Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 32
A 63-year-old woman presents to the clinic with recurrent midepigastric pain during the last 3 months. She reports some relief shortly after eating, but then the discomfort returns. She has tried various over-the-counter medications without relief. She also reports feeling tired and has had to increase the amount of ibuprofen needed for relief of her arthritis. She denies nausea, vomiting, and diarrhea. On examination she is found to have mild midepigastric tenderness and guaiac positive stool. A complete blood count revealed a microcytic anemia and normal white blood cell count, consistent with iron deficiency. The patient was referred to a gastroenterologist who performed an upper gastrointestinal (GI) endoscopy that identified gastric ulcers. He stated that he suspected that the ibuprofen, a nonsteroidal anti-inflammatory drug (NSAID) was the causative agent and suggested switching from ibuprofen to a coxib, such as celecoxib.
![]() What is the likely biochemical etiology of the disorder?
What is the likely biochemical etiology of the disorder?
![]() Why do coxibs generally have a lower incidence of upper GI problems than other NSAIDs?
Why do coxibs generally have a lower incidence of upper GI problems than other NSAIDs?
![]() What is the major difference between aspirin and other NSAIDs with regard to platelet function?
What is the major difference between aspirin and other NSAIDs with regard to platelet function?
ANSWERS TO CASE 32:
NSAID-Associated Gastritis
Summary: A 63-year-old woman with arthritis taking an NSAID with recent onset of epigastric pain relieved with food, guaiac positive stools, and iron deficiency anemia. Endoscopic examination reveals gastric ulcers.
• Biochemical etiology: Primarily, NSAID inhibition of a gastric enzyme (cyclooxygenase [COX]-1) required for synthesis of prostaglandins that have a protective effect on the gastric mucosa. A contributory factor is direct mucosal damage due to the acidic chemistry of NSAIDs.
• Decreased gastric side effects with coxibs: Traditional NSAIDs, such as ibuprofen and aspirin, inhibit both COX-1 and COX-2. The coxibs are selective inhibitors of COX-2, allowing continued production of protective prostaglandins by gastric COX-1.
• Difference between aspirin and other NSAIDs: Aspirin covalently modifies platelet COX-1, thus irreversibly blocking thromboxane formation and reducing platelet function for the lifespan of the affected platelet (platelets cannot synthesize new proteins). The inhibitory action of other NSAIDs on platelet COX-1 is not covalent and is eventually reversed when the agents’ blood levels decline.
CLINICAL CORRELATION
NSAIDs, also known as prostaglandin synthesis inhibitors or COX inhibitors, can induce upper GI irritation or ulcers. The NSAIDs include a wide variety of medications including aspirin, ibuprofen, naproxen, and indomethacin. These medications are used for pain, inflammation, dysmenorrhea, headache, arthritis, or fever. These compounds act as anti-inflammatory and antipyretic agents by inhibiting COX catalysis by prostaglandin H synthase (PGHS). PGHS has 2 isoenzymes: PGHS-1 (or COX-1) is generally a basal enzyme found in various tissues including platelets and gastric mucosa, while PGHS-2 (or COX-2) is an inducible enzyme typically expressed in response to cytokines and mitogens at sites of inflammation or cell proliferation.
Older NSAIDs, such as aspirin and ibuprofen, inhibit both COX-1 and -2, but a newer class of NSAIDs, called coxibs, is selective for COX-2 inhibition. By sparing production of cytoprotective prostaglandins by mucosal COX-1, coxibs have fewer problems with irritation and ulceration in the upper GI tract.
Thromboxane produced by platelet COX-1 in concert with a downstream enzyme is prothrombotic, so aspirin and other NSAIDs cause platelet dysfunction and increase bleeding time. Aspirin is unusual in that it causes covalent, irreversible inhibition of the COX protein, whereas other NSAIDs have noncovalent, reversible actions. Thus, because they cannot synthesize more COX protein, platelets are irreversibly affected by aspirin but only temporarily affected by other NSAIDs. Low-dose aspirin is often used in antithrombotic prophylaxis.
The prothrombotic and vasoconstrictive actions of COX-1-derived thromboxane in the vasculature are opposed by an antithrombotic and vasodilative prostaglandin, prostacyclin, that originates from COX-2 in vascular endothelial cells. Thus, the COX-2 selective coxibs tend to decrease prostacyclin levels in the vasculature without reducing the thromboxane levels. This tendency is thought to explain the small but significant increase in cardiovascular risk that led to withdrawal of 2 coxibs from the US market.
APPROACH TO:
Prostaglandin Metabolism
OBJECTIVES
1. Describe the biosynthetic and cell signaling pathways involving prostanoids.
2. Distinguish between the pathophysiologic roles of the 2 PGHS isoforms.
3. Cite the pharmacologic targets of NSAIDs and the characteristics that distinguish coxibs and aspirin from other NSAIDs.
DEFINITIONS
EICOSANOIDS: Oxygenated lipid-signaling molecules containing 20 carbons derived from polyunsaturated fatty acids released from membrane phospholipids by the action of phospholipase A2. These include the prostanoids produced by the cyclooxygenase pathway and the leukotrienes produced by the lipoxygenase pathway.
PROSTANOIDS: Oxygenated lipid signaling molecules derived from polyunsaturated fatty acids released from membrane phospholipids by the action of phospholipase A2. Prostanoids include prostaglandins, prostacyclin, and thromboxanes.
PROSTAGLANDIN: An oxygenated lipid-signaling molecule that has a 5-member ring system that is derived from arachidonic acid and other 20-carbon polyunsaturated fatty acids. The prostaglandins are hormone-like molecules that regulate cellular events near the area in which they are synthesized.
THROMBOXANE: An oxygenated lipid-signaling molecule that has a 6-member ring system derived from arachidonic acid and other 20-carbon polyunsaturated fatty acids. Thromboxanes are involved in platelet aggregation as well as vasoconstriction and bronchoconstriction and lymphocyte proliferation.
PGH SYNTHASE: Prostaglandin H synthase; the enzyme that catalyzes the formation of prostaglandin H from C20 polyunsaturated acids. PGH synthase has 2 activities; the cyclooxygenase activity introduces the 5-membered ring into the polyunsaturated fatty acid while also introducing an endoperoxide between carbons 9 and 10 and a hydroperoxide at carbon 15. The peroxidase activity reduces the hydroperoxide to a hydroxyl group using glutathione as the source of reducing equivalents. PGH synthase exists in 2 isoforms, PGHS-1 and PGHS-2. PGHS-1 is the “basal” isoform and is expressed constitutively, whereas PGHS-2 is the inducible isoform and has been implicated in cell proliferation and inflammation.
NSAID: Nonsteroidal anti-inflammatory drug; NSAIDs inhibit the COX activity of PGH synthase, thus inhibiting the production of prostaglandins and thromboxanes.
COXIBS: A class of NSAIDs that is selective for inhibition of the cyclooxygenase activity of PGHS-2, with weaker action against the PGHS-1 cyclooxygenase.
DISCUSSION
Prostanoids are oxygenated lipid-signaling molecules derived from polyunsaturated fatty acids. The major prostanoids synthesized from the prototypical polyunsaturated fatty acid, arachidonic acid, are prostaglandin (PG) D2, PGE2, PGF2a, PGH2, PGI2 (also known as prostacyclin), and thromboxane (TX) A2. The prostanoid-signaling cascade begins with an external stimulus, most often the binding of a ligand to a cell surface receptor that activates one or more phospholipases A2. The latter are enzymes that release arachidonic acid from its esterified form in membrane phospholipids such as phosphatidylethanolamine and phosphatidylinositol. Arachidonate is converted to PGH2by one of the isoforms of PGH synthase (PGHS-1 or -2), enzymes localized to the endoplasmic reticulum membrane and the nuclear envelope.
In turn, PGH2 is metabolized to the prostanoid lipid signals (PGD2, PGE2, PGF2a, PGH2, PGI2, or TXA2) by one of the secondary enzymes named for the individual prostanoid produced (Figure 32-1). The type of prostanoid produced is determined by which downstream enzyme is present; usually one downstream enzyme predominates in a given cell. For example, the prominent secondary enzyme in platelets is thromboxane synthase, whereas vascular endothelial cells feature prostacyclin (PGI) synthase. Prostanoid signaling molecules usually exit the cell that produces them to act on G-protein coupled receptors on the surface of the same cell or cells nearby (termed autocrine or paracrine actions). Some prostanoids may be further metabolized to ligands for a subset of nuclear receptors, the peroxisome proliferator-activated receptors (PPARs). The active prostanoids are rapidly converted to inactive metabolites by enzymes present in a variety of cells. As a result, prostanoid signaling molecules have very short half-lives in the circulation and are not hormones in the conventional sense.
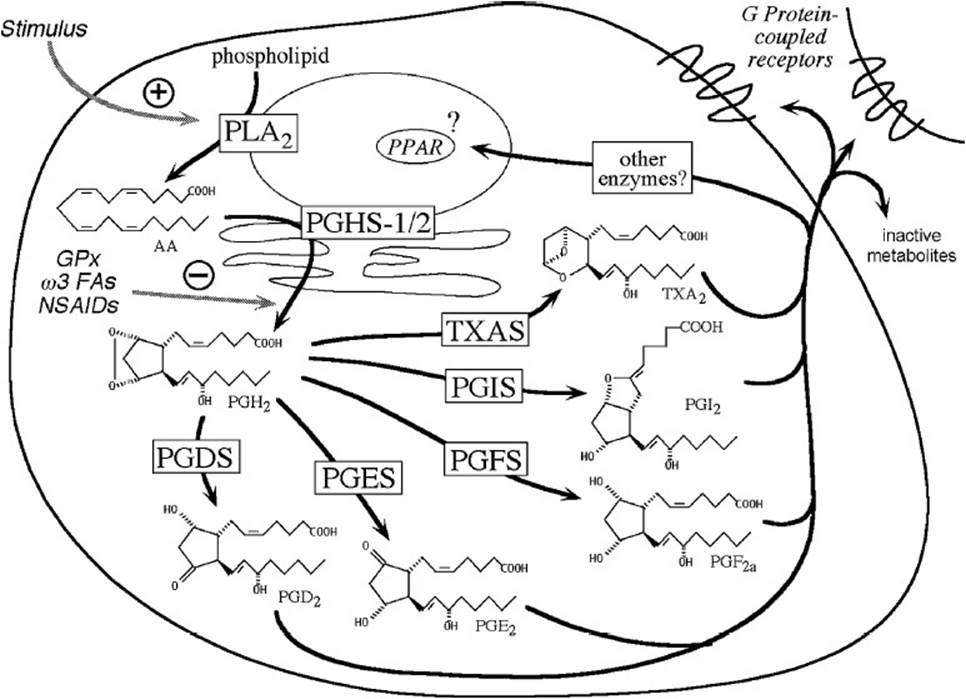
Figure 32-1. Diagram illustrating cell-signaling pathways involving prostanoids.
Conversion of arachidonate to PGH2 is a key regulatory step in prostanoid biosynthesis. Each PGHS isoform catalyzes 2 separate reactions (Figure 32-2). The first reaction (arachidonate → PGG2) involves insertion of 2 molecules of oxygen and cyclization of the fatty acid backbone. This step is catalyzed by the COX activity of PGHS-1 or -2; it is these COX activities (also called COX-1 and COX-2) that are inhibited by NSAIDs. The second step (PGG2→PGH2) involves the reduction of the hydroperoxide on C15 to an alcohol and is catalyzed by the peroxidase activity of PGHS-1 or -2.
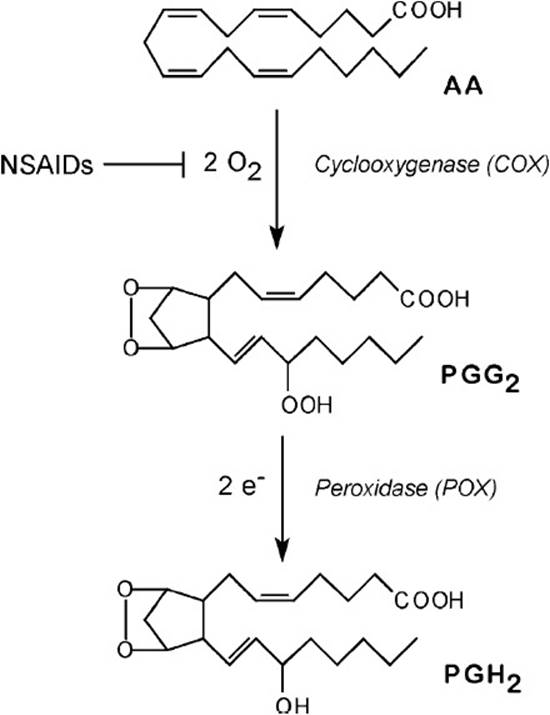
Figure 32-2. Steps in conversion of arachidonic acid to PGH2 by PGH synthase. A cosubstrate, indicated by e-, is required to furnish 2 reducing equivalents for the peroxidase reaction.
Although both PGHS isoforms have cyclooxygenase and peroxidase activities and are structurally similar proteins, they have very distinct pathophysiologic functions. Many cells, including platelets and gastric mucosal cells, have moderate levels of the “basal” isoform, PGHS-1. Functions attributed to PGHS-1 include regulating hemostasis and vascular tone, renal function, and maintaining gastric mucosal integrity. A smaller number of cells, such as macrophages, vascular endothelial cells, and fibroblasts, dramatically upregulate levels of the “inducible” isoform, PGHS-2, in response to cytokines or mitogens. PGHS-2 has been implicated in cell proliferation, inflammation, carcinogenesis, and parturition.
Many COX inhibitors have been developed and their structures are quite varied (Figure 32-3). All known inhibitors compete with fatty-acid substrate for binding to the cyclooxygenase site on the enzyme. Aspirin was one of the earliest NSAIDs discovered and is now widely used as an analgesic and antiinflammatory agent. More recently, aspirin has emerged as a very useful antithrombotic agent because of its action against platelet COX activity. Aspirin shows both archetypal modes of COX inhibition. The first mode involves rapid reversible binding of inhibitor (I) at the COX site of the enzyme (E) to form an EI complex; the second mode involves a slower conversion of EI to a higher affinity complex, EI′ (Equation 1).
![]()
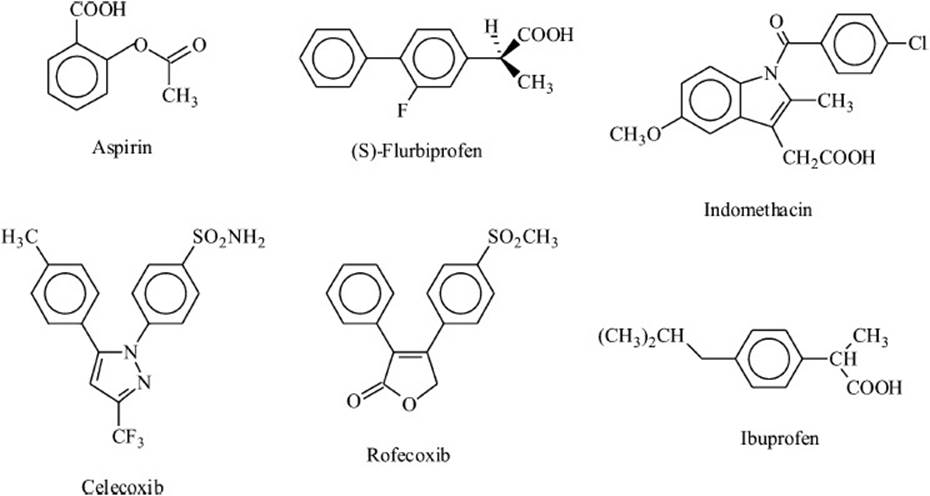
Figure 32-3. Structures of some cyclooxygenase inhibitors.
EI and EI′ cannot bind fatty acid and thus neither can catalyze the cyclooxygenase reaction. For aspirin, conversion of EI to EI′ is accompanied by covalent modification of the protein, making the transition irreversible. Formation of the EI′ complex produces a more powerful COX inhibition because the inhibitor is not readily displaced by substrate and because inhibition persists even when free inhibitor is removed. Flurbiprofen and indomethacin form EI′ complexes with both PGHS-1 and -2, although they do not covalently modify either protein. Ibuprofen forms only EI complexes with both PGHS isoforms. The coxibs (such as celecoxib and rofecoxib) derive their exquisitely selective inhibition of PGHS-2 cyclooxygenase from their ability to form noncovalent EI′ complexes with PGHS-2 and not with PGHS-1. This selectivity has made coxibs useful for anti-inflammatory and antiproliferative therapy with reduced GI adverse events, but it also makes them ineffective as antiplatelet agents and, consequently, can increase cardiovascular risks.
COMPREHENSION QUESTIONS
32.1 The coxibs, including celecoxib, are a recently developed class of NSAIDs. The coxibs show anti-inflammatory actions without affecting platelet function. These effects of the coxibs are best attributed to selective inhibition of which of the following?
A. The cytosolic isozyme of phospholipase A2 (cPLA2)
B. The COX activity of the “basal” prostaglandin H synthase isozyme (PGHS-1)
C. The COX activity of the “inducible” PGHS-2
D. The microsomal isozyme of prostaglandin E synthase (mPGES-1)
32.2 Prostaglandins comprise a family of oxygenated lipid signaling molecules derived from polyunsaturated fatty acids such as arachidonic acid. They are involved in regulating a number of cellular processes. Some of the prostaglandins act to increase vasodilation and levels of cAMP in cells, whereas others increase vasoconstriction and bronchoconstriction and smooth-muscle contraction. In the conversion of arachidonic acid to prostaglandins, the oxygenation step is accomplished by the enzyme that synthesizes which of the following compounds?
A. Prostaglandin D2
B. Prostaglandin E2
C. Prostaglandin F2α
D. Prostaglandin H2
E. Prostaglandin I2
32.3 Signaling via prostanoids begins by interaction of the prostanoid with its receptor. The receptor involved is usually located in which part of the cell?
A. Plasma membrane of a cell near the cell making the prostanoid
B. Nucleus of a cell in a different organ from the cell making the prostanoid
C. Endoplasmic reticulum of the cell making the prostanoid
D. Lysosomes of a cell circulating in the blood
E. Golgi of a cell circulating in the blood
ANSWERS
32.1 C. The coxibs were designed to inhibit the activity of the inducible form of PGH synthase so as not to inhibit the constitutive production of prostaglandins and thromboxanes. They inhibit the first step in the process, which is catalyzed by the COX activity of PGH synthase-2.
32.2 D. The first step in the synthesis of prostaglandins and thromboxanes is the reaction catalyzed by the cyclooxygenase activity of PGHS. This reaction causes the cyclization of the fatty acid while at the same time introducing an unstable endoperoxide between carbons 9 and 10 and a hydroperoxide at carbon 15 to produce PGG2, which is rapidly reduced to PGH2 by the peroxidase activity of PGHS.
32.3 A. The prostanoids have a wide variety of physiologic effects, but they regulate these processes locally by binding to a receptor on the plasma membrane of a cell close to where the prostanoid was synthesized. Binding of the prostanoid to the receptor usually activates a GTP-binding protein, which acts to activate (or inhibit) adenylate cyclase or the phosphatidylinositol cascade.
BIOCHEMISTRY PEARLS
![]() Conversion of arachidonate to PGH2 via COX catalysis is a key regulatory step in prostanoid biosynthesis.
Conversion of arachidonate to PGH2 via COX catalysis is a key regulatory step in prostanoid biosynthesis.
![]() Aspirin has emerged as a very useful antithrombotic agent because of its action against platelet COX activity.
Aspirin has emerged as a very useful antithrombotic agent because of its action against platelet COX activity.
![]() The coxibs (such as celecoxib) derive their exquisitely selective inhibition of COX-2 cyclooxygenase from their ability to form noncovalent EI′ complexes with COX-2 and not with COX-1.
The coxibs (such as celecoxib) derive their exquisitely selective inhibition of COX-2 cyclooxygenase from their ability to form noncovalent EI′ complexes with COX-2 and not with COX-1.
![]() The selectivity of coxibs for COX-2 over COX-1 gives them anti-inflammatory and antiproliferative actions with reduced GI adverse events, but it also makes them ineffective as antiplatelet agents, thus increasing the risks of myocardial infarction and stroke.
The selectivity of coxibs for COX-2 over COX-1 gives them anti-inflammatory and antiproliferative actions with reduced GI adverse events, but it also makes them ineffective as antiplatelet agents, thus increasing the risks of myocardial infarction and stroke.
REFERENCES
Blobaum AL, Marnett LJ. Structural and functional basis of cyclooxygenase inhibition. J Med Chem. 2007;50(7):1425-1441.
Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548): 1871-1875.
Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116(1):4-15.
Parfitt JR, Driman DK. Pathological effects of drugs on the gastrointestinal tract: a review. Hum Pathol. 2007;38(4):527-536.