA, which does not bind tetracycline.
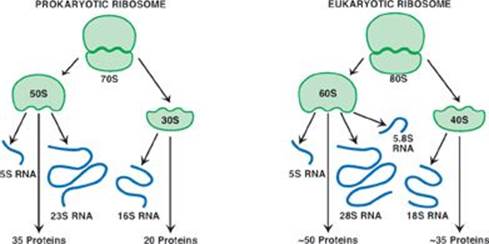
CASE 3: Blood on the Toothbrush with Vitamin C Deficiency
Q1. Answer = C. Vitamin C (ascorbic acid) functions as a coenzyme in the hydrolylation of proline and lysine in the synthesis of collagen, a fibrous protein of the extracellular matrix. Vitamin C reduces dietary iron from the ferric (Fe3+) to the ferrous (Fe2+) form that is required for absorption. With a deficiency of vitamin C, uptake of dietary iron is impaired and results in a microcytic, hypochromic anemia. As a water-soluble vitamin, vitamin C is not stored. Cross-linking of collagen by lysyl oxidase requires copper, not vitamin C.
Q2. Answer = A. An inability to absorb vitamin B12 leads to pernicious anemia and is most commonly caused by decreased production of intrinsic factor (IF) by the parietal cells of the stomach (see Figure). Vitamins D and A, in complex with their receptors, bind to DNA and alter gene expression. Thiamine (vitamin B1) is a coenzyme in the oxidative decarboxylation of pyruvate and α-ketoglutarate and, therefore, is important in energy metabolism in most cells. Methotrexate inhibits dihydrofolate reductase, the enzyme required to reduce dihydrofolate to tetrahydrofolate (THF), the functional coenzyme form of folate. This results in decreased availability of THF. It is pyridoxine (vitamin B6) as pyridoxal phosphate (PLP) that is the coenzyme for most reactions involving amino acids. (Note that tetrahydrobioptrin is required by aromatic amino acid hydroxylases.)
Q3. Nutritional anemias are characterized by either increased red blood cell (RBC) size (folate and B12 deficiencies) or decreased RBC size (iron and vitamin C deficiencies). In hemolytic anemias, such as is seen in glucose 6-phosphate dehydrogenase and pyruvate kinase deficiencies and in sickle cell anemia, RBC size typically is normal and RBC number is decreased.
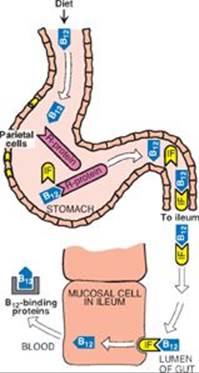
CASE 4: Rapid Heart Rate, Headache, and Sweating with a Pheochromocytoma
Q1. Answer = D. Degradation of both epinephrine and norepinephrine involves methylation by catechol-O-methytransferase (COMT) that produces normetanephrine from norepinephrine and metanephrine from epinephrine (see Figure). Both of these products are deaminated to vanillylmandelic acid by monoamine oxidase (MAO). The substrate for the synthesis of the catecholamines is tyrosine, which gets hydroxylated to 3,4-dihydroxyphenylalanine (DOPA) by tetrahydrobiopterin-requiring tyrosine hydroxylase. DOPA is converted to dopamine by a pyridoxal phosphate–requiring decarboxylase. (Note that most carboxylases require biotin.) Norepinephrine is converted to epinephrine by methylation, and S-adenosylmethionine provides the methyl group.
Q2. Answer = A. Norepinephrine released from the sympathetic nervous system functions as a neurotransmitter that acts on postsynaptic neurons and causes, for example, increased heart rate. It is also released from the adrenal medulla and, along with epinephrine, functions as a counterregulatory hormone that results in mobilization of stored fuels (for example, glucose and triacylglycerols). These actions are mediated by the binding of norepinephrine to adrenergic receptors, which are G protein–coupled receptors of the plasma membrane, and not to nuclear receptors like those of steroid hormones or membrane tyrosine kinase receptors like that of insulin.
Q3. Septic shock is vasodilatory hypotension (low blood pressure caused by blood vessel dilation) resulting from the production of large amounts of nitric oxide by inducible nitric oxide synthase (iNOS) in response to infection. Norepinephrine bound to receptors on smooth muscle cells causes vasoconstriction and, thus, raises blood pressure.
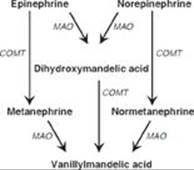
CASE 5: Sun Sensitivity with Xeroderma Pigmentosum
Q1. Answer = D. Pyrimidine dimers are the characteristic DNA lesion caused by ultraviolet (UV) radiation. Their repair involves the excision of an oligonucleotide containing the dimer and replacement of that oligonucleotide, a process known as nucleotide excision repair. (See Figure for a representation of the process in prokaryotes.) DNA repair systems are found in prokaryotes and eukaryotes. Nothing is “error free,” but the homologous-recombination method of double-strand break repair is much less prone to error than is the nonhomologous–end-joining method because any DNA that was lost is replaced. Mismatched-base repair involves identification and repair of the newly synthesized (daughter) strand. In prokaryotes, the extent of strand methylation is used to discriminate between the strands. Base excision repair, the mechanism by which uracil is removed from DNA, utilizes a glycosylase to remove the base, creating an apyrimidinic or apurinic (AP) site. The sugar-phosphate is then removed by the actions of an endo- and exonuclease.
Q2. Answer = A. All replication requires an RNA primer because DNA polymerases (pols) cannot initiate DNA synthesis. The chromatin of eukaryotes gets decondensed (relaxed) for replication. Relaxation can be accomplished, for example, by acetylation via histone acetyltransferases (HATs). Prokaryotes have more than one DNA pol. For example, pol III extends the RNA primer with DNA, and pol I removes the primer and replaces it with DNA. Replication is initiated at specific locations (one in prokaryotes, many in eukaryotes) that are recognized by proteins (for example, DnaA in prokaryotes). Deoxynucleoside monophosphates (dNMPs) are joined by a phosphodiester bond that links the 3-hydroxyl group of the last dNMP added with the 5-phosphate group of the incoming nucleotide, thereby forming a 3’→5’ phosphodiester bond as pyrophosphate is released.
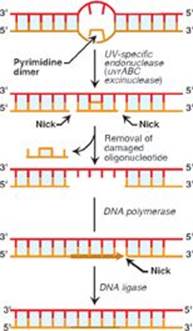
Q3. Proofreading occurs during replication in the S phase of the cell cycle and involves the 3ʹ→5ʹ exonuclease activity possessed by some DNA pols (see Figure). Repair can occur independently of replication and, therefore, can be performed outside of the S phase.
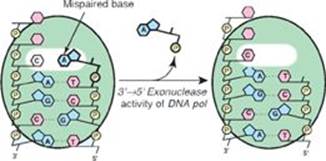
CASE 6: Dark Urine and Yellow Sclerae with Glucose 6-Phosphate Dehydrogenase Deficiency
Q1. Answer = B. Glutathione in its reduced form (G-SH) is an important antioxidant. The enzyme glutathione peroxidase reduces hydrogen peroxide (a reactive oxygen species) to water as glutathionine is oxidized (G-S-S-G). Reduced nicotinamide adenine dinucleotide phosphate (NADPH)-requiring glutathionine reductase regenerates the reduced, functional form of glutathione (see Figure A). The NADPH is supplied by the oxidative reactions of the pentose phosphate pathway (see Figure B), which is regulated by the availability of NADPH at the glucose 6-phosphate dehydrogenase (G6PD)-catalyzed step (the first step). Deficiency of G6PD occurs in all cells, but the effects are seen in red blood cells where the pentose phosphate pathway is the only source of NADPH. The pathway involves two irreversible oxidative reactions, each of which generates NADPH. The NADPH is used in reductive processes such as fatty acid synthesis (not oxidation) as well as steroid hormone and cholesterol synthesis.
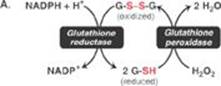
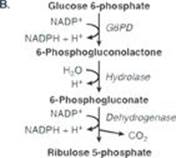
Q2. Answer = A. Jaundice (icterus) refers to the yellow color of the skin, nail beds, and sclerae that results from the deposition of bilirubin when the bilirubin level in the blood is elevated (hyperbilirubinemia; see Image C). Bilirubin has low solubility in aqueous solutions, and its solubility is increased by conjugation with uridine diphosphate (UDP)-glucuronic acid in the liver, forming bilirubin diglucuronide or conjugated bilirubin (CB). In hemolytic conditions, such as G6PD deficiency, both conjugated and unconjugated bilirubin (UCB) are increased, but it is UCB that is found in the blood. CB is sent into the intestine. Phototherapy is used to treat unconjugated hyperbilirubinemia because it converts bilirubiin to isomeric forms that are more water soluble. Bilirubin is the product of heme degradation in cells of the reticuloendothelial system, particularly in the liver and the spleen. The porphyrias are pathologies of heme synthesis and, therefore, are not characterized by hyperbilirubinemia.
C. 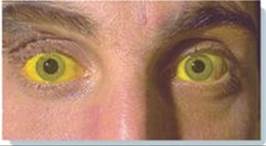
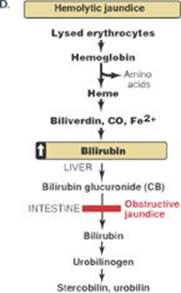
Q3. With hemolysis, more bilirubin is produced and conjugated. CB is sent to the intestine where it is converted to urobilinogen, some of which is reabsorbed, enters the portal blood, and travels to the kidney. Because the source of urinary urobilinogen is intestinal urobilinogen, urinary urobilinogen will be low in obstructive jaundice because intestinal urobilinogen will be low as a result of the obstruction of the common bile duct (see Figure D).
CASE 7: Joint Pain with Gout
Q1. Answer = F. Salvage of the purine bases hypoxanthine and guanine to the purine nucleotides inosine monophosphate (IMP) and guanosine monophosphate (GMP) by hypoxanthine-guanine phosphoribosyltransferase (HGPRT) requires 5-phosphoribosyl-1-pyrophosphate (PRPP) as the source of the ribose 1-phosphate. Salvage decreases the amount of substrate available for degradation to uric acid. Therefore, a deficiency in salvage results in hyperuricemia (see figure). Noncompetitive inhibitors such as oxypurinol have no effect on the Michaelis constant (Km) but decrease the apparent maximal velocity (Vmax). Colchicine is an anti-inflammatory drug. It has no effect on the enzymes of purine synthesis or degradation. Glutamine (not glutamate) is a nitrogen source for purine ring synthesis. In purine nucleotide synthesis, the purine ring system is constructed on the ribose 5-phosphate provided by PRPP. Allopurinol and its metabolite, oxypurinol, inhibit xanthine oxidase of purine degradation. The amidotransferase is an enzyme of purine synthesis. Its activity is decreased by purine nucleotides and increased by PRPP.
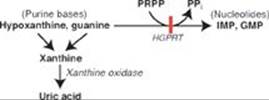
Q2. Answer = B. Methotrexate inhibits dihydrofolate reductase, decreasing the availability of N5,N10-methylene tetrahydrofolate needed for synthesis of deoxythymidine monophosphate (dTMP) from deoxyuridine monophosphate (dUMP); see Figure. Carbamoyl phosphate synthetase (CPS) II is the regulated enzymic activity of pyrimidine biosynthesis in humans. CPS I is an enzyme of the urea cycle. Orotic aciduria is a rare pathology of pyrimidine synthesis caused by a deficiency in one or both enzymic activities of bifunctional uridine monophosphate synthase. Pyrimidine nucleotide synthesis, like purine synthesis and salvage, requires PRPP.
Q3. Increased activity of PRPP synthetase results in increased synthesis of PRPP. This results in an increase in purine nucleotide synthesis beyond need. The excess purine nucleotides get degraded to uric acid, thereby causing hyperuricemia.
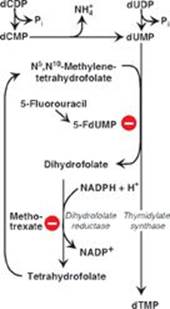
CASE 8: No Bowel Movement with Cystic Fibrosis
Q1. Answer = A. The clinical manifestations of cystic fibrosis (CF) are the consequence of chloride retention with increased water absorption that causes mucus on an epithelial surface to be excessively thick and sticky. The result is pulmonary and gastrointestinal problems such as respiratory infection and impaired exocrine and endocrine pancreatic functions (pancreatic insufficiency). Impaired endocrine pancreatic function can result in diabetes with associated hyperglycemia. The genetic testing technique described, and one used in the diagnosis of CF, is the use of allele-specific oligonucleotides (ASOs). Some mutations do result in increased degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, but degradation is initiated by tagging the protein with ubiquitin. Frameshift mutations alter the reading frame through the addition or deletion of nucleotides by a number not divisible by three. The ΔF509 mutation is caused by the loss of three nucleotides that code for phenylalanine at position 509 in the CFTR protein and, therefore, is not a frameshift mutation.
Q2. Answer = A. Targeting of proteins destined to function as components of the plasma membrane is an example of cotranslational targeting. It involves the initiation of translation on cytosolic ribosomes; the recognition of the amino (N)-terminal signal sequence in the protein; the movement of the protein-synthesizing complex to the outer face of the membrane of the endoplasmic reticulum (ER); and the continuation of protein synthesis such that the protein is threaded into the lumen of the ER, packaged into vesicles that travel to and through the Golgi, and eventually fuse with the plasma membrane. The N-terminal signal sequence is removed by a peptidase in the lumen of the ER. Mannose 6-phosphate is the signal that posttranslationally targets proteins to the matrix of the lysosome where they function as acid hydrolases.
Q3. The pancreatic insufficiency seen in some patients with CF results in a decreased ability to digest food, and digestion is required for absorption. Dietary fats move through the intestine and are excreted in the stool, which is foul-smelling and bulky and may float (see Figure). Patients are at risk for malnutrition and deficiencies in fat-soluble vitamins. Oral supplementation of pancreatic enzymes is the treatment.
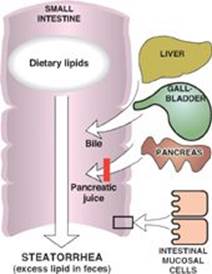
CASE 9: Hyperammonemia with a Urea Cycle Defect
Q1. Answer = B. Argininosuccinate lyase (ASL) cleaves argininosuccinate to arginine and fumarate. The increase in argininosuccinate and citrulline and the decrease in arginine seen in the patient indicate a deficiency in ASL (see Figure below). With arginase deficiency, arginine would be increased, not decreased. Additionally, with arginase deficiency, the hyperammonemia would be less severe. Deficiency of argininosuccinate synthetase (ASS) would also cause an increase in citrulline, but argininosuccinate would be low to absent. Deficiency of carbamoyl phosphate synthetase (CPS) I is characterized by low levels of arginine and citrulline. Deficiency of ornithine transcarbamoylase (OTC), the only X-linked enzyme of the urea cycle, would result in low levels of arginine and citrulline and elevated levels of urinary orotic acid. [Note: The orotic acid is elevated because the carbamoyl phosphate (CP) substrate of OTC is being used in the cytosol as a substrate for pyrimidine synthesis.]
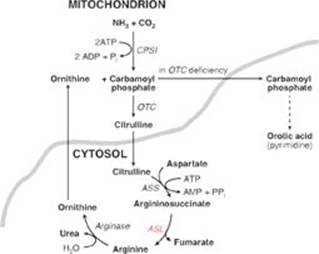
Q2. Arginine supplementation is helpful because the arginine will be hydrolyzed to urea plus ornithine by arginase. The ornithine will be combined with carbamoyl phosphate to form citrulline (see Figure above). With ASL (and ASS) deficiency, citruline accumulates and is excreted, thereby carrying waste nitrogen out of the body.
Q3. Answer = D. In individuals with milder (partial) deficiencies in the enzymes of the urea cycle, hyperammonemia may be triggered by physiologic stress (for example, an illness or prolonged fasting) that decreases the insulin-to-counterregulatory hormone ratio. [Note: The degree of the hyperammonemia is usually less severe than that seen in the neonatal onset forms.] The shift in the ratio results, in part, in skeletal muscle proteolysis, and the amino acids that are released get degraded. Degradation involves transamination by pyridoxal phosphate–requiring aminotransferases that generate the α-keto acid derivative of the amino acid plus glutamate. The glutamate undergoes oxidative deamination to α-ketoglutarate and ammonia (NH3) by glutamate dehydrogenase (GDH): see Figure. [Note: GDH is unusual in that it uses both nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP) as coenzymes.]
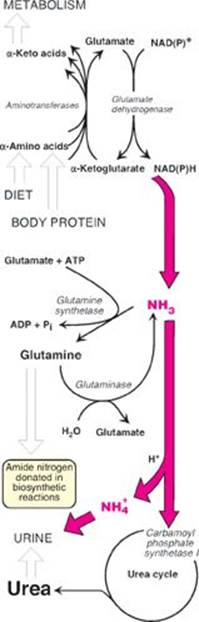
The ammonia (toxic) can be transported to the liver as glutamine and alanine. The glutamine is generated by the amination of glutamate by adenosine triphosphate–requiring glutamine synthetase. In the liver, the enzyme glutaminase removes the ammonia, which can be converted to urea by the urea cycle (see Figure). Glutamine, then, is a nontoxic vehicle of ammonia transport in the blood. Alanine is generated in skeletal muscle from the catabolism of the branched-chain amino acids. In the liver, alanine is transaminated to pyruvate (used in gluconeogenesis) and glutamate. Thus, alanine carries nitrogen to the liver for conversion to urea. Therefore, defects in the urea cycle would result in an elevation in ammonia, glutamine, and alanine. The elevated ammonia drives respiration, and the hyperventilation causes a rise in pH (respiratory alkalosis). [Note: Hyperammonemia is toxic to the nervous system. Although the exact mechanisms are not completely understood, it is known that the metabolism of large amounts of ammonia to glutamine (in the astrocytes of the brain) results in osmotic effects that cause the brain to swell. Additionally, the rise in glutamine decreases the availability of glutamate, an excitatory neurotransmitter.]
CASE 10: Swollen, Painful Calf with Deep Venous Thrombosis
Q1. Answer = D. Thrombin, a serine protease, is activated by the prothrombinase complex of factor (F)Xa + FVa. Once formed, activated thrombin proteolytically activates components of the extrinsic (FVII) and intrinsic (FXI, FVIII) pathways, generating FXa. Thrombin can also activate FV, FI, and FXIII of the common pathway (see Figure below). Hemophilia A is caused by a deficiency in FVIII. FIX deficiency results in hemophilia B. FIII, also known as tissue factor (TF), is a transmembrane glycoprotein of the vascular endothelium. It functions as an accessory protein and not a protease. Formation of the platelet plug is primary hemostasis, and formation of the fibrin meshwork is secondary hemostasis. Vitamin K is required for the activation (g-carboxylation) of FII, FVII, FIX, and FX but not for FI (fibrinogen).
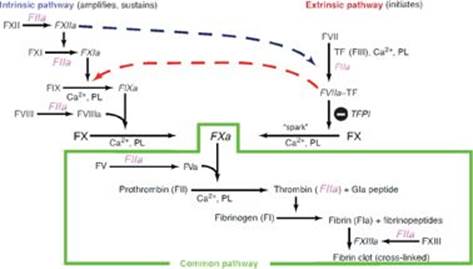
Q2. Answer = C. FV Leiden is a mutant form of FV that is resistant to proteolysis by the activated protein C complex. Decreased ability to degrade FV allows continued production of activated thrombin and leads to an increased risk of clot formation or thrombophilia. Antithrombin III (ATIII) and protein S are proteins of anticoagulation. Increased, not decreased, production of prothrombin would result in thrombophilia. Deficiency of von Willebrand factor causes a coagulopathy or a deficiency in clotting through effects on FVIII and platelets.
Q3. Heparin and warfarin are anticoagulants. Heparin, a glycosaminoglycan, increases the affinity of ATIII for thrombin. Binding of ATIII removes thrombin from the blood and prevents it from converting fibrinogen to fibrin. Warfarin, a synthetic analog of vitamin K, inhibits vitamin K epoxide reductase and prevents the regeneration of the functional hydroquinone form of the vitamin that is required for the γ-carboxylation of glutamate residues to γ-carboxyglutamate (Gla) residues in FII, FVII, FIX, and FX (see Figures).