Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 1
A 15-year-old African-American girl presents to the emergency department with complaints of bilateral thigh and hip pain. The pain has been present for 1 day and is steadily increasing in severity. Acetaminophen and ibuprofen have not relieved her symptoms. She denies any recent trauma or excessive exercise. She does report feeling fatigued and has been having burning with urination along with urinating frequently. She reports having similar pain episodes in the past, sometimes requiring hospitalization. On examination, she is afebrile (without fever) and in no acute distress. No one in her family has similar episodes. Her conjunctiva and mucosal membranes are slightly pale in coloration. She has nonspecific bilateral anterior thigh pain with no abnormalities appreciated. The remainder of her examination is normal. Her white blood cell count is elevated at 17,000/mm3, and her hemoglobin level is decreased at 7.1 g/dL. Urinalysis demonstrated an abnormal number of numerous bacteria.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the molecular genetics behind this disorder?
What is the molecular genetics behind this disorder?
![]() What is the pathophysiologic mechanism of her symptoms?
What is the pathophysiologic mechanism of her symptoms?
ANSWERS TO CASE 1:
Sickle Cell Disease
Summary: A 15-year-old African-American girl with recurrent bilateral thigh and hip pain, anemia, and symptoms and laboratory evidence of a urinary tract infection.
• Most likely diagnosis: Sickle cell disease (pain crisis).
• Biochemical mechanism of disease: Single amino acid substitution on hemoglobin β chain, inherited in an autosomal recessive fashion (1 in 12 African Americans in United States are carriers of the trait).
• Pathophysiologic mechanism of symptoms: The sickled red blood cells cause infarction of the bone, lung, kidney, and other tissue from vasoocclusion.
CLINICAL CORRELATION
This 15-year-old girl’s description of her pain is typical of a sickle cell pain crisis. Many times, infection is a trigger, most commonly pneumonia or urinary tract infection. This case is consistent with urinary tract infection, indicated by her symptoms of urinary frequency and burning with urination (dysuria). Her white blood cell count is elevated in response to the infection. The low hemoglobin level is consistent with sickle cell anemia. Because she is homozygous (both genes coding for sickle hemoglobin), both her parents have sickle cell trait (heterozygous) and, thus, do not have symptoms. The diagnosis can be established with hemoglobin electrophoresis. Treatment includes searching for an underlying cause of crisis (infection, hypoxia, fever, excessive exercise, and extreme changes in temperature), administration of oxygen, intravenous fluids for hydration, pain management, and consideration of blood transfusion.
APPROACH TO:
Sickle Cell Disease
OBJECTIVES
1. Understand the primary, secondary, tertiary, and quaternary levels of protein structure.
2. Describe the structure of hemoglobin and its role in oxygen binding and dissociation.
3. Describe the mechanism that amino acid substitution results in sickle cell hemoglobin.
DEFINITIONS
ALLOSTERIC EFFECTORS: Molecules that bind to enzymes or protein carriers at sites other than the active- or ligand-binding site. On binding, allosteric effectors either positively or negatively affect the enzymatic activity or capability of the protein to bind its ligand.
GLOBIN: The globular proteins that are the polypeptide components of myoglobin and hemoglobin. They contain a hydrophobic pocket that holds the heme prosthetic group.
GLOBIN FOLD: The 3-dimensional (3D) structure of the proteins that is common to myoglobin and the subunits of hemoglobin.
HEME: A porphyrin ring that has a Fe+2 ion coordinately bound in the center of the molecule. Heme binds oxygen in hemoglobin and myoglobin and serves as an electron carrier in the cytochromes.
HEMOGLOBIN: The tetrameric protein in high concentration in red blood cells that binds oxygen in the capillaries of the lungs and delivers it to peripheral tissues. Each globin subunit contains a heme group that binds oxygen when the iron atom is in the ferrous (+2) oxidation state.
MYOGLOBIN: A protein having a single globin polypeptide with a bound heme group. It is primarily located in muscle cells and stores oxygen for times when a high demand exists for energy.
PROTEIN PRIMARY (I°) STRUCTURE: The amino acid sequence of the protein, listed from the amino-terminal amino acid to the carboxy-terminal amino acid.
PROTEIN SECONDARY (II°) STRUCTURE: The local 3D spatial arrangement of amino acids close to one another in the primary sequence. α-Helices and β-sheets are the predominant secondary structures in proteins.
PROTEIN TERTIARY (III°) STRUCTURE: The overall 3D structure of a single polypeptide chain, including positions of disulfide bonds. Noncovalent forces such as hydrogen bonding, electrostatic forces, and hydrophobic effects are also important.
PROTEIN QUATERNARY (IV°) STRUCTURE: The overall 3D arrangement of polypeptide subunits in a multisubunit protein.
DISCUSSION
The activity of a given protein is dependent on proper folding of its polypeptide chain to assume a defined 3D structure. The importance of protein folding to molecular medicine is emphasized by the fact that many disease-causing mutations do not directly affect the active- or ligand-binding site of proteins, but instead cause local or global alterations in protein structure or disrupt the folding pathway such that the native protein fold is not achieved, or undesirable interactions with other proteins are promoted. The molecular defect that changes adult hemoglobin (HbA) to sickle hemoglobin (HbS), leading to sickle cell anemia, is a classic example of mutations that affect protein structure.
Protein structure is typically classified as consisting of 4 levels: primary (I°), secondary (II°), tertiary (III°), and quaternary (IV°). Primary structure is the sequence of amino acids in the protein. Secondary structure is the local 3D spatial arrangement of amino acids that are close to one another in the primary sequence. α-Helices and β-sheets compose the majority of secondary structures in all known proteins. Tertiary structure is the spatial arrangement of amino acid residues that are far apart in the linear primary sequence of a single polypeptide chain, and it includes disulfide bonds and noncovalent forces. These noncovalent forces include hydrogen bonding, which is also the primary stabilization force for the formation of α-helices and β-sheets, electrostatic interactions, van der Waals forces, and hydrophobic effects. Quaternary structure is the manner in which subunits of a multisubunit protein are arranged with respect to one another.
Normal hemoglobin has 4 subunits called globins. HbA has 2 β (α1 and α2) and 2 α (β1 and β 2) globin chains. Each globin chain has an associated heme prosthetic group, which is the site of oxygen binding and release. All globin chains have similar primary sequences. The secondary structure of globin chains consists of approximately 75% α-helix. The similar primary sequence promotes a similar tertiary structure in all globins that is called the globin fold, which is compact and globular in overall conformation. The quaternary structure of HbA can be described as a dimer of α1β1 and α2β2 dimers. The αβ dimers move relative to one another during the binding and release of oxygen.
Hemoglobin must remain soluble at high concentrations within the red blood cell to support normal oxygen binding and release properties. This is made possible by a distribution of amino acid side chains in which hydrophobic residues are sequestered in the interior core of the folded globin subunits, while hydrophilic residues dominate the water-exposed surface of the globin fold. The disk-shaped heme prosthetic group is inserted into a hydrophobic pocket formed by the globin.
Hemoglobin and the homologous monomeric protein myoglobin (Mb) both bind and release oxygen as a function of the surrounding concentration, or partial pressure of oxygen. A plot of percent saturation of hemoglobin or Mb with oxygen versus the partial pressure of oxygen is called the oxygen dissociation curve. Unlike Mb, which has a simple hyperbolic oxygen dissociation curve typical of ligand binding to a monomeric protein, the quaternary structure of HbA allows it to bind oxygen with positive cooperativity, giving a sigmoidal oxygen dissociation curve (Figure 1-1). Essentially, binding of oxygen to one HbA subunit increases the affinity of binding to other subunits in the tetramer, thereby shifting the equilibrium between oxy and deoxy forms. The net effect of this cooperativity is that HbA releases oxygen, whereas Mb globin would be most saturated with oxygen at the partial pressure of oxygen normally found in resting peripheral tissues. The quaternary structure of HbA also allows it to respond to 2,3-bisphosphoglycerate, carbon dioxide, and hydrogen ion, all of which are heterotropic negative allosteric effectors of oxygen binding.
Sickle cell anemia is due to a particular defect in the HBB gene. The HBB gene, located on the short arm of chromosome 11, codes for the formation of the globin-β gene product of 146 amino acids. A single base mutation in the coding region at amino acid position 6 in the protein results in the conversion of a GAG codon to GTG, leading to the nonconservative substitution of a hydrophobic valine residue for a hydrophilic glutamate in the protein, which is now called HbS. This conversion brings about the profound structural changes in the deoxygenated HbS described below. Because sickle cell anemia is an autosomal recessive disease, an individual must carry 2 copies of the HbS variant to develop the symptoms of the disease. Individuals with 1 variant (HbS) and 1 normal (HbA) allele are carriers and are generally free of symptoms. If one parent has sickle cell trait (HbAS) and the other parent is homozygous for HbA, then no offspring will have sickle cell disease, but some may be carriers (HbAS) and some homozygous for hemoglobin A (HbAA). By contrast, if both parents have sickle trait, there is a 1 out of 4 chance that the child will be homozygous for HbA (HbAA), a 2 out of 4 chance that the offspring will be heterozygous (HbAS) and carry sickle cell trait, and a 1 out of 4 chance that the offspring will have sickle cell disease (HbSS), as illustrated in Figure 1-1.
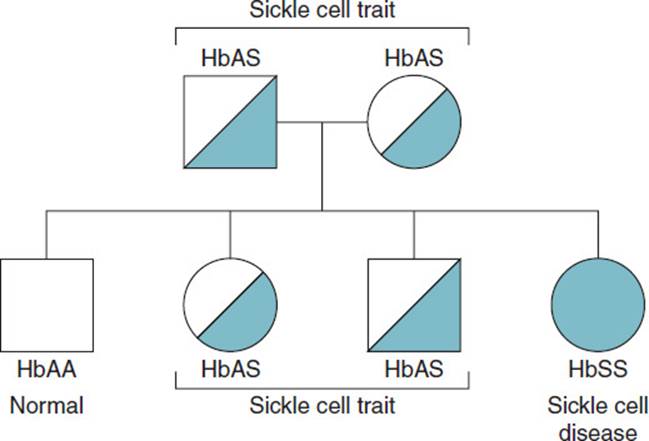
Figure 1-1. Genetics of the sickle cell disease when both parents are heterozygous for HbS (sickle cell trait).
The intrinsic oxygen-binding properties of HbA and HbS are the same; however, the solubility of deoxy HbS is reduced because exposure of Val-6 at the surface of the β-chain leads to a hydrophobic interaction with hydrophobic residues on another β-chain. Because hemoglobin is present at very high concentrations in the red blood cell, deoxy HbS will polymerize and precipitate inside the cell. The precipitate takes the form of elongated fibers because of the association of complementary hydrophobic surfaces on the β- and α-chains of deoxy HbS. At oxygen saturations found in arterial blood, the oxy HbS predominates and HbS does not precipitate because Val-6 of the β-chain is not exposed to the surface.
The tendency for deoxy HbS to precipitate is why clinical manifestations of sickle cell anemia are brought on by exertion and why treatment includes oxygen administration. The stiff fibrous precipitate causes the red blood cell to deform into the characteristic sickle shape and makes the normally malleable cell susceptible to hemolysis.
Although sickle cell disease may be the most widely known condition resulting from a variant in the HBB gene, other conditions are also recognized. Hemoglobin C is produced when the glutamate at position 6 is changed to a lysine residue. Hemoglobin SC disease occurs when an individual has 1 HbS and 1 HbC allele, resulting in symptoms similar to sickle cell anemia, and the severity of the condition may range from mild to equivalent to sickle cell disease. However, 2 hemoglobin C alleles have a milder resultant disease that causes anemia (hemoglobin C disease). Hemoglobin E results when amino acid position 26 of the β-globin gene is changed from glutamic acid to lysine. When an individual carries both HbS and HbE, symptoms may be as severe as in sickle cell disease. Hemoglobin S and hemoglobin C variants are more common in West African lineages, while hemoglobin E variants are more common in those with Southeast Asian lineages.
COMPREHENSION QUESTIONS
1.1 A newly married African-American couple, both from families having histories of good health, is about to have a child. If the incidence of the sickle cell trait is approximately 1 in 12 among persons of African descent in the United States, then what is the chance that the woman will give birth to a child affected by sickle cell disease?
A. 1 in 12
B. 1 in 24
C. 1 in 96
D. 1 in 288
E. 1 in 576
1.2 A 25-year-old African-American man with sickle cell anemia, who has been hospitalized several times for painful sickle cell crises, has successfully been free of these crises since he has been on hydroxyurea therapy. Treatment with hydroxyurea results in which of the following?
A. An increase in the oxygen affinity of HbS
B. An increase in the levels of hemoglobin F (HbF) in red blood cells
C. A decreased cooperativity in oxygen binding by HbS
D. A post-translational modification of HbS that prevents polymerization
E. A decreased ability of HbS to bind 2,3-bisphosphoglycerate (2,3-BPG)
1.3 A pregnant woman is able to transfer oxygen to her fetus because fetal hemoglobin has a greater affinity for oxygen than does HbA. Why is the affinity of fetal hemoglobin for oxygen higher?
A. The tense form of hemoglobin is more prevalent in the circulation of the fetus.
B. There is less 2,3-BPG in the fetal circulation compared with maternal circulation.
C. Fetal hemoglobin binds 2,3-BPG with fewer ionic bonds than HbA.
D. The Bohr effect is enhanced in the fetus.
E. The oxygen-binding curve of fetal hemoglobin is shifted to the right.
ANSWERS
1.1 E. Because sickle cell anemia has an autosomal recessive inheritance pattern and the incidence in the United States to persons of African descent is approximately 1 in 12, each parent has a 1 in 12 chance of being a carrier. Because it is an autosomal recessive gene, the couple’s offspring would have a 1 in 4 chance of being homozygous if both parents were also carriers. Thus, the chance of having a child with the sickle cell trait is 1 in 576 (or [1/12][1/12][1/4]).
1.2 B. By inhibiting the enzyme ribonucleotide reductase, hydroxyurea has been shown to increase the levels of fetal hemoglobin (HbF, α2γ2) by mechanisms not fully understood. The increase in HbF concentrations has the effect of decreasing HbS levels in the red blood cell. The increased concentration of HbF disrupts the polymerization of HbS and decreases the incidence of sickle cell crises. Hydroxyurea does not affect the oxygen affinity or cooperativity of oxygen binding of HbS, nor does it react with HbS to cause a post-translational modification or affect 2,3-BPG binding.
1.3 C. In HbA 2,3-BPG binds to ionized residues in the interface between the 2 β-chains, thus decreasing the oxygen affinity. The γ-chains of fetal hemoglobin have fewer ionized residues in the corresponding interface and bind 2,3-BPG with less affinity, which allows for a greater binding affinity for oxygen.
BIOCHEMISTRY PEARLS
![]() The quaternary structure of HbA allows it to bind oxygen with positive cooperativity, giving a sigmoidal oxygen dissociation curve.
The quaternary structure of HbA allows it to bind oxygen with positive cooperativity, giving a sigmoidal oxygen dissociation curve.
![]() Sickle cell anemia results from the nonconservative substitution of valine for glutamate at the sixth residue of the β chain of hemoglobin.
Sickle cell anemia results from the nonconservative substitution of valine for glutamate at the sixth residue of the β chain of hemoglobin.
![]() Precipitation of HbS is more likely to occur from exertion and deoxygenation. Therefore, treatment consists of oxygen and hydration.
Precipitation of HbS is more likely to occur from exertion and deoxygenation. Therefore, treatment consists of oxygen and hydration.
![]() The stiff fibrous precipitate of HbS causes the red blood cell to deform into the characteristic sickle shape and makes the normally malleable cell susceptible to hemolysis.
The stiff fibrous precipitate of HbS causes the red blood cell to deform into the characteristic sickle shape and makes the normally malleable cell susceptible to hemolysis.
REFERENCES
Schultz RM, Liebman MN. Proteins II: structure-function relationships in protein families. In: Devlin TM, ed. Textbook of Biochemistry with Clinical Correlations. 5th ed. New York: Wiley-Liss; 2002.
Weatherall DJ, Beaudet AL, Vogelstein B, et al. The hemoglobinopathies. In: Scriver CR, Beaudet AL, Sly W, et al, eds. The Metabolic & Molecular Basis of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001.