Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 10
A 4-day-old male infant presents to the emergency department with vomiting and increasing lethargy. The patient was healthy at birth and experienced no delivery complications. The infant’s feeding patterns have deteriorated over the previous 2 days and the parents deny any trauma. Upon examination, the patient is afebrile and his urine has a sweet odor. Alternating muscular hypotonia and hypertonia is observed. Computed tomography of the head reveals cerebral edema. Lumbar puncture results show clear cerebrospinal fluid (CSF), containing 4 WBC per high power field, no RBC, 45 mg% of protein, 46 mg% of sugar, and no bacteria in Gram stain smears. The CSF culture is negative. Ketone bodies and keto acids are detected in the urine. Serum analysis reveals elevated levels of branched-chain amino acids.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the molecular mechanism behind this disorder?
What is the molecular mechanism behind this disorder?
![]() What is the pathophysiologic mechanism of his symptoms?
What is the pathophysiologic mechanism of his symptoms?
ANSWERS TO CASE 10:
Maple Syrup Urine Disease
Summary: A 4-day-old male infant with a history of poor feeding, signs consistent with encephalopathy, and laboratory evidence of metabolic dysfunction.
• Most likely diagnosis: Maple syrup urine disease (MSUD)
• Molecular mechanism of disease: Mutation in one or more of the subunits of the branched-chain α-ketoacid dehydrogenase complex (BCKD), inherited in an autosomal recessive manner.
• Pathophysiologic mechanism of symptoms: The dysfunctional enzyme complex is unable to degrade branched-chain amino acids (BCAAs), resulting in an accumulation of, among others, leucine. This leads to acute hyperosmolarity and cerebral edema.
CLINICAL CORRELATION
MSUD is an autosomal recessive disorder caused by a dysfunctional BCKD unable to degrade BCAAs such as leucine, isoleucine, and valine. Elevated levels of leucine in the plasma and organs effects regulation of cellular volume leading to cerebral edema and the other neurologic problems (hypotonia/hypertonia). The characteristic urine odor is the result of elevated isoleucine metabolite found in the urine.
MSUD classically appears newborns; however, there are some forms of the disease that may present later. The diagnosis is confirmed by elevated plasma BCAAs and their metabolites. Newborn screening is also available immediately after birth. Treatment involves diet modification (limiting intake of BCAAs) and early recognition of metabolic decompensation with appropriate intervention.
APPROACH TO:
Maple Syrup Urine Disease
OBJECTIVES
1. Describe the normal structure and function of the BCKD complex.
2. Explain how structure and function of the BCKD complex are altered in MSUD.
3. Relate the function of amino acid ratio maintenance in the brain to pathologic changes in MSUD.
DEFINITIONS
AUTOSOMAL MUTATION: A genetic mutation that equally affects males and females. The chromosome affected is not a sex chromosome.
BRANCHED-CHAIN AMINO ACID (BCAA): An amino acid having aliphatic side-chains with a branch (a carbon atom bound to more than two other carbon atoms). Among the proteinogenic amino acids, there are 3 BCAAs: leucine, isoleucine, and valine.
BRANCHED-CHAIN α-KETO ACID DEHYDROGENASE COMPLEX (BCKD): Branched-chain α-keto acid dehydrogenase is a multienzyme complex associated with the inner membrane of mitochondria, and functions in the catabolism of branched-chain amino acids. The complex consists of multiple copies of 3 components: branched-chain α-keto acid decarboxylase (E1), lipoamide acyltransferase (E2), and lipoamide dehydrogenase (E3).
FOUNDER MUTATION: In genetics, a founder mutation is a mutation that appears in the DNA of one or more individuals who are founders of a distinct population. Founder mutations originate with changes that occur in the DNA and are passed down to other generations.
KINASE: An enzyme that catalyzes the transfer of a phosphate group or another high-energy molecular group to an acceptor molecule. Each of these kinases is named for its receptor, such as acetate kinase, fructokinase, or hexokinase.
PHOSPHATASE: Any of a group of enzymes capable of catalyzing the hydrolysis of esterified phosphoric acid, with liberation of inorganic phosphate, found in practically all tissues, body fluids, and cells, including erythrocytes and leukocytes.
TRANSAMINASE: Any of a group of enzymes that catalyze the transfer of an amino group from an amino acid to an acceptor keto-acid, resulting in the formation of a new amino acid and the conversion of the original amino acid to a keto-acid; also termed aminotransferase.
DISCUSSION
MSUD is caused by a defect in BCAA (leucine, isoleucine, and valine) metabolism. These amino acids are normally degraded, through a sequence of reactions, to acetyl-CoA, acetoacetate, and succinyl-CoA. The second step in this process is catalyzed by the mitochondrial BCKD, which converts an α-keto acid to an acyl-CoA via oxidative decarboxylation. BCKD is a multienzyme complex consisting of 6 subunits. The complex contains 4 enzymatic subunits (E1 [α and β], E2, and E3), BCKD kinase, and BCKD phosphatase. Phosphorylation of the complex inactivates it and, conversely, it is activated by dephosphorylation. The genes encoding the enzymatic components are the BCKA decarboxylase (E1) α subunit gene, BCKA decarboxylase (E1) β subunit gene, dihydrolipoyl transacylase (E2) subunit gene, and lipoamide dehydrogenase (E3) subunit gene. The complex utilizes the coenzymes thiamine pyrophosphate, lipoamide, FAD, and the terminal oxidizing agent id NAD+ (Figure 10-1).
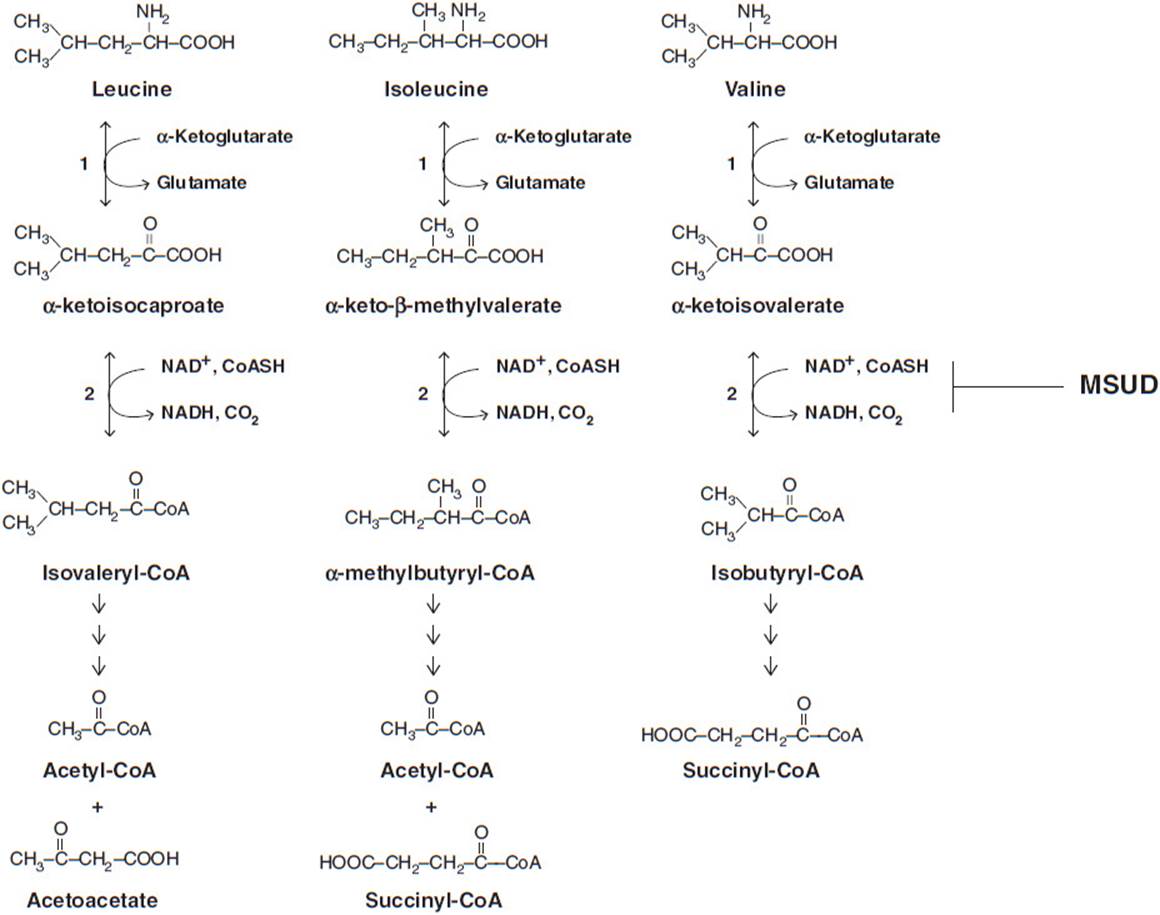
Figure 10-1. Abbreviated BCAA degradation pathway depicting the loss of function of the BCKD complex in the second step of the pathway observed in cases of MSUD. Steps 1 (BCAA transaminase) and 2 (BCKD complex) are common for leucine, valine and isoleucine. BCAA = branched-chain amino acid, BCKD = branched-chain α-ketoacid dehydrogenase complex, MSUD = maple syrup urine disease.
MSUD is caused by an inherited autosomal mutation in either E1α (MSUD type 1A), E1β (MSUD type 1B), or E2 (MSUD type 2). Mutations may also occur in the E3 subunit but cause a different phenotype. The incidence rate in the general population is estimated to be 1:185,000 live births though it can increase to 1:176 live births when considering the Old World Mennonite population. A founder mutation in B CKDHA (E1 α) is believed to be prevalent in this group, with a classic MSUD carrier frequency of 1 in 10. This founder mutation results from a tyrosine to asparagine mutation in the E1 component that prevents the proper interaction of the α and β subunits. In the absence of this interaction, the normal tetrameric structure of the E1 subunit is disrupted, rendering an enzyme with decreased or no functional activity.
MSUD can be classified as classic, intermediate or intermittent and the criteria for classification depend largely on the level of enzyme activity. Classic cases of MSUD have little to no residual BCKDH activity (0%-2% of normal activity) and present with symptoms within a few days of birth. Intermediate and intermittent forms of the disease typically have some level of enzyme activity (3%-30% and 5%-20%, respectively). Patients with intermediate MSUD may appear normal in the neonatal period but present with the same phenotypic symptoms either in infancy or later (typically between the ages of 5 months and 7 years). Intermittent MSUD patients usually develop normally and only display symptoms during times of physiologic stress.
The lack of function of the BCKD complex results in accumulation of the BCAAs and their keto-acid derivatives. As these products build up, they become toxic to organs and, in particular, the brain. Elevated levels of leucine interfere with transport of other large neutral amino acids across the blood-brain barrier, reducing the availability of these required amino acids. This cerebral amino acid deficiency negatively affects brain growth and hinders the synthesis of neurotransmitters (dopamine, serotonin, norepinephrine, and histamine) required for cell-to-cell communication. Leucine is considered neurotoxic because it promotes a neurochemical syndrome that disturbs brain protein accretion, neurotransmitter synthesis, cell volume regulation, neuron growth, and myelin synthesis. Additionally, the cerebral accumulation of α-ketoisocaproic acid has been implicated in the inhibition of the mitochondrial electron transport chain, leading to the elevated cerebral lactate level observed in acute MSUD encephalopathy.
Treatment of MSUD involves limiting dietary intake of branched-chain amino acids, particularly leucine. Unlike leucine, elevated blood concentrations of valine and isoleucine seem to be more easily withstood. Restriction of dietary protein, which naturally contains less valine and isoleucine than leucine, can lead to deficiencies of valine and isoleucine even when leucine concentrations are elevated. Deficiencies of isoleucine and valine can be even more problematic for the brain, since all of the BCAAs use the same transporter to cross the blood–brain barrier. Thus, careful monitoring of BCAA levels in blood is imperative, and supplementation of valine and isoleucine may be required.
COMPREHENSION QUESTIONS
10.1 One hallmark of MSUD is the presence of ketones in the urine. A quick screen involves the addition of dinitrophenylhydrazine (2-DNP) to the patient’s urine sample. A positive result is indicated by the formation of a yellow-white precipitate. The type of reaction responsible for this product can best be described as which of the following?
A. Isomerization reaction
B. Condensation reaction
C. Combustion reaction
D. Acid/base reaction
10.2 In addition to MSUD, there are a few additional inborn errors of metabolism that also present with neonatal encephalopathy. These include hyperketosis syndromes such as β-ketothiolase deficiency, urea cycle defects, glycine encephalopathy, and propionic or methylmalonic academia. Which of the following tests would allow the definitive diagnosis of MSUD?
A. Urine protein analysis
B. Cranial computed tomography
C. CSF culture
D. Plasma amino acid analysis
10.3 A healthy couple of Old World Mennonite descent is preparing to have a child. Given that the carrier incidence of the trait is 1 in 10, what is the probability that their child will have MSUD?
A. 1 in 10
B. 1 in 100
C. 1 in 400
D. 1 in 200
ANSWERS
10.1 B. This reaction can be described as a condensation reaction, with 2 molecules joining together with a loss of water. In this case, the reaction is as follows:
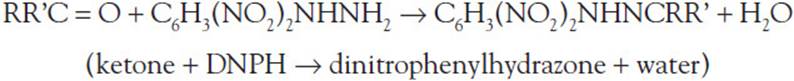
The other types of reactions do not apply. In an isomerization reaction, the structural arrangement of a compound is changed but its net atomic composition remains the same. A combustion reaction is a type of redox reaction in which a combustible material combines with an oxidizer to form oxidized products and generate heat. An acid-base reaction is type of double displacement reaction that occurs between an acid and a base. The H+ion in the acid reacts with the OH– ion in the base to form water and an ionic salt.
10.2 D. In cases of MSUD the levels of plasma BCAAs will be elevated. This will not be true for the other metabolic disorders described. Urine protein analysis and CSF culture would be negative for these disorders and would not allow for differentiation. Conversely, cranial computed tomography scans in each type of case could show signs of cerebral edema.
10.3 C. MSUD is inherited in an autosomal recessive manner, meaning that in order to be affected, the offspring would need to be homozygous for the trait. One in 4 offspring would have this genotype. Given the approximate incidence rate of the trait in the Mennonite population, each parent has a probability of 1/10 to be a carrier. Therefore, the probability that the child is affected is (1/10) × (1/10) × (1/4) = 1/400.
BIOCHEMISTRY PEARLS
![]() The BCAAs are degraded, in part, by the multienzyme complex BCKD.
The BCAAs are degraded, in part, by the multienzyme complex BCKD.
![]() MSUD results from a mutation in one of the enzymatic subunits of this enzyme.
MSUD results from a mutation in one of the enzymatic subunits of this enzyme.
![]() Symptoms of MSUD are caused by an accumulation of the BCAAs and branched-chain keto-acids.
Symptoms of MSUD are caused by an accumulation of the BCAAs and branched-chain keto-acids.
REFERENCES
Bodamer O, Hahn S, Tepas E. Overview of Maple Syrup Urine Disease. Up-to-date. August 8, 2012.
Chuang DT, Chuang JL, Wynn RM. Lessons from genetic disorders of branched-chain amino acid metabolism. J Nutr. 2006;136:243S-249S.
Chuang DT, Wynn RM, Shih VE. Maple syrup urine disease (branched-chain ketoacidosis). In: Valle D, et al, eds. Scriver’s The Online Metabolic & Molecular Bases of Inherited Disease. McGraw-Hill; accessed June 12, 2012.
Strauss KA, Puffenberger EG, Morton DH. Maple syrup urine disease. In: Gene Reviews. [Internet.] Seattle (WA): University of Washington, Seattle; Initial Posting: January 30, 2006; Last Update: December 15, 2009.
Voet D, Voet JG. Amino acid metabolism. Biochemistry. 3rd ed. City, NJ: John Wiley & Sons, Inc; 2004.