Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 20
A 21-year-old primigravid woman at 35 weeks gestation presents to the hospital with nausea, vomiting, and malaise over the last several days. Patient has also noticed that her eyes were turning yellow in color. Her prenatal course has otherwise been unremarkable. On examination she is found to have elevated blood pressure, proteinuria, increased liver function tests, prolonged clotting studies, hyperbilirubinemia, hypofibrinogenemia, and hypoglycemia. Pelvic ultrasonography identified a viable intrauterine pregnancy of approximately 35 weeks gestation. After admission, the mother underwent emergent cesarean delivery, and she subsequently developed worsening hypoglycemia and coagulopathy and went into hepatic coma with renal failure. After reviewing all the laboratory results and her clinical picture, the patient was diagnosed with acute fatty liver of pregnancy (AFLP).
![]() What is an associated biochemical disorder?
What is an associated biochemical disorder?
![]() What is the etiology of the hypoglycemia?
What is the etiology of the hypoglycemia?
ANSWERS TO CASE 20:
Acute Fatty Liver in Pregnancy
Summary: 21-year-old woman at 35 weeks gestation with malaise, nausea and vomiting, jaundice, elevated blood pressure, elevated liver function tests, coagulopathy, hypoglycemia, and subsequently hepatic coma and renal failure. She has been diagnosed with AFLP.
• Associated biochemical defect: Fetal deficiencies of long chain 3-hydroxyacyl-coenzyme A dehydrogenase (LCHAD).
• Cause of hypoglycemia: Decreased liver glycogen after liver undergoes fatty infiltration and subsequent liver failure. Histology reveals swollen hepatocytes in which the cytoplasm is filled with microvesicular fat.
CLINICAL CORRELATION
AFLP is a poorly understood condition affecting only pregnant women with the clinical manifestations of hypoglycemia, liver failure, metabolic acidosis, renal failure, and coagulopathy. Affected patients may become jaundiced or encephalopathic from liver failure, usually reflected by an elevated ammonia level. Profound hypoglycemia is common. The mortality rate is approximately 10% to 15%. Management is delivery, with supportive measures such as magnesium sulfate to prevent seizures, replacement of blood or clotting factors, and management of blood pressure. The pathophysiology may be related to fetal deficiencies of LCHAD.
APPROACH TO:
Glycogen and Carbohydrate Metabolism
OBJECTIVES
1. Know about glycogen storage, synthesis, and degradation.
2. Be familiar with the regulation of glycogen synthesis and degradation.
3. Understand the role of liver glycogen in carbohydrate metabolism.
DEFINITIONS
GLUCOSE TRANSPORTER ISOFORM 2 (GLUT 2): A facilitative glucose transporter present in the liver, the β-cells of the pancreas, and the basolateral surface of intestinal epithelial cells.
ADENYLATE CYCLASE: The enzyme that, when activated by hormones binding to receptors, catalyzes the cyclization of adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP) with the release of pyrophosphate.
BRANCHING ENZYME: 1,4-α-Glucan branching enzyme; an enzyme that removes an oligosaccharide of about seven glucosyl residues from the nonreducing end of a glycogen chain and transfers it to another chain, creating an α(1→6) glycosidic bond.
DEBRANCHING ENZYME: A bifunctional enzyme that catalyzes 2 reactions in the degradation of glycogen. It transfers a trisaccharide from the nonreducing end of a 4 glucosyl residue branch of the glycogen molecule to the nonreducing end of the same or adjacent glycogen molecule (oligo-1,4–1,4-glucantransferase activity). It also hydrolyzes the α(1→6) linkage of the remaining glucosyl residue of the branch, thus releasing free glucose (amylo-1,6-glucosidase activity).
GLYCOGEN: The storage form of glucose in tissues. It is a large polysaccharide composed of glucose residues in primarily α(1→4) glycosidic linkages with some α(1→6) branch points.
GLYCOGENESIS: The synthesis of glycogen from glucose 1-phosphate.
GLYCOGENOLYSIS: The breakdown of glycogen to glucose 1-phosphate (and some small amount of free glucose).
GLYCOGEN PHOSPHORYLASE: The enzyme that causes the release of glucose 1-phosphate from glycogen. It accomplishes this by catalyzing a phosphorolysis of glucosyl residues from glycogen; that is, it breaks the α(1→4) glycosidic bonds by adding inorganic phosphate and releasing glucose 1-phosphate.
GLYCOGEN SYNTHASE: The enzyme that causes the addition of glucosyl residues to a growing glycogen molecule using uridine diphosphate (UDP)-glucose and releasing inorganic pyrophosphate.
DISCUSSION
AFLP is a rare (occurring in 1 in 7000 to 16,000 deliveries) but potentially fatal disease that typically develops during the third trimester. Patients most commonly present with clinical and laboratory evidence of acute hepatic failure with decreased hepatic metabolic activity. Hypoglycemia, nausea and vomiting, jaundice, general malaise, elevated blood pressure, disseminated intravascular coagulation, hemorrhage, infection, and encephalopathy are the most common clinical findings. The etiology of the syndrome is not clear, although recent reports have linked some cases of AFLP with a fetal inborn error in fatty acid metabolism. For the majority of cases in which an inborn error in the fetus does not appear to play a role, the cause of the disease is unknown.
One of the primary functions of the liver is to maintain blood glucose levels. When blood glucose levels are high following a meal, the liver takes in glucose via the high capacity, insulin-insensitive glucose transporter GLUT 2 and converts it into glycogen for storage, metabolizes it to pyruvate by glycolysis, or uses it to produce NADPH and pentoses for biosynthetic processes via the pentose phosphate pathway. When blood glucose levels drop after fasting or vigorous exercise, the liver mobilizes its glycogen stores in response to glucagon and epinephrine and exports glucose into the blood. As glycogen levels are depleted, the liver begins to synthesize glucose via gluconeogenesis. In addition to a source of carbons to synthesize glucose, which it obtains from either lactate or the breakdown of amino acids, the liver also needs a source of energy in the form of ATP. β-Oxidation of fatty acids provides the reducing equivalents (NADH and FADH2), by which ATP is synthesized through the action of the electron transport system and oxidative phosphorylation.
The storage form of glucose is glycogen, which is stored in both muscle and liver. However, the function of stored glycogen is different in these 2 tissues. Muscle uses glycogen as a fuel reserve to provide ATP for its own needs, whereas the liver uses stored glycogen as a reservoir for glucose to maintain blood glucose levels. When blood glucose concentrations drop, glucagon and epinephrine are released into the bloodstream and bind to glucagon and epinephrine receptors on hepatocytes. The binding of the hormones to the receptors activates adenylate cyclase producing 3′,5′-cAMP. When cAMP binds to cAMP-dependent protein kinase (PKA), it is activated and is able to phosphorylate target proteins. This leads to activation of glycogen phosphorylase, the enzyme primarily responsible for mobilizing glucose from glycogen. Phosphorylase, which is stabilized by pyridoxal phosphate (vitamin B6), catalyzes the phosphorolysis of glycogen; it cleaves the 1,4-glycosidic bond of a terminal glucose residue from the nonreducing end of the glycogen molecule using inorganic phosphate (Figure 20-1). The products are glucose 1-phosphate and glycogen that is shorter by 1 glucose residue.
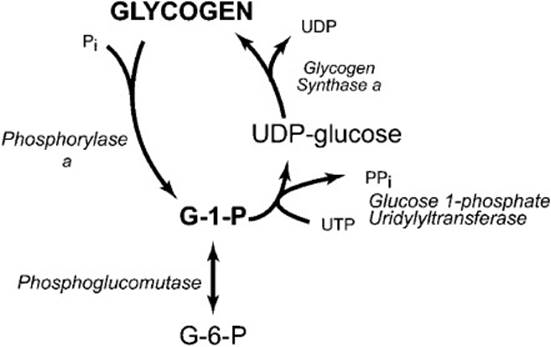
Figure 20-1. Reactions involved in the synthesis and breakdown of glycogen.
Mobilization of glycogen stores also requires the participation of a debranching enzyme because phosphorylase ceases to cleave α-1,4-glycosidic linkages 4 glucosyl residues from an α-1,6-branch site. The debranching enzyme has 2 catalytic activities: a transferase activity and a glucosidase activity. The transferase activity of the enzyme removes an oligosaccharide composed of the terminal 3 glucosyl residues from the 4 residue branch and transfers it to a free 4-hydroxyl group of the terminal glucosyl residue of another branch. The remaining glucosyl residue that is in an α-1,6-glucosidic linkage is then hydrolyzed by the glucosidase activity to release free glucose.
Glucose 1-phosphate released from glycogen by phosphorylase is converted to glucose 6-phosphate by phosphoglucomutase. Glucose 6-phosphatase, which is only present in liver and other gluconeogenic tissues, hydrolyzes the phosphate to produce free glucose. Glucose is then exported from the liver via the GLUT 2 transporter to increase the blood glucose concentration.
Following a meal, glycogen concentrations within the liver rise rapidly to high levels; this can be up to 10% of the wet weight of the liver. Glucose in the blood is transported into the hepatocyte by the GLUT 2 transporter and is converted to glucose 6-phosphate by glucokinase. Phosphoglucomutase then catalyzes the readily reversible reaction that converts glucose 6-phosphate to glucose 1-phosphate. The glucose 1-phosphate is further activated to UDP-glucose by glucose 1-phosphate uridylyltransferase in a reaction that consumes uridine triphosphate (UTP) and produces inorganic pyrophosphate. This reaction is thermodynamically favored by the hydrolysis of pyrophosphate by pyrophosphatase, which also makes the formation of UDP-glucose an irreversible reaction. Glycogen synthase catalyzes the addition of a glucosyl residue to a glycogen molecule using UDP-glucose as the substrate, forming an α(1→4) glycosidic bond and releasing UDP. Because glycogen synthase cannot create an α(1→6) linkage, an additional enzyme is required to form branches. When a chain of at least 11 glucosyl residues has been synthesized, 1,4-α-glucan branching enzyme removes a chain of about 7 glucosyl residues and transfers it to another chain, creating an α(1→6) glycosidic bond. This new branch point must be at least 4 glucosyl residues away from another branch point.
Because the synthesis and mobilization of glycogen together form a potential futile cycle, the competing processes must be regulated to prevent waste of ATP/UTP. This is accomplished by hormonal as well as allosteric controls. The enzymatic cascade, which is promulgated when glucagon or epinephrine bind to their respective receptors on the liver cells, is presented in Figure 20-2. The cAMP that is produced by activation of adenylate cyclase binds to PKA and activates it so that it can phosphorylate its target proteins. These include phosphorylase kinase, glycogen synthase, and inhibitor 1. Phosphorylation of glycogen synthase converts it to an inactive form, whereas phosphorylation of phosphorylase kinase and inhibitor 1 activate them. The phosphorylated inhibitor 1 strongly binds to protein phosphatase 1, but it is a poor substrate and is slowly hydrolyzed. Although the phosphorylated inhibitor 1 is bound to the phosphatase, it will inhibit it from acting on other phosphorylated proteins. Thus, while protein phosphatase 1 is inhibited, those proteins that are activated by phosphorylation remain active, and those that are inhibited by phosphorylation stay in their inactive form.
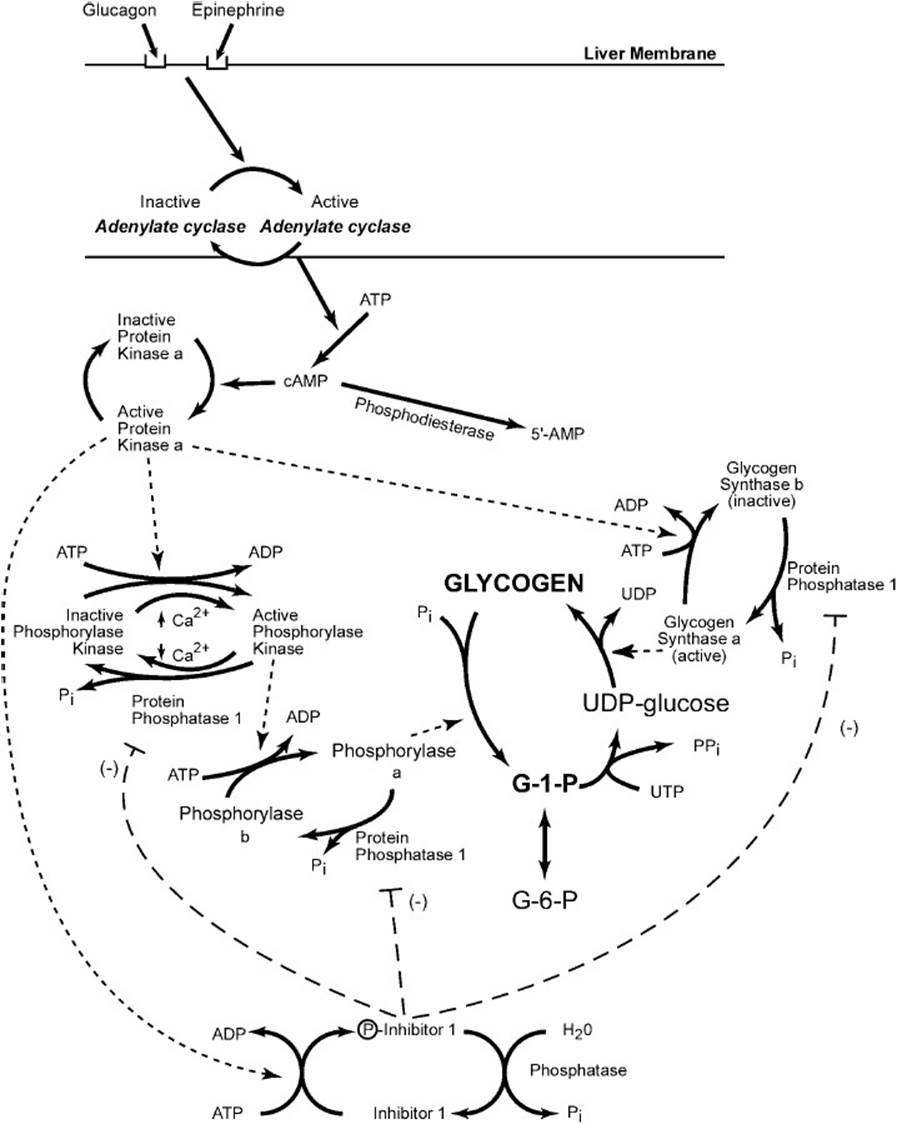
Figure 20-2. The mobilization of glycogen in the liver in response to hormonal signals. Binding of the hormones glucagon, epinephrine, or both causes the activation of adenylate cyclase resulting in the production of cyclic adenosine monophosphate, which activates protein kinase A (PKA). By phosphorylation reactions, PKA inactivates glycogen synthase, activates a cascade that results in active glycogen phosphorylase, and produces an active inhibitor of protein phosphatase 1.
Phosphorylation of phosphorylase kinase partially activates it so that it can phosphorylate phosphorylase b to its active form. Phosphorylase kinase is also partially activated by Ca2+; full activation is obtained when it both binds Ca2+ and is phosphorylated. Conversion of phosphorylase b to phosphorylase a enables glucose 1-phosphate to be released from glycogen. Thus, glucagon and epinephrine start a cascade that mobilizes glucose from glycogen and at the same time inhibits the storage of glucose as glycogen.
When blood glucose levels are elevated, insulin is secreted from the pancreatic cells. When insulin binds to hepatic insulin receptors, it results in the activation of a complex series of kinases that leads to the activation of protein phosphatase 1. Protein phosphatase 1 dephosphorylates phosphorylase kinase, phosphorylase, and inhibitor 1, thus inactivating them and inhibiting the phosphorolysis of glycogen. It also dephosphorylates glycogen synthase, converting it to its active form and enabling the storage of glucose as glycogen. In addition, the liver form of phosphorylase a is inhibited by elevated intracellular concentrations of glucose. Thus insulin favors the storage of glycogen and inhibits its mobilization.
Although the etiology of the AFLP is unclear, it does appear to be a defect affecting mitochondrial processes. Liver biopsy usually will show mitochondrial disruption and microvesicular fat deposits, indicating decreased β-oxidation of fatty acids. The fatty acids, since they cannot be efficiently oxidized in the mitochondria, are converted to triglycerides, which build up in the hepatocyte. The fat infiltration decreases the amount of glycogen that can be stored and mobilized to maintain blood glucose levels. Gluconeogenesis is also depressed because ATP is not available from the oxidation of fatty acids. Thus, blood glucose levels decline.
As noted above, there have been reports that link some cases of AFLP with a defect in fatty acid metabolism in the fetus. These include fetal deficiencies of long chain 3-hydroxyacyl-coenzyme A dehydrogenase (LCHAD), carnitine-palmitoyl transferase 1 (CPT 1), and medium chain acylcoenzyme A dehydrogenase (MCAD). The mechanism by which defective fetal fatty acid oxidation causes maternal illness is not known. However, since the fetus uses primarily glucose metabolism for its energy needs, it is likely that toxic products from the placenta, which does use fatty acid oxidation, cause the maternal liver failure.
COMPREHENSION QUESTIONS
For questions 20.1 and 20.2 refer to the following case:
A female infant appeared normal at birth but developed signs of liver disease and muscular weakness at 3 months. She had periods of hypoglycemia, particularly on awakening. Examination revealed an enlarged liver. Laboratory analyses following fasting revealed ketoacidosis, blood pH 7.25, and elevations in both alanine transaminase (ALT) and aspartate transaminase (AST) levels. Administration of glucagon following a carbohydrate meal elicited a normal rise in blood glucose, but glucose levels did not rise when glucagon was administered following an overnight fast. Liver biopsy revealed an increase in the glycogen content (6% of wet weight).
20.1 In which of the following enzymes is a genetic deficiency most likely for this patient?
A. Branching enzyme
B. Debranching enzyme
C. Glucose-6-phosphatase
D. Glycogen synthase
E. Muscle phosphorylase
20.2 To prevent the frequent episodes of hypoglycemia, which of the following dietary supplements would be most appropriate for this patient?
A. Casein (milk protein)
B. Fish oil
C. Fructose
D. Lactose
E. Uncooked cornstarch
20.3 A 17-year-old boy presents complaining of an inability to perform strenuous exercise without bringing on painful muscle cramps and weakness. He indicated that mild to moderate exercise resulted in no problems. When he was administered an ischemic exercise test, his serum lactate concentrations did not significantly increase. A deficiency in which of the following enzymes is most likely the cause of the patient’s muscle cramps?
A. Carnitine palmityl transferase II
B. Glucose-6-phosphatase
C. Glycogen phosphorylase
D. Glycogen synthase
E. Very long chain acyl-CoA dehydrogenase
20.4 A 23-year-old man has been vigorously working on the yard and begins to feel slightly light-headed from hypoglycemia. He drinks a can of soda and is aware of the competition for the glucose to be stored in his liver as glycogen versus used as energy in his muscles. What is the best explanation regarding the fate of the glucose in the soda?
A. The lower Km of hexokinase compared with the Km of glucokinase will tilt the glucose toward glycolysis.
B. The bolus of glucose via the soda will lead to a higher glucose level, inducing storage of the glucose into glycogen in the liver.
C. The muscle is using high levels of glucose, leading to an increased level of glucose 6-phosphate thus inhibiting glucokinase.
D. The glucose will be equally used by muscle for metabolism and liver for glycogen storage.
ANSWERS
20.1 B. Definitive diagnosis would await analysis of the glycogen structure and enzyme activities, but the hepatomegaly, increased liver glycogen content, fasting hypoglycemia, and muscle weakness are consistent with Cori disease type 3 glycogen storage disease. The increase in glycogen content results from an inability to degrade glycogen beyond the limit dextrin of phosphorylase. A deficiency in the debranching enzyme leaves glycogen with short outer branches.
20.2 E. Because fasting hypoglycemia results from an inability to break down glycogen past the limit dextrin of phosphorylase, a patient with type 3 glycogen storage disease should be given frequent meals high in carbohydrates. Uncooked cornstarch is an effective supplement because it is slowly digested, and therefore the glucose is released slowly into the bloodstream, helping to maintain blood glucose concentrations.
20.3 C. Although a deficiency in a number of enzymes can result in exercise intolerance, the lack of an increase in serum lactate following ischemic exercise points to an inability to a defect in the breakdown of glycogen in the muscle. The muscle depends on glycogenolysis for intense exercise, and fatigue rapidly ensues when glycogen is depleted. Patients with a deficiency in the muscle isoform of glycogen phosphorylase (McArdle disease) can tolerate mild to moderate exercise, but get muscle cramps with strenuous exercise as a consequence of the lack of glycogenolysis in the muscle cell.
20.4 A. Hexokinase is found in most tissues, and because of the very low Km (substrate concentration at which the enzyme achieves half maximal velocity) for glucose, it is designed to work maximally to provide ATP for tissue even at low levels of glucose. Hexokinase is inhibited by glucose 6-phosphate and is most active with low levels of glucose 6-phosphate. Glucokinase found in the liver has a high Km for glucose and is very active after a meal. The glucose in the soda would likely be used for ATP production.
BIOCHEMISTRY PEARLS
![]() The etiology of the syndrome is not clear, although recent reports have linked some cases of AFLP with a fetal inborn error in fatty acid metabolism.
The etiology of the syndrome is not clear, although recent reports have linked some cases of AFLP with a fetal inborn error in fatty acid metabolism.
![]() Following a meal, glycogen concentrations within the liver rise rapidly to high levels; this can be up to 10% of the wet weight of the liver.
Following a meal, glycogen concentrations within the liver rise rapidly to high levels; this can be up to 10% of the wet weight of the liver.
![]() Liver insufficiency may be associated with hypoglycemia.
Liver insufficiency may be associated with hypoglycemia.
REFERENCES
Castro M, Fassett MJ, Telfer RB, et al. Reversible peripartum liver failure: A new perspective on the diagnosis, treatment, and cause of acute fatty liver of pregnancy, based on 28 consecutive cases. Am J Obstet Gynecol. 1999;181(2):389-395.
Rakheja D, Bennett MJ, Rogers BB. Long-chain L-3-hydroxyacyl-coenzyme A dehydrogenase deficiency: a molecular and biochemical review. Lab Invest. 2002;82(7):815-824.
Roe CR, Ding J. Mitochondrial fatty acid oxidation disorders. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Metabolic and Molecular Basis of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001:2297-2326.
Saudubray J-M, Charpentier C. Clinical phenotypes: diagnosis/algorithms. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Metabolic and Molecular Basis of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001:1327-1403.
Yang Z, Yamada J, Zhao Y, et al. Prospective screening for pediatric mitochondrial trifunctional protein defects in pregnancies complicated by liver disease. JAMA. 2002;288(17):2163-2166.