Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 29
A 6-year-old boy is brought to his pediatrician with profound mental retardation. Upon questioning the mother, several members of the parents’ families have had mental retardation. She also has noticed that he appears very active and restless, frequently getting into trouble in school. On examination, the boy appears restless and his speech is significantly delayed and incomprehensible. The boy has coarse facial features but otherwise has a normal examination. Laboratory tests reveal an elevated level of heparan sulfate.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the inheritance pattern of this disorder?
What is the inheritance pattern of this disorder?
![]() What are some other causes of mucopolysaccharidoses (MPS)?
What are some other causes of mucopolysaccharidoses (MPS)?
ANSWERS TO CASE 29:
Sanfilippo Syndrome
Summary: A 6-year-old boy with severe mental retardation, family history of mental retardation, language delays, and behavioral problems.
• Diagnosis: Sanfilippo syndrome
• Inheritance pattern: Autosomal recessive
• Other causes: Hunter syndrome, Hurler syndrome, or Morquio syndrome
CLINICAL CORRELATION
Excessive accumulation of proteins, nucleic acids, carbohydrates, and lipids can result from deficiency of 1 or more lysosomal hydrolases. Lysosomal storage diseases are classified by the stored material. Accumulation of glycosaminoglycans results in MPS. Common causes of this disorder include Hunter syndrome, Hurler syndrome, and Sanfilippo syndrome. Sanfilippo syndrome is inherited in an autosomal recessive pattern and clinically evident by profound mental retardation, lack of normal developmental milestones, and significant language delay. Sanfilippo syndrome results in an excess of heparan sulfate and can be caused by a variety of enzyme deficiencies.
APPROACH TO:
Lysosomal Degradation of Glycosaminoglycans
OBJECTIVES
1. Understand the structural roles of glycosaminoglycans and proteoglycans.
2. Describe how glycosaminoglycans and proteoglycans are synthesized.
3. Describe the biochemical pathways needed for glycosaminoglycan catabolism.
4. Explain why lysosomal enzyme deficiencies and glycosaminoglycan accumulation result in clinical signs/symptoms.
DEFINITIONS
ENDOGLYCOSIDASE: An enzyme that hydrolyzes an interior glycosidic bond between 2 sugars in a polysaccharide or oligosaccharide to produce 2 smaller oligosaccharides.
EXOGLYCOSIDASE: An enzyme that hydrolyzes the glycosidic bond between the terminal 2 sugars of an oligosaccharide or polysaccharide releasing the terminal sugar from the nonreducing end of the polymer, thus leaving it 1 sugar shorter.
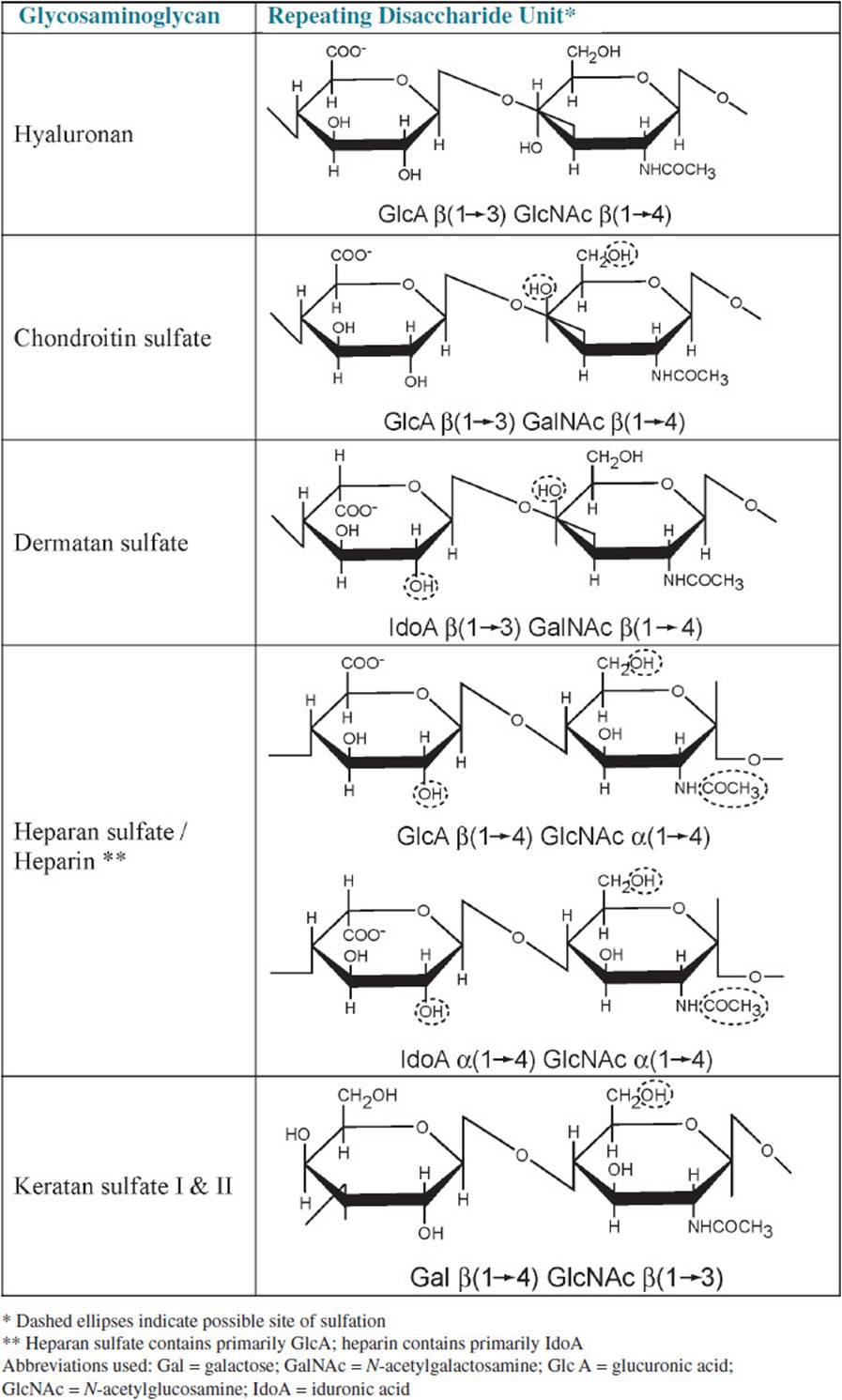
GLYCOSAMINOGLYCAN (GAG): Formerly known as mucopolysaccharide; a heteropolysaccharide composed of a repeating disaccharide of an N-acetylated hexosamine (glucosamine or galactosamine) and an acidic hexose (glucuronic acid or iduronic acid); this repeating disaccharide unit is frequently sulfated in 1 or more positions; most glycosaminoglycans are covalently linked to a core protein in structures known as proteoglycans.
HYALURONAN: Formerly known as hyaluronic acid; a glycosaminoglycan composed of alternating residues of glucuronic acid and N-acetylglucosamine. Hyaluronan is not sulfated nor is it covalently linked to a protein.
MUCOPOLYSACCHARIDOSIS: A genetic disorder involving a lack of a lysosomal enzyme required for the degradation of glycosaminoglycans leading to buildup of glycosaminoglycans in the lysosome and increased excretion of glycosaminoglycan fragments in urine.
SULFATASE: An enzyme that catalyzes the hydrolysis of sulfate ester bond releasing free inorganic sulfate from the substrate.
DISCUSSION
The extracellular matrix that surrounds and binds certain types of cells is composed of numerous components, including fibrous structural proteins, such as various collagens, adhesive proteins like laminin and fibronectin, and proteoglycans that form the gel into which the fibrous structural proteins are embedded. Proteoglycans are very large macromolecules consisting of a core protein to which many long polysaccharide chains called glycosaminoglycans are covalently bound. Due to the high negative charge of the glycosaminoglycans, the proteoglycans are very highly hydrated, a property that allows the proteoglycans to form a gel-like matrix that can expand and contract. The proteoglycans are also effective lubricants.
The glycosaminoglycans (GAGs; formerly called mucopolysaccharides) are long, linear polymers of repeating disaccharide units containing an acidic sugar (glucuronic acid or iduronic acid) and a hexosamine (glucosamine or galactosamine, both usually N-acetylated). The exception to this general structure is keratan sulfate, which has galactose in place of the acidic hexose. Table 29-1 lists the various types of GAGs and the structures of their repeating disaccharide units. The GAGs are often highly sulfated, which increases their negative charge and their ability to bind water molecules. All of the GAGs except hyaluronan are covalently linked to one of approximately 30 different core proteins to form proteoglycans. The core protein is synthesized on the rough endoplasmic reticulum and transferred to the Golgi where nucleoside diphosphate–activated acidic and amino sugars are alternately added to the nonreducing end of the growing polysaccharide by glycosyltransferases, resulting in the characteristic repeating disaccharide structure common to the GAGs. Hyaluronan, which is not sulfated nor covalently linked to a core protein, is synthesized at the plasma membrane by hyaluronan synthases. Hyaluronan synthases are integral membrane proteins that catalyze the alternate addition of UDP-glucuronate and UDP-N-acetylglucosamine to the reducing end of the growing hyaluronan polymer at the inner surface of the plasma membrane as it extrudes the nonreducing end of the GAG into the extracellular space. Hyaluronan can then assemble into large macromolecular complexes with other proteoglycans, which are noncovalently attached to hyaluronan by link proteins.
Table 29-1 • GLYCOSAMINOGLYCAN COMPOSITION
Degradation of proteoglycans during normal turnover of the extracellular matrix begins with proteolytic cleavage of the core protein by proteases in the extracellular matrix, which then enters the cell via endocytosis. The endosomes deliver their content to the lysosomes, where the proteolytic enzymes complete the degradation of the core proteins and an array of glycosidases and sulfatases hydrolyze the GAGs to monosaccharides. The lysosomes contain both endoglycosidases, which hydrolyze the long polymers into shorter oligosaccharides, and exoglycosidases that cleave individual acidic- or aminosugars from the GAG fragments.
Lysosomal catabolism of GAGs proceeds in a stepwise manner from the non-reducing end, as is shown with the degradation of heparan sulfate in Figure 29-1. If the terminal sugar is sulfated, then the sulfate bond must be hydrolyzed by a specific sulfatase before the sugar can be removed. When the sulfate has been removed, a specific exoglycosidase then hydrolyzes the terminal sugar from the nonreducing end of the oligosaccharide, thus leaving it 1 sugar shorter. Degradation continues in this stepwise fashion, alternating between removal of sulfates by sulfatases and cleavage of the terminal sugars by exoglycosidases. If removal of a sulfate leaves a terminal glucosamine residue, then it must first be acetylated to N-acetylglucosamine because the lysosome lacks the enzyme required to remove glucosamine. This is accomplished by an acetyltransferase that uses acetyl-CoA as the acetyl group donor. When the glucosamine residue has been N-acetylated it can be hydrolyzed by α-N-acetylglucosaminidase, allowing the continuation of the stepwise degradation of the GAG.
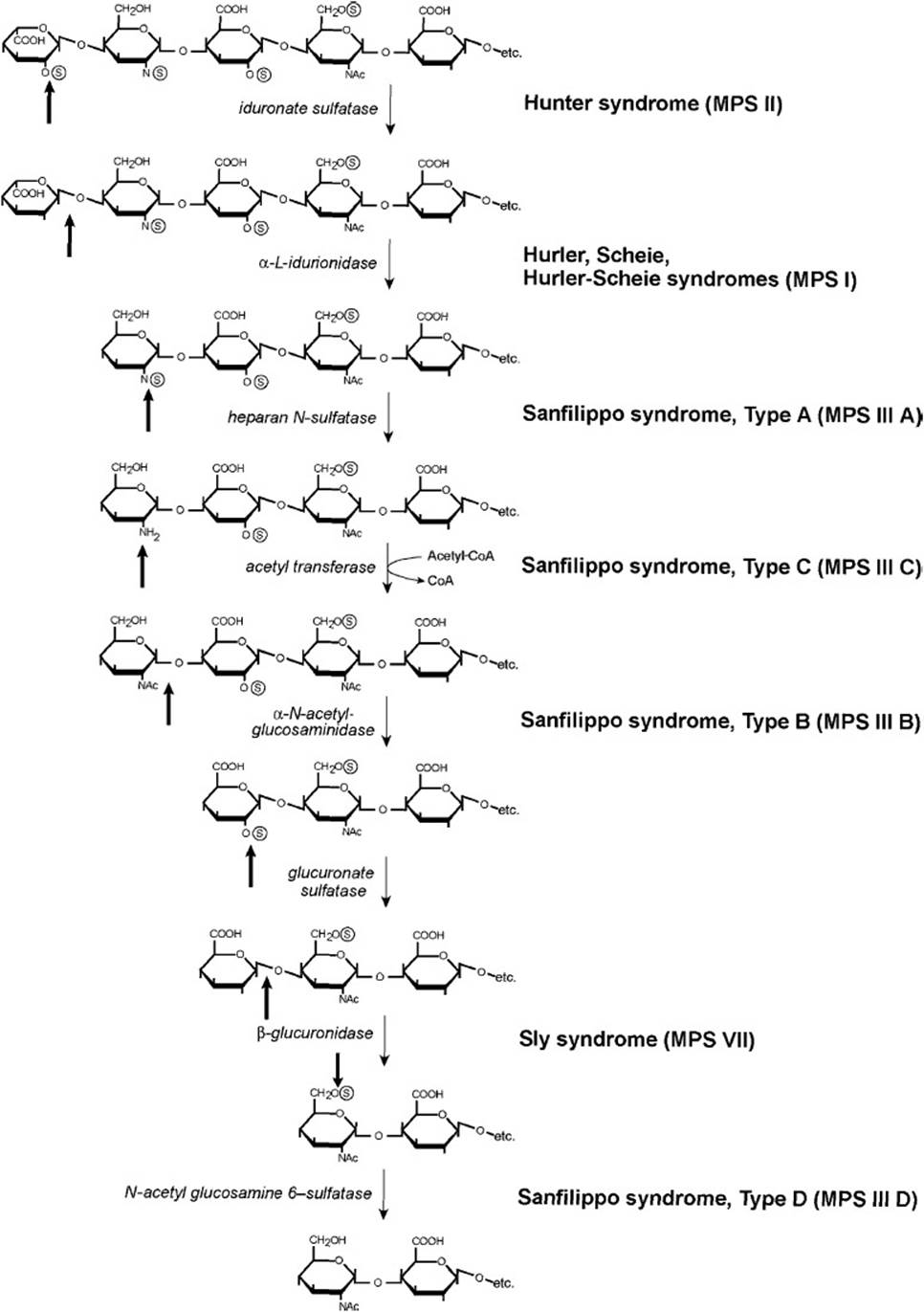
Figure 29-1. Lysosomal degradation of heparan sulfate. Dark arrows indicate the site of action of the enzyme, which is listed in italics. Mucopolysaccharidoses (MPS) due to a deficiency of the indicated enzymes are listed in bold to the right of the enzyme.
Disease states known as Mucopolysaccharidoses (MPS) occur when there is a genetic deficiency of the enzymes involved in the lysosomal breakdown of the GAGs. A deficiency of any of these enzymes can lead to the accumulation of partially degraded GAGs in lysosomes and increased urinary excretion of GAG fragments. Histologic examination of affected cells shows large vacuoles, which are lysosomes engorged with partially degraded GAGs. Because GAGs are present throughout the body, deficiencies of enzymes that degrade them affect bone, connective tissues, and other organs.
MPS is classified into 7 clinical types and all are transmitted by autosomal recessive inheritance except for Hunter syndrome (MPS II, iduronate sulfatase deficiency), which is an X-linked disorder. Diagnosis of the specific disorder is made by measuring the specific enzyme activities in leukocytes or cultured skin fibroblasts. Because it takes some time for the GAGs to accumulate, individuals with a mucopolysaccharidosis usually have a period of normal development before symptoms appear. Depending on the type and severity of the disorder, physical symptoms can include coarse facial features, dwarfism and deformities of the skeleton, cardiovascular impairments, hepatosplenomegaly, neurologic deficits, and mental retardation. As shown in Figure 29-1, Sanfilippo syndrome can be caused by a deficiency of 1 of 4 different enzymes that degrade heparan sulfate. Although the facial coarsening is mild in individuals with Sanfilippo syndrome, the accumulation of heparan sulfate in lysosomes leads to severe neurologic and mental impairment that result in death usually by the end of the second decade of life.
COMPREHENSION QUESTIONS
29.1 A 5-year-old boy is seen by a pediatrician because his parents are concerned about his aggressive behavior, hyperactivity, and loss of language skills. He also has recently become increasingly unsteady on his feet and has experienced a recent seizure. Slight facial feature coarsening is noted. In which of the following processes is this child most likely to have a disorder?
A. Mobilization of glycogen
B. Gluconeogenesis
C. Salvage of purine bases
D. Degradation of glycosaminoglycans
E. Cholesterol metabolism
29.2 A 15-month-old white girl is brought to the pediatrician because of recurrent upper-respiratory tract infection. During the physical examination, the girl is noted to have a short stature, some clouding of the corneas, coarse facial features, and an enlarged tongue. She also appears to have some hearing loss and other developmental delays. The pediatrician suspects the child has a mucopolysaccharidosis. Which of the following is she least likely to have?
A. Hurler syndrome (MPS I)
B. Hunter syndrome (MPS II)
C. Morquio syndrome (MPS IV)
D. Sly syndrome (MPS VI)
E. Sanfilippo syndrome (MPS III)
29.3 A 3-year-old boy with coarse facial features, progressive loss of motor skills, hepatosplenomegaly and chronic diarrhea is suspected of having Hunter syndrome (MPS II). Which of the following monosaccharide residues would be expected to be found at the nonreducing end of glycosaminoglycans in this patient’s urine?
A. N-Acetylglucosamine
B. N-Acetylgalactosamine
C. Glucuronate
D. Iduronate
E. Iduronate 2-sulfate
ANSWERS
29.1 D. The patient is exhibiting the classic symptoms of Sanfilippo syndrome, which is a deficiency in 1 of 4 different lysosomal enzymes that breakdown glycosaminoglycans leading to the buildup of heparan sulfate and dermatan sulfate in lysosomes.
29.2 B. All the types of MPS are transmitted by autosomal recessive inheritance except Hunter syndrome (MPS II), a deficiency in iduronate sulfatase that is X-linked recessive. Because Hunter syndrome is X-linked, it is almost exclusively seen in males. Because this patient is female, she would not be expected to have an X-linked disorder.
29.3 E. Because this patient is suspected of having Hunter syndrome, a deficiency in iduronate sulfatase, iduronate 2-sulfate would be expected to be present at the nonreducing end of glycosaminoglycans found in this patient’s urine. A deficiency of iduronate sulfatase would prevent the sulfate ester bond of iduronate 2-sulfate residues from being hydrolyzed and further degradation of the glycosaminoglycan would be halted.
BIOCHEMISTRY PEARLS
![]() Often enzyme deficiencies are inherited as autosomal recessive disorders, so that both chromosomes are defective for the individual to be affected.
Often enzyme deficiencies are inherited as autosomal recessive disorders, so that both chromosomes are defective for the individual to be affected.
![]() MPS occur when there is a genetic deficiency of the enzymes involved in the lysosomal breakdown of the glycosaminoglycans.
MPS occur when there is a genetic deficiency of the enzymes involved in the lysosomal breakdown of the glycosaminoglycans.
![]() In Sanfilippo syndrome, the accumulation of heparan sulfate in lysosomes leads to severe neurologic and mental impairment that result in death usually by the end of the second decade of life.
In Sanfilippo syndrome, the accumulation of heparan sulfate in lysosomes leads to severe neurologic and mental impairment that result in death usually by the end of the second decade of life.
REFERENCES
Bittar T, Washington ER III. Mucopolysaccharidosis. http://emedicine.medscape.com/article/1258678-overview.
Baloghova J, Schwartz RA, Baranova Z. Mucopolysaccharidoses Types I-VII. http://emedicine.medscape.com/article/1115193-overview.
Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001.