Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 12
A 25-year-old Mediterranean woman presents to her obstetrician at 12-weeks gestation for her first prenatal visit. This is her first pregnancy, and she is concerned about her baby and the risk of inheriting a “blood” disease like others in her family. The patient reports a personal history of mild anemia but nothing as severe as her brother who required frequent transfusions and died at the age of 10 years. The patient was told by her physician that she did not need to take iron supplementation for her anemia. Patient denies having any anemic symptoms. Her physical examination is consistent with a 12-week pregnancy and ultrasonography confirmed an intrauterine pregnancy at 12-weeks gestation. The patient’s hemoglobin (Hb) level shows a hypochromic, microcytic (small-sized red cell) anemia (Hb, 9 g/dL) and Hb electrophoresis demonstrated increased Hb A2 level (4.0%) and increased fetal Hb level, a pattern consistent with β-thalassemia minor. The patient underwent chorionic villus sampling to assess whether the fetus was affected, and the diagnosis returned in several hours.
![]() What is the molecular genetics behind this disorder?
What is the molecular genetics behind this disorder?
![]() What was the likely test and what is the biochemical basis?
What was the likely test and what is the biochemical basis?
ANSWERS TO CASE 12:
Thalassemia/Oligonucleotide Probe
Summary: A 25-year-old Mediterranean pregnant woman has a history of asymptomatic mild hypochromic, microcytic anemia, elevated Hb A2 and F on electrophoresis. Her brother had severe hemolytic disease that required transfusions and ultimately caused his premature death at the age of 10 years. She is diagnosed with β-thalassemia minor.
• Molecular genetics: Impaired production of β-globin peptide chain. Numerous mutations have been identified in the production of RNA, including in the promoter region and splice junctions.
• Likely test: Oligonucleotide probe. After chorionic villus sampling is performed, a radioactive probe can be used and hybridized with specific genetic mutations in fetus DNA, allowing for prompt detection and prenatal diagnosis.
CLINICAL CORRELATION
Anemia is the abnormally low level of Hb or red blood cell mass, which has the potential of limiting the delivery of oxygen to tissue. By far, the most common cause of anemia is iron deficiency, leading to small volume of red blood cells (microcytic). Another common cause of microcytic anemia is thalassemia. Certain ethnicities have higher incidences of thalassemia (eg, Mediterranean, East Asian descent).
This patient is of Mediterranean descent, making thalassemia more likely. Furthermore, the microcytic (small red blood cell size) anemia in the face of elevated Hb A2 and F is consistent with β-thalassemia minor. Patients with β-thalassemia major (Cooley anemia) typically have severe anemia requiring frequent transfusions and shortened life expectancy. Infants will appear healthy after birth, but as the HbF levels fall, the infant becomes severely anemic. Females with β-thalassemia major who survive beyond childhood are usually sterile.
APPROACH TO:
Oligonucleotide Probes
OBJECTIVES
1. Know about the use of oligonucleotide probes for detection of mutations.
2. Know how oligonucleotide segments are synthesized.
3. Be familiar with common mutations that cause thalassemias (substitutions in TATA box, mutations in splice junction, and changes in stop codon).
DEFINITIONS
ERYTHROPOIESIS: Erythropoiesis is the development of mature red blood cells containing Hb (erythrocytes) from pluripotential stem cells via a linear cascade.
FRAMESHIFT MUTATION: Insertion or deletion of a number of nucleotides that are not divisible by 3 into a coding sequence, thereby causing an alteration in the reading frame of the entire sequence downstream of the mutation.
LOCUS CONTROL REGION: Regulatory region which is believed to regulate transcription by opening and remodeling chromatin structure. It may also have enhancer activity.
PROMOTER SEQUENCE: A regulatory region present at a short distance upstream from the 5′ end of a transcription start site that acts as the binding site for RNA polymerase to initiate transcription. The TATA box and CAAT box are DNA consensus sequences in the promoter region.
THALASSEMIA: A group of genetic disorders that is characterized by the absence of or reduced synthesis of 1 or more of the 4 globin chains in Hb. The sequelae can range from benign to fatal, depending on the severity of the decrease in the globin chain.
TRANSPOSONS: These are segments of DNA that can move around to different positions in the genome of a single cell. In the process, they may cause mutations and increase (or decrease) the amount of DNA in the genome. These mobile segments of DNA are sometimes called “jumping genes.”
DISCUSSION
Thalassemia is a common condition in the Mediterranean region, and its clinical features were described as early as 1925. The term thalassemia is coined from the Greek word, which means “the sea.” Mendelian transmission of thalassemia was discovered in 1938. Later, it became apparent that thalassemia is not a single disease but a group of genetic disorders, all of which arise from abnormalities in Hb synthesis. The thalassemias are characterized by the absence or reduced synthesis of 1 or more of the 4 globin chains of Hb. There are 4 different genetic loci that control the formation of α-, β-, γ-, and δ-chains of Hb. Fetal Hb (HbF) is formed from 2 α- and 2 γ-chains. In adults, the γ-chain is replaced by the β- and δ-chains, which combine with 2 α-chains to form HbA (α2β2, approximately 97%) and HbA2 (α2γ2, approximately 3%; Figure 12-1). In the late 1970s, the α-gene was found to be localized to chromosome 16, whereas the β-, γ-, and δ-genes were clustered on chromosome 11. Thus, although thalassemia does not occur in the fetus since the β-globin genes are activated only after birth, prenatal diagnosis is possible.
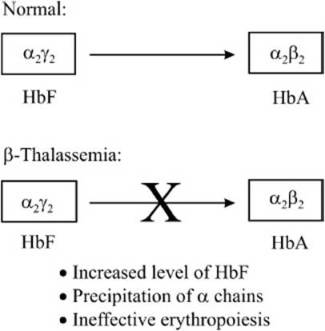
Figure 12-1. Maturation of hemoglobin from the fetal form to the adult form. In β-thalassemias, because of a defect in the β-globin gene, the γ-chain cannot be effectively replaced with the β-chain.
Thalassemias are named according to the affected globin chain: Thus, a disorder of the α-chain would be an α-thalassemia, and a disorder of the β-chain would be a β-thalassemia. There are 2 major forms of thalassemia, one in which the β-globin chain is not produced (β0) and another in which there is significantly reduced level of β-globin chain (β+). The positive category includes variants with unusually high levels of HbF or HbA2, variants with normal levels of HbA2, silent or benign variants, those that are inherited in a dominant fashion, and variants that are not linked to the β-gene cluster.
A severe decrease in β-globin levels leads to the precipitation of the α-chain, which, in turn, causes a defect in the maturation of the erythroid precursor, and erythropoiesis, thus reducing red cell survival. The profound anemia in the affected individual stimulates the production of erythropoietin leading to the expansion of bone marrow and subsequent skeletal deformities. The hyperplasia of the bone marrow induces increased iron absorption leading to the deposition of iron in tissues. If the concentration of iron in the tissues becomes too high, then this can lead to organ failure and death if appropriate therapeutic steps are not taken.
There are at least 200 β-thalassemia alleles that have been characterized to date. β-Thalassemias are caused primarily by point mutations within the β-globin gene and the neighboring flanking region. There are 2 exceptions that are attributed to larger deletions ranging from 290 base pairs (bp) to more than 60 kbp. One is a family of upstream deletions, which down regulates the locus control region, and the other affects the promoter region of β-globingene. The promoter region deletions typically include the messenger RNA (mRNA) cap site, TATA box, and CAAT elements. Apart from deletions, there are insertions of 6 to 7 kbp retrotransposons in the β-globin gene that decrease the transcript level to 15% in comparison with the control. In addition, there are also a few thalassemia mutations that segregate independently of the β-globin gene, such as those involving the trans-acting regulatory factors that affect the expression of the β-globin gene. One can thus broadly classify the different classes of mutations which result in β-thalassemia as follows:
1. Transcription (eg, deletion, insertion)
2. mRNA processing (eg, cryptic splice sites, consensus sequence, poly A addition site)
3. Translation (initiation, nonsense, frameshift mutations)
4. Post-translation stability (highly unstable β-chain)
5. Determinants unlinked to the β-globin gene cluster
The most common point mutations occur in the CAAT or TATA box in the promoter sequence. The single base substitution A to G at position –29 in individuals of African descent leads to a mild form of the disease, whereas the substitution A to C in those of Chinese descent leads to thalassemia major. In addition, a single nucleotide substitution in the GT/AG splice junction can lead to a misspliced mRNA that does not allow the translation of a functional β-globin chain. The C to T mutation at position –101 causes an extremely mild deficit in β-globin gene. The effect is so mild that it is a “silent” or benign mutation in heterozygotes. All the point mutations that prevent the translation of the β-globin mRNA (eg, changing the start codon ATG to GTG, or TGT to the stop codon TGA) or a single nucleotide frame shift lead to the β0-phenotype.
There are several biochemical techniques that can be used to accurately determine the genetic defect that results in a particular thalassemia. Many of these techniques make use of the polymerase chain reaction (PCR), which amplifies the DNA and allows detection of multiple mutations at once.
The amplification refractory mutation system (ARMS) is a technique in which the target DNA is amplified using a common primer and either mutation specific primer for β-thalassemia or the correct sequence primer to the target. This method provides quick screening of the DNA to detect if the patient carries the mutant gene or not. When both the primers are used in the same PCR reaction, they compete to amplify the target, and the technique is termed competitive oligonucleotide priming, and the primers are labeled with different fluorescent dyes to allow detection.
A PCR restriction enzyme analysis takes advantage of the occurrence of approximately 40 thalassemia mutations that either introduce or remove a restriction endonuclease site. The PCR amplified target sequence is digested using restriction enzymes (enzymes that cleave DNA at particular nucleotide sequences) and the pattern of fragmentation on an agarose gel defines the presence or absence of a particular mutation.
Radiolabeled single stranded DNA oligonucleotide probes are allowed to anneal with the target strand in an oligonucleotide hybridization analyses. Typically, the target DNA is fixed onto nitrocellulose or nylon membrane. The probe forms stable duplexes with target sequences from the heterogenous mixture of many sequences in genomic DNA and is detected by autoradiography using x-ray film. In populations that have predominantly one common mutation, a very successful and efficient hybridization analysis is the PCR allele-specific oligonucleotide assay. This requires that the sequence of the common mutation is known. The amplified genomic DNA is transferred onto a nylon membrane and probed using an allele-specific oligonucleotide, that is, one that is complementary to the sequence of the DNA that contains the mutation. The membrane is probed both with oligonucleotides having the mutant and the correct DNA sequence of the β-globin gene. The genotype of the DNA is determined by the presence or the absence of the hybridization signal. Recently, microchips, which are an array of oligonucleotides immobilized on a glass plate, have been used to detect thalassemias. Fluorescent-labeled DNA from an individual is hybridized onto this microchip and the reaction is monitored with a fluorescent microscope. This technique allows the study of a specific mutation using several hundred oligonucleotides simultaneously. It is highly sensitive with a low background; however, the resolving power is low.
Using 1 or a more of the DNA analysis tools described above, effective prenatal detection of various mutations in the β-globin gene cluster has been successfully achieved. Although gene therapy for genetic diseases is in its infancy, hopefully the future will bring effective treatment options for patients affected with thalassemia.
COMPREHENSION QUESTIONS
12.1 Micawley Talltwin is a 7-year-old child star brought to his pediatrician by his parents after they noticed that he felt very fatigued. They also noted that his abdomen seemed to be enlarged. Examination reveals an enlarged spleen. Further history reveals that he has been taking vitamins and iron supplements over the last few months. Laboratory tests show a microcytic anemia and elevated iron levels in tissues. Which of the following conditions is most consistent with the findings in this patient?
A. Aplastic anemia
B. Cooley anemia
C. Pernicious anemia
D. Thalassemia major
E. Thalassemia minor
12.2 After diagnosing the patient in question 12.1 and ascertaining that he is not agranulocytic, his physician prescribes subcutaneous infusion of deferoxamine, an iron chelator, and monitors him for several weeks. What is his condition most likely to be on reexamination?
A. Iron levels decrease, but he remains anemic.
B. Iron levels decrease, and anemia recovers.
C. He develops irreversible and severe agranulocytosis.
D. Iron and vitamin C levels decrease.
12.3 An electrophoretic analysis of Hb level in the patient in questions 12.1 and 12.2 indicates that, although there is a decrease in the relative amount of the β-chain with respect to the α-chain, both the β- and the α-chains migrate at the same position as normal chains. His anemia is most likely caused by which of the following?
A. Defect in an enzyme involved in heme synthesis
B. Point mutation in the coding region of the gene coding for the β-chain
C. Frameshift mutation in the coding region of the gene coding for the β-chain
D. Mutation in the promoter of the β-chain gene
E. Mutation in the structural gene of the β-chain
12.4 As a medical geneticist, you analyze the DNA of the patient from question 12.1 and find that he is homozygous for thalassemia. Assuming the disease is autosomal recessive, what can you deduce about the genotype of Mr. and Mrs. Talltwin?
A. Dad Talltwin is a carrier of the disease and Mom Talltwin is normal.
B. Mom Talltwin is a carrier of the disease and Dad Talltwin is normal.
C. Dad Talltwin is homozygous and Mom Talltwin is normal.
D. Mom Talltwin is homozygous and Dad Talltwin is normal.
E. Both Mom and Dad Talltwin are carriers of the disease.
ANSWERS
12.1 E. The correct diagnosis is thalassemia minor because the patient had been asymptomatic until the age of 7 years. If he had thalassemia major or Cooley anemia, then he would have exhibited symptoms as early as his first birthday. Pernicious anemia leads to a macrocytic or megaloblastic anemia, whereas aplastic anemia is characterized by normal sized erythrocytes.
12.2 A. The iron chelator helps in excreting iron but has no role in increasing red blood cell production to counteract anemia.
12.3 D. Because the β-chain is decreased with respect to the α-chain, it is most likely that there is a mutation that decreases the expression of the β-chain gene, in which a mutation in the promoter region could result. A point mutation in the β-chain leading to an amino acid substitution could lead to changes in electrophoretic mobility but would not alter the levels of expression. A frameshift mutation in the β-chain would result in decreased β-chain on the electrophoregram.
12.4 E. Thalassemia is autosomal recessive; therefore, Micawley Talltwin can get the disease only if both the parents were to be heterozygotes (or a carrier) of that particular trait.
BIOCHEMISTRY PEARLS
![]() The thalassemias are characterized by the absence or reduced synthesis of 1 or more of the 4 globin chains of Hb.
The thalassemias are characterized by the absence or reduced synthesis of 1 or more of the 4 globin chains of Hb.
![]() Thalassemias are named according to the affected globin chain; thus, a disorder of the α-chain would be an α-thalassemia and a disorder of the β-chain would be a β-thalassemia.
Thalassemias are named according to the affected globin chain; thus, a disorder of the α-chain would be an α-thalassemia and a disorder of the β-chain would be a β-thalassemia.
![]() The ARMS is a technique in which the target DNA is amplified using a common primer and either mutation specific primer for β-thalassemia or the correct sequence primer to the target.
The ARMS is a technique in which the target DNA is amplified using a common primer and either mutation specific primer for β-thalassemia or the correct sequence primer to the target.
![]() PCR restriction enzyme analysis takes advantage of the occurrence of approximately 40 β-thalassemia mutations that either introduce or remove a restriction endonuclease site.
PCR restriction enzyme analysis takes advantage of the occurrence of approximately 40 β-thalassemia mutations that either introduce or remove a restriction endonuclease site.
![]() Radio-labeled single stranded DNA oligonucleotide probes are allowed to anneal with the target strand in an oligonucleotide hybridization analyses.
Radio-labeled single stranded DNA oligonucleotide probes are allowed to anneal with the target strand in an oligonucleotide hybridization analyses.
![]() With 1 common mutation, a very successful and efficient hybridization analysis is the PCR allele-specific oligonucleotide assay. This requires that the sequence of the common mutation is known.
With 1 common mutation, a very successful and efficient hybridization analysis is the PCR allele-specific oligonucleotide assay. This requires that the sequence of the common mutation is known.
REFERENCES
Kanavakis E, Traeger-Synodino J, Vrettou C, et al. Prenatal diagnosis of the thalassaemia syndromes by rapid DNA analytical methods. Mol Hum Reprod. 1997;3(6):523-528.
Weatherall DJ, Clegg, JB. The Thalassaemia Syndromes. 4th ed. London: Blackwell Science; 2001.