Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 44
A 21-year-old healthy male college student went to celebrate his birthday with some friends at a bar. His friends convinced him to have his first beer since he just turned 21 years of age. After consuming the beer, he began to experience intense, worsening abdominal pain that was nonspecific in location and described as cramping. Nausea and vomiting then ensued and he was taken to the emergency department. Upon arrival to the emergency department, he was found to be very anxious with hallucinations. He was noted to be hypertensive, tachycardic, and diaphoretic. Peripheral neuropathy was also noticed on examination. Initial laboratory test revealed a normal complete blood count, drug screen, and EtOH level. Serum and urine aminolevulinic acid (ALA) and porphobilinogen (PBG) levels were both found to be elevated.
![]() What is the likely diagnosis?
What is the likely diagnosis?
![]() What is the underlying biochemical problem?
What is the underlying biochemical problem?
ANSWERS TO CASE 44:
Porphyria (Acute Intermittent Porphyria)
Summary: A 21-year-old healthy male patient with sudden onset abdominal pain, nausea and vomiting, hypertension, tachycardia, and peripheral neuropathy after consumption of his first alcoholic beverage. Further testing revealed elevated levels of both serum and urine ALA and PBG.
• Diagnosis: Porphyria (likely acute intermittent porphyria, ie, variegate)
• Biochemical problem: Enzymatic deficiency in heme biosynthetic pathway
CLINICAL CORRELATION
Porphyrias are inherited disorders in the heme biosynthetic pathway. Porphyrias are classified as either hepatic or erythropoietic depending on primary site of accumulation. Inheritance is usually autosomal dominant. Patients often are asymptomatic unless exposed to factors that increase production of porphyrias (drugs, alcohol, sunlight). Erythropoietic etiologies primarily present with photosensitivity. Hepatic porphyrias can present with primarily neurovisceral symptoms, which may include abdominal pain, nausea and vomiting, tachycardia and hypertension, peripheral neuropathy, and mental symptoms (hallucinations, anxiety, seizures). Diagnosis is confirmed with elevated levels of ALA and PBG in the urine and serum. Specific tests can be performed to detect which enzyme is deficient (ie, variegate porphyria is caused by deficiency in the protoporphyrinogen oxidase [PPO] enzyme). Treatment is supportive with avoidance of triggers in the future.
OBJECTIVES
1. Describe the biosynthesis of heme.
2. Explain why certain triggers (such as EtOH) cause increases in ALA and PBG levels.
3. Explain why treatment with intravenous heme or hematin is effective.
DEFINITIONS
AMINOLEVULINIC ACID SYNTHASE (ALAS): Mitochondrial matrix enzyme that catalyzes the rate-limiting synthesis of ALA via condensation of succinyl-CoA and glycine.
ALA DEHYDRATASE (ALAD): Cytosolic enzyme that catalyzes the asymmetric condensation of 2 molecules of ALA to form PBG.
AUTONOMIC NEUROPATHY: Autonomic nervous disruption or deregulation affecting the cardiovascular, urogenital, gastrointestinal systems. Symptoms include abdominal pain, nausea and vomiting, tachycardia, and hypertension (also known as visceral neuropathy).
COPROPORPHYRINOGEN OXIDASE (CPO): Mitochondrial enzyme that catalyzes specifically the conversion of coproporphyrinogen III to protoporphyrinogen IX.
FERROCHELATASE: Assists in the insertion of the ferrous iron into the protoporphyrin IX; the final step in heme synthesis.
HEMATIN: Compound similar in structure to heme, except that the iron is in the ferric state (tetracoordinated with the pyrrole nitrogens and a hydroxyl group); can restore negative regulation of ALAS.
HEME: An essential metalorganic cofactor, consisting of 1 ferrous iron atom coordinated within a tetrapyrrole ring, protoporphyrin IX.
PORPHOBILINOGEN DEAMINASE (PBGD): Cytosolic enzyme that processes 6 PBG molecules through a hexapyrrole adduct to catalyze the formation of a free linear tetrapyrrole, hydroxymethylbilane (also known as uroporphyrinogen I synthase).
PORPHYRIA: Any of a number of diseases characterized by derangement in porphyrin metabolism; many are caused by genetic defects in the biosynthetic enzymes.
PROTOPORPHYRINOGEN OXIDASE (PPO): Catalyzes the oxidation of protoporphyrinogen IX to produce protoporphyrin IX.
PYRIDOXAL PHOSPHATE: Coenzyme active derivative of vitamin B6.
UROPORPHYRINOGEN (URO) III COSYNTHASE: Cytosolic enzyme that catalyzes formation of the URO III isomer from hydroxymethylbilane.
URO DECARBOXYLASE (UROD): Cytosolic enzyme that catalyzes the removal of the carboxyl groups from the side chains of both URO isoforms converting them to their respective coproporphyrinogens (ie, COPRO I and COPRO III).
DISCUSSION
The cofactor heme is required for numerous processes throughout the body. Most importantly, the heme iron facilitates systemic oxygen transfer via hemoglobin, participates in mitochondrial electron transport, and mediates oxidative drug metabolism in the liver through various cytochromes P450. All cellular tissues are capable of synthesizing heme, but expression of the pathway enzymes and levels of intermediates are greatest in erythropoietic and hepatic tissues due to high demand for heme incorporation into hemoglobin and cytochromes, respectively. Heme is a metalorganic compound, consisting of 1 ferrous iron atom coordinated within a tetrapyrrole ring, protoporphyrin IX. Protoporphyrin IX is derived from 8 molecules each of succinyl-CoA and glycine. Heme is a relatively planar molecule and highly stabilized by strong resonance throughout the tetrapyrrole ring system. The structure of heme is shown in Figure 44-1.
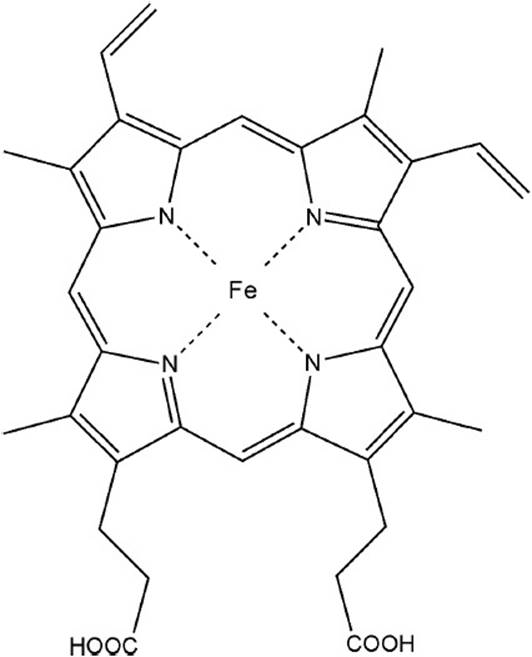
Figure 44-1. Structure of heme.
The biosynthetic pathway includes eight steps, shown in Figure 44-2. The first step is the rate-limiting condensation reaction between succinyl-CoA and glycine to form δ-ALA. This reaction is catalyzed by a mitochondrial matrix enzyme, ALA synthase (ALAS), and requires the cofactor pyridoxal phosphate. The ALAS protein is produced in the cytosol but remains unfolded or inactive until it is directed to the mitochondrial matrix where its N-terminal signaling sequence is cleaved. In addition, this enzyme catalyzes the primary regulatory step in heme biosynthesis and is negatively regulated by any accumulation of free heme in the mitochondrial matrix. In the next step of the pathway, ALA dehydratase catalyzes the asymmetric condensation of 2 molecules of ALA to form PBG. Through the addition of water and the removal of 4 amino groups, PBG deaminase produces the linear tetrapyrrole intermediate, hydroxymethylbilane (HMB) from 4 molecules of PBG. At this point, HMB can close either in an enzyme-independent manner to form uroporphyrinogen (URO) I, or in an enzyme-dependent manner through uroporphyrinogen III cosynthase to form uroporphyrinogen III, the intermediate that will ultimately lead to heme formation. URO I and URO III differ in the order of the carboxymethyl and carboxyethyl substituents around the tetrapyrrole ring. In the last cytosolic steps, URO decarboxylase (UROD) recognizes either isomer URO I or III and removes specific carboxyl groups leaving coproporphyrinogen I or III, respectively. Coproporphyrinogen oxidase (CPO) acts exclusively on the type III isomer of coproporphyrinogen; after its substrate enters the mitochondrion CPO catalyzes the conversion of COPRO III to protoporphyrinogen III. This intermediate is oxidized by PPO to form protoporphyrin IX. In the final step of heme synthesis, the enzyme ferrochelatase inserts a ferrous iron into water-insoluble protoporphyrin IX. The excess protoporphyrin IX not converted to heme is removed via biliary excretion into the intestine.
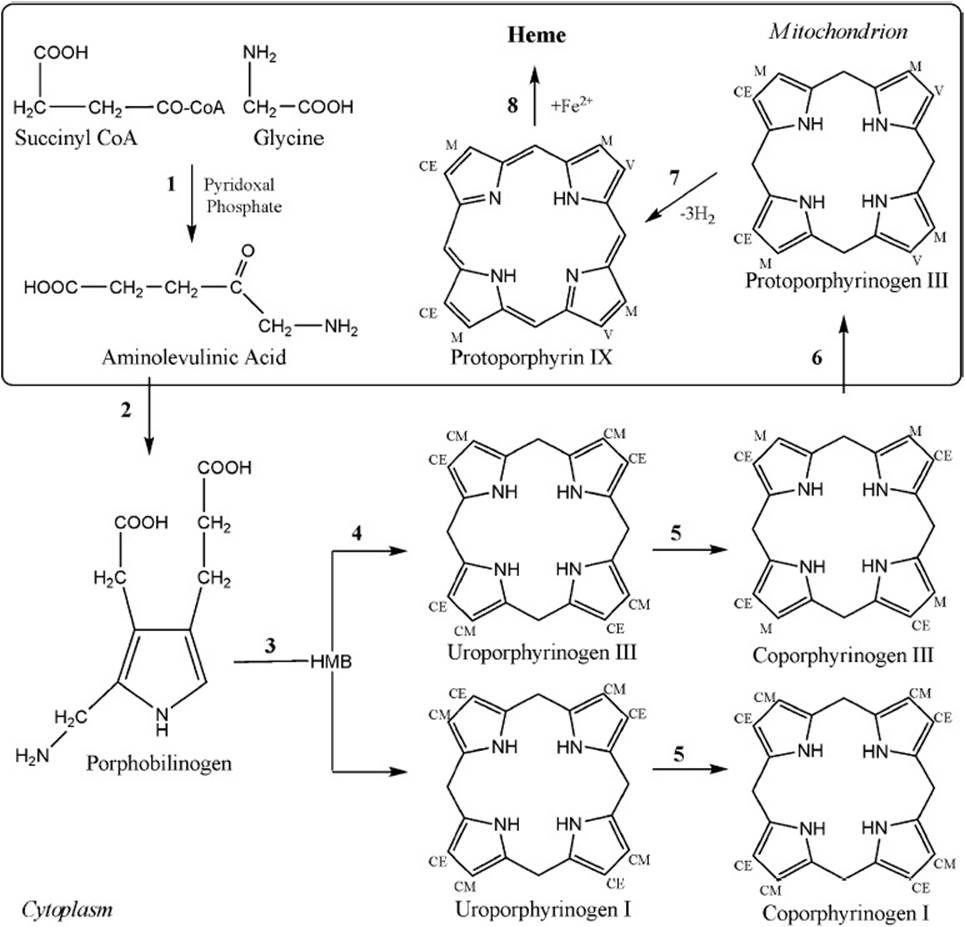
Figure 44-2. Heme biosynthetic pathway involving (1) ALAS, (2) ALAD, (3) PBGD, (4) UROS, (5) UROD, (6) CPO, (7) PPO, and (8) ferrochelatase. CE = carboxyethyl, CM = carboxymethyl, HMB = hydroxymethylbilane, M = methyl, V = vinyl.
The porphyrias are a group of inherited disorders resulting from specific enzymatic defects of the heme biosynthetic pathway. Figure 44-3 shows the heme biosynthetic pathway and characteristics of porphyrias associated with each step. A derangement in any of the involved enzymes can result in an overproduction or backlog of heme precursors prior to the deficient enzymatic step. All of the intermediate compounds in this pathway are potentially toxic. The disorders are divided into 2 broad groups, erythropoietic and hepatic porphyrias, based on the primary source of precursor accumulation. Furthermore, the porphyrias are also categorized based on the appearance of distinctive acute or chronic symptoms. Pressure to overcome the metabolic “roadblock” contributes to the rapid accumulation of precursors in acute attacks. Early pathway intermediates—that is, ALA and PBG are associated with acute neurovisceral symptoms—whereas later intermediates (which undergo porphyrinogen-porphyrin conversions upon exposure to light) cause skin photosensitivity through free-radial damage. The porphyrins can be detected and identified by spectrofluorometry based on characteristic excitation and emission wavelengths for each porphyrin. For example, serum uroporphyrin levels can be determined by detection at 615 nm after excitation at 395 to 398 nm, while protoporphyrin is detected at 626 nm. The excretion pathway of the porphyrins is determined by their water solubility. The first possible porphyrin byproduct of the heme biosynthetic pathway, uroporphyrin is by far the most water-soluble, while protoporphyrin the least soluble. Accordingly, uroporphyrin is excreted predominantly in urine, coproporphyrin in urine and in bile, and protoporphyrin exclusively in bile.
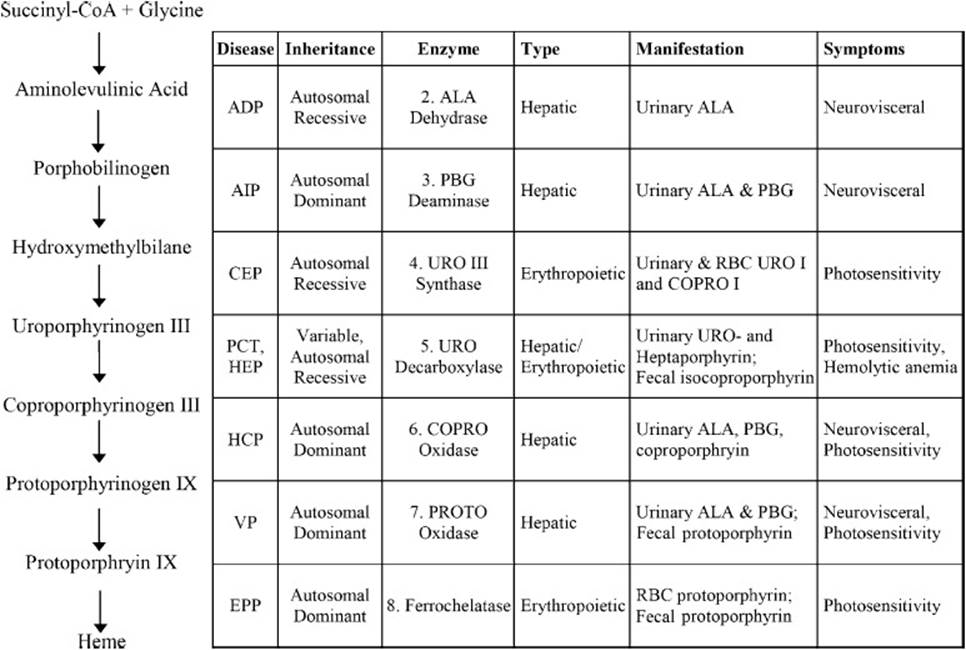
Figure 44-3. Heme biosynthetic pathway and characteristics associated with specific enzyme-deficiency porphyrias. ADP = ALA dehydratase deficiency porphyria, AIP = acute intermittent porphyria, CEP = congenital erythropoietic porphyria, EPP = erythropoietic protoporphyria, HCP = hereditary coproporphyria, HEP = hepatoerythropoietic porphyria, PCT = porphyria cutanea tarda, VP = variegate porphyria.
In most cases, these disorders are inherited in an autosomal dominant manner where the individual carries one normal allele and one loss-of-function allele. Under normal circumstances, the wild type allele allows for expression of enough functional enzymes to meet the individual’s requirement for heme (symptom-free carriers). However, certain environmental triggers, for example, drugs, alcohol, steroids, fasting, trauma, and/or high stress can increase this demand beyond a level that can be compensated by the single functional allele. Upon exposure to the trigger(s), patients with acute hepatic porphyrias can shift from a compensated phase to a decompensated latent phase (increased precursor production and excretion without symptoms) or to a clinically manifest stage (marked by abdominal, peripheral neurologic, cardiovascular, and psychiatric symptoms).
Alcohol ingestion induces increased activity of ALAS significantly in the liver and moderately in peripheral tissues. This effect is mediated by alleviating the negative regulation of ALAS by free heme. Mitochondrial concentrations of free heme are diminished by increased heme utilization and/or reduced activity of downstream pathway enzymes. In the liver, the demand for heme is exacerbated by requirement for heme incorporation into alcohol-eliminating cytochromes P450. Upon increased activity of ALAS, a genetic defect in either ALAD or PBGD would cause the rapid accumulation of ALA or both ALA and PBG, resulting in ALAD deficiency porphyria(very rare) or acute intermittent porphyria, respectively. During an attack, excess ALA and PBG produced in the liver are secreted into systemic circulation and are later excreted in the urine. In circulation, these neurotoxic compounds have the greatest effect on the autonomic and peripheral nervous systems resulting in the peripheral neuropathy and neurovisceral symptoms, that is, abdominal pain, nausea and vomiting, tachycardia, and hypertension.
Alternatively, patients with cutaneous porphyrias, for example, porphyria cutanea tarda or hereditary coproporphyria, chronic symptoms develop as a result of sun exposure (400 nm radiation). The excess porphyrins accumulated in the skin can absorb light energy and transfer it to damaging chemical reactions, such as peroxidation of membrane lipids. This manifests as thickening of the dermal vessel walls leading to damage of the epidermal basement membrane, excessive fragility, blistering, and scarring. In variegate porphyria, patients can present with neurovisceral symptoms (acute symptoms are identical but often milder than those of acute intermittent porphyria [AIP]) and/or cutaneous symptoms.
Porphyrias are diagnosed after demonstration and biochemical identification of the increased porphyrin precursor(s). Acute porphyrias are often misdiagnosed, and attacks can be fatal. The intravenous administration of heme or hematin can aid in reestablishing the negative regulation of ALAS during acute attacks. With ALAS activity attenuated, the precursor backlog at the enzymatic deficient step can begin to return to manageable levels, and the patient’s condition can shift back to the compensated, symptom-free phase. After diagnosis of acute porphyria is confirmed, strict avoidance of environmental triggers can prevent acute attacks. Most patients with acute porphyrias can lead a normal life; however, complications such as hypertension, chronic renal failure, and hepatoma can become problematic.
COMPREHENSION QUESTIONS
For questions 44-1 and 44-2 refer to the following case:
A 30-year-old white woman enters the emergency department complaining of nausea, severe abdominal pain, and prolonged constipation. She appeared distraught and was sweating. She described beginning an extremely low calorie diet within the past 2 months in an attempt to lose weight. Physical examination determined rapid heart rate, moderate hypertension, and weakness in the extremities. In addition, mild dermatitis/blistering is noted on her hands as well as scarring on her face.
44.1 You suspect porphyria. Which biochemical laboratory test(s) would be sufficient to determine the type of porphyria?
A. Urinary ALA and PBG
B. Urinary and fecal PBG and porphyrins
C. Porphyrin spectrofluorometry (plasma scan)
D. None of the above is sufficient alone
44.2 Laboratory tests revealed elevated levels of urinary PBG and coproporphyrin and plasma fluorescence emission at 626 nm. Which type of porphyria does the patient have and what is the most likely biochemical explanation?
A. Acute intermittent porphyria; PBGD heterozygous enzyme deficiency causing PBG backlog
B. Porphyria cutanea tarda; UROD homozygous enzyme deficiency causing uroporphyrin backlog
C. Variegate porphyria; PPO heterozygous deficiency causing protoporphyrin IX backlog
D. Variegate porphyria; PPO homozygous deficiency causing protoporphyrin IX backlog
44.3 The second enzyme in the heme pathway, ALAD, is very sensitive to inhibition by heavy metals (eg, lead). Which of the following test results would distinguish lead-based poisoning from acute intermittent porphyria?
A. Decreased serum and urinary ALA and PBG levels
B. Increased serum and urinary ALA and PBG levels
C. Increased serum and urinary ALA levels, decreased serum and urinary PBG levels
D. Decreased serum and urinary ALA levels, increased serum and urinary PBG levels
ANSWERS
44.1 C. Given the patient’s symptoms (neurologic and cutaneous), the main porphyrias to consider are hereditary coproporphyria and variegate porphyria. Both hereditary coproporphyria and variegate porphyria show elevated ALA and PBG levels during acute attacks; therefore, urine and serum tests for these molecules would not distinguish between these two possiblities. Similarly, both disorders show coproporphyrin in stool. In this case, spectrofluorometric assay alone to distinguish between coproporphyrin and protoporphyrin in plasma would be sufficient.
44.2 C. Variegate porphyria patients present with the same, but generally milder, symptoms as AIP. The emission wavelength at 626 nm is characteristic of protoporphyrin IX; the compound is responsible for the skin sensitivity not observed in AIP. Porphyria cutanea tarda can also be ruled out because the characteristic fluorescence emission for uroporphyrin is 615 nm. Homozygous deletion of PPO is unlikely because the patient’s symptoms were triggered by stress in the form of caloric restriction. Patients with homozygous deficiency in the heme synthesis enzymes show early onset and more severe symptoms.
44.3 C. An increase in urine/serum ALA levels without concomitant increase in PBG indicates disrupted activity of ALAD. ALA alone can cause the same neurovisceral symptoms as ALA and PBG together cause in AIP. ALAD-deficiency porphyria is an extremely rare autosomal recessive disorder.
BIOCHEMISTRY PEARLS
![]() The porphyrias are a group of inherited disorders resulting from specific enzymatic defects of the heme biosynthetic pathway.
The porphyrias are a group of inherited disorders resulting from specific enzymatic defects of the heme biosynthetic pathway.
![]() The major types of porphyria are each caused by mutations in one of the genes required for heme production.
The major types of porphyria are each caused by mutations in one of the genes required for heme production.
![]() The first step in heme formation is the rate-limiting condensation reaction between succinyl-CoA and glycine to form δ-ALA. This reaction is catalyzed by the mitochondrial matrix enzyme ALAS.
The first step in heme formation is the rate-limiting condensation reaction between succinyl-CoA and glycine to form δ-ALA. This reaction is catalyzed by the mitochondrial matrix enzyme ALAS.
![]() Forms of porphyria include ALAS-deficiency porphyria, acute intermittent porphyria, congenital erythropoietic porphyria, erythropoietic protoporphyria, hepatoerythropoietic porphyria, hereditary coproporphyria, porphyria cutanea tarda, and variegate porphyria.
Forms of porphyria include ALAS-deficiency porphyria, acute intermittent porphyria, congenital erythropoietic porphyria, erythropoietic protoporphyria, hepatoerythropoietic porphyria, hereditary coproporphyria, porphyria cutanea tarda, and variegate porphyria.
REFERENCES
Awad W. Iron and heme metabolism. In: Devlin TM, ed. Textbook of Biochemistry with Clinical Correlations. 7th ed. New York: Wiley-Liss; 2010.
Doss MO, Kuhnel A, Gross U. Alcohol and porphyrin metabolism. Alcohol. 2000;35(2):109-125.
Kauppinen R. Porphyrias. Lancet. 2005;365(9455):241-252.
Sassa S. Modern diagnosis and management of the porphyrias. Br J Haematol. 2006;135(3):281-292.