Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 38
A 1-year-old girl is brought to her pediatrician’s office with concerns about her development. She had an uncomplicated birth outside the United States at term. The mother reports that the baby is not achieving the normal milestones for a baby of her age. She also reports an unusual odor to her urine and some areas of hypopigmentation on her skin and hair. On examination, the girl is noted to have some muscle hypotonia and microcephaly. The urine collected is found to have a “mousy” odor.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the biochemical basis of the hypopigmented skin and hair?
What is the biochemical basis of the hypopigmented skin and hair?
ANSWERS TO CASE 38:
Phenylketonuria
Summary: A 1-year-old girl born outside the United States with developmental delays, hypotonia, hypopigmentation, and foul smelling urine.
• Likely Diagnosis: Phenylketonuria (PKU)
• Biochemical basis of hypopigmentation: Phenylalanine is competitive inhibitor of tyrosinase (key enzyme in melanin synthesis)
CLINICAL CORRELATION
Elevated phenylalanine can be caused by a variety of different enzyme deficiencies resulting in impaired conversion of phenylalanine to tyrosine. The most common deficiency is in phenylalanine hydroxylase (autosomal recessive) resulting in the classic picture of PKU. Two other enzyme deficiencies leading to PKU include dihydropteridine reductase and 6-pyruvoyl-tetrahydropterin synthase, an enzyme in the biosynthetic pathway of tetrahydrobiopterin. With PKU, the baby appears normal at birth but then fails to reach normal developmental milestones. If unrecognized, the child will develop profound mental retardation and impairment of cerebral function. A mousy odor of the skin, hair, and urine can often be detected clinically. Areas of hypopigmentation develop secondary to the disruption of melanin synthesis. In the United States, all children are screened for PKU in hopes to prevent the serious life-long complications. Treatment consists of dietary modifications with limitation of phenylalanine intake and supplementation of tyrosine. The diagnosis of PKU and initiation of diet modification must be implemented prior to 3 weeks of age to prevent mental retardation and the other classic signs of PKU.
APPROACH TO:
Phenylketonuria
OBJECTIVES
1. Describe the biochemical conversion of phenylalanine to tyrosine.
2. Describe the biochemical events that occur when conversion of phenylalanine to tyrosine is inhibited.
DEFINITIONS
HYPOPIGMENTATION: Lack of color in skin or hair due to the absence or low quantity of the skin and hair pigment melanin, a product of tyrosine (and phenylalanine) metabolism.
TETRAHYDROBIOPTERIN: A 4-electron-reduced form of the reducing agent biopterin required to supply electrons to phenylalanine hydroxylase for conversion of phenylalanine to its hydroxylated product, the amino acid tyrosine.
PHENYLKETONURIA: The presence of elevated amounts of phenylketones—primarily phenylpyruvate—in urine; a primary indication of disturbance of phenylalanine metabolism resulting from elevated transamination of phenylalanine due to reduction in phenylalanine hydroxylation to tyrosine.
DISCUSSION
Phenylketonuria is a disease readily diagnosable in childhood, and it is important for an optimal clinical outcome for it to be diagnosed as early as possible. Laboratory tests can be performed in the neonatal period and now genetic testing can identify the trait before birth.
Phenylketonuria obtains from an elevated level of phenylpyruvate in the urine of the patient. As shown in Figure 38-1, phenylpyruvate is the cognate α-ketoacid of the amino acid phenylalanine. It is formed by transaminating phenylalanine and α-ketoglutarate to yield glutamate and phenylpyruvate. This reaction is freely reversible; therefore, it is driven by elevated concentrations of reactants or products. For large scale conversion of phenylalanine to phenylpyruvate an increase in the concentration of phenylalanine must occur to drive the transamination reaction toward the formation of phenylpyruvate. Such an elevated phenylalanine level will be reflected in the blood as hyperphenylalaninemia, which is defined as a plasma phenylalanine level above 120 μmol/L. The most likely cause is a disturbance in the phenylalanine hydroxylase reaction. Disease states resulting from disturbances of the phenylalanine hydroxylase reaction can be found (1) at the level of the enzymes in the reaction reflected in absent or altered proteins, (2) at the metabolic level reflected in cognate effects on other metabolic processes, and (3) at the cognitive level reflected in changes in brain function and mental retardation.
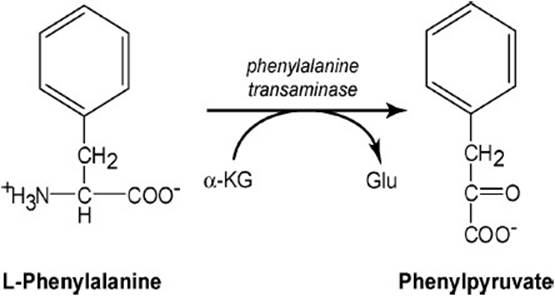
Figure 38-1. Transamination of phenylalanine to yield phenylpyruvate, a phenylketone.
Phenylalanine Hydroxylation Reaction and Its Components. As shown in Figure 38-2, the phenylalanine hydroxylation reaction is the pathway whereby dietary phenylalanine can be converted into tyrosine relieving some of the dietary requirement for tyrosine. It is catalyzed by phenylalanine hydroxylase, a monooxygenase requiring molecular oxygen and a specific 2-electron donor tetrahydrobiopterin. One atom of molecular oxygen appears in the product tyrosine as a hydroxyl group in the para position, the remaining oxygen atom appearing in the product water.
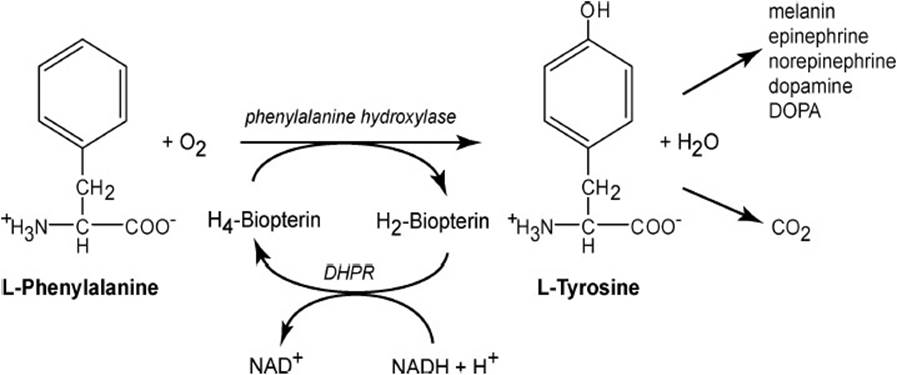
Figure 38-2. Normal conversion of phenylalanine into the amino acid tyrosine catalyzed by phenylalanine hydroxylase.
The provision of reducing equivalents to phenylalanine hydroxylase is dependent on reduction of dihydrobiopterin by NADH catalyzed by the enzyme dihydropteridine reductase (Figure 38-2). This reduction is dependent on the availability of biopterin and therefore on the biopterin synthetic pathway. Thus, any genetic or protein folding defect in either dihydropteridine reductase or the biopterin biosynthetic enzymes would compromise the efficacy of phenylalanine hydroxylation to tyrosine resulting in hyperphenylalaninemia and also phenylketonuria resulting from increase transamination of phenylalanine to phenylpyruvate.
The principal enzyme in this pathway is phenylalanine hydroxylase. The gene for phenylalanine hydroxylase is located on chromosome 12 at band region q 23.2 and comprises 100 kb of genomic DNA. Several hundred alleles causing disease states have been recognized for this gene, more than 60% of which are classified as missense alleles. European and Chinese populations show 1 order of magnitude higher incidence than persons of African descent. This population specific expression of disease causing alleles may explain the wide range of incidence reported for this disease entity (5-350 cases/million live births).
Fates of Tyrosine
Tyrosine can be degraded by oxidative processes to acetoacetate and fumarate which enter the energy generating pathways of the citric acid cycle to produce CO2 and ATP as indicated in Figure 38-2. Tyrosine can be further metabolized to produce various neurotransmitters such as dopamine, epinephrine, and norepinephrine. Hydroxylation of tyrosine by tyrosine hydroxylase produces dihydroxyphenylalanine (DOPA). This enzyme, like phenylalanine hydroxylase, requires molecular oxygen and tetrahydrobiopterin. As is the case for phenylalanine hydroxylase, the tyrosine hydroxylase reaction is sensitive to perturbations in dihydropteridine reductase or the biopterin synthesis pathway, any one of which could lead to interruption of tyrosine hydroxylation, an increase in tyrosine levels, and an increase in transamination of tyrosine to form its cognate α-keto acid, para-hydroxyphenylpyruvate, which also would appear in urine as a contributor to phenylketonuria.
Tyrosine is also the precursor to melanin formation in melanocytes, the first step of which is catalyzed by tyrosinase as shown in Figure 38-3. This reaction is a 2-step reaction in which DOPA is an intermediate in the formation of dopaquinone. Ring closure of the alanine portion of dopaquinone forms a pyrrole ring and subsequent reactions give rise to the melanins, the primary dark pigment associated with skin color being eumelanin. The absence of tyrosinase gives rise to classic albinism. Phenylalanine is a competitive inhibitor with tyrosine for tyrosinase. Thus in a situation wherein phenylalanine hydroxylase activity is deficient, not only does the α-keto cognate transamination product phenylpyruvate increase but so does the level of phenylalanine. Thus, excess phenylalanine inhibits tyrosinase and melanin formation resulting in hypopigmentation of skin and hair in affected persons.
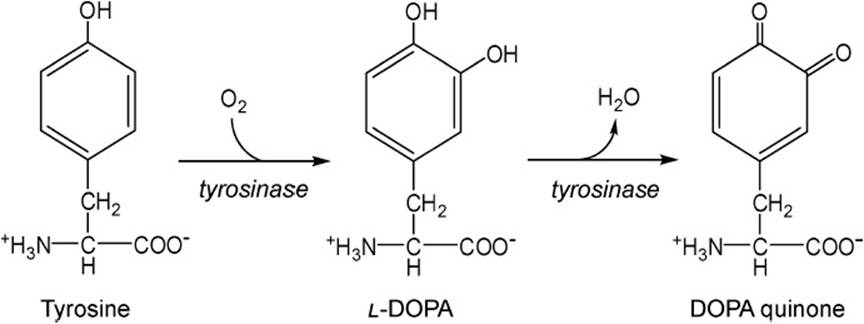
Figure 38-3. Conversion of tyrosine to dopaquinone by the enzyme tyrosinase, a Cu+2-dependent enzyme in melanocytes.
COMPREHENSION QUESTIONS
38.1 A 3-month-old boy presents with elevated levels of phenylalanine, para-hydroxyphenylpyruvate, and phenylpyruvate in the serum. His skin color is pale. Your differential diagnosis is PKU. Which of the following would be consistent in such a case?
A. Elevated levels of homogentisic acid in the serum
B. Deficiency in vitamin B12 (cobalamin)
C. Elevated levels of pyridoxal phosphate in the serum
D. Urine in the boy’s diaper smells like fresh maple syrup
E. Phenylalanine hydroxylase activity is only 2% of normal
38.2 A 1-year-old girl presents at your clinic the day after you saw the 3-month-old boy. The symptoms are the same so you order a test on phenylalanine hydroxylase to confirm your diagnosis of phenylketonuria. To your surprise, the phenylalanine hydroxylase activity is well within the normal range. Which of the following might you check next to support your diagnosis?
A. Tyrosine: α-ketoglutarate transaminase
B. Tyrosinase
C. Homogentisic acid oxidase
D. Dihydropteridine reductase
E. Dopamine hydroxylase
38.3 Skin color is the aggregate result of the expression of a number of genes modified by ethnic origin and genetic inheritance. Hypopigmentation may be caused by which of the following?
A. Excess formation of melanin
B. Excess phenylalanine in the serum and tissues
C. Hyposecretion of melatonin
D. Excessive stimulation of tyrosinase
E. Low levels of para-hydroxyphenylpyruvate
ANSWERS
38.1 E. The correct response is very low levels of phenylalanine hydroxylase, a key enzyme in the metabolic sequelae of phenylketonuria—ie, elevated phenylalanine, phenylpyruvate, and para-hydroxyphenylpyruvate in blood. Homogentisic acid is an intermediate in the breakdown of tyrosine to fumarate and acetoacetate. Vitamin B12 is required in the metabolism of branched-chain amino acids not phenylalanine. The α-keto acids of the branched chain amino acids produce the maple-syrup odor.
38.2 D. The correct response is dihydropteridine reductase. This enzyme reduces dihydrobiopterin to tetrahydrobiopterin the obligate electron donor for phenylalanine hydroxylase. Tyrosinase is the first enzyme on the pathway to melanin. Dopamine hydroxylase and tyrosine transaminase are enzymes on other tyrosine metabolic tracts. Homogentisic acid oxidase is an enzyme on the pathway of tyrosine to fumarate and acetoacetate.
38.3 B. Excess phenylalanine inhibits tyrosinase the first step toward melanin production, thus resulting in hypopigmentation. Excess melanin leads to hyperpigmentation. Melatonin is a hormone involved in the sleep cycle. Excessive stimulation of tyrosinase would lead to more melanin and therefore hyperpigmentation. Para-hydroxyphenylpyruvate means less transamination and perhaps more tyrosine converted to melanin and hyperpigmentation.
BIOCHEMISTRY PEARLS
![]() PKU is an autosomal recessive disorder of amino acid metabolism affecting approximately 1 out of 10,000 of infants in North America.
PKU is an autosomal recessive disorder of amino acid metabolism affecting approximately 1 out of 10,000 of infants in North America.
![]() It is most often due to deficiency of the enzyme phenylalanine hydroxylase, which causes the accumulation of harmful metabolites, including phenylketones.
It is most often due to deficiency of the enzyme phenylalanine hydroxylase, which causes the accumulation of harmful metabolites, including phenylketones.
![]() The gene for phenylalanine hydroxylase is located on chromosome 12 at band region q 23.2.
The gene for phenylalanine hydroxylase is located on chromosome 12 at band region q 23.2.
![]() If untreated, PKU may lead to mental retardation, seizures, psychoses, eczema, and a distinctive “mousy” odor.
If untreated, PKU may lead to mental retardation, seizures, psychoses, eczema, and a distinctive “mousy” odor.
![]() PKU is a disease readily diagnosable in childhood and it is important for an optimal clinical outcome for it to be diagnosed as early as possible.
PKU is a disease readily diagnosable in childhood and it is important for an optimal clinical outcome for it to be diagnosed as early as possible.
REFERENCES
Devlin TM, ed. Textbook of Biochemistry with Clinical Correlations. 7th ed. New York: Wiley-Liss; 2010.
Scriver CR, Beaudet AL, Sly WS, et al. The Metabolic and Molecular Basis of Inherited Disease. 8th ed, New York: McGraw Hill; 2001:1667-1776.