Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION IV. Structure, Function, & Replication of Informational Macromolecules
Chapter 38. Regulation of Gene Expression
P. Anthony Weil, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Explain that the many steps involved in the vectorial processes of gene expression, which range from targeted modulation of gene copy number, to gene rearrangement, to transcription, to mRNA processing and transport from the nucleus, to translation, to protein post-translational modification and degradation, are all subject to regulatory control, both positive and negative. Changes in any, or multiple of these processes, can increase or decrease the amount and/or activity of the cognate gene product.
Explain that the many steps involved in the vectorial processes of gene expression, which range from targeted modulation of gene copy number, to gene rearrangement, to transcription, to mRNA processing and transport from the nucleus, to translation, to protein post-translational modification and degradation, are all subject to regulatory control, both positive and negative. Changes in any, or multiple of these processes, can increase or decrease the amount and/or activity of the cognate gene product.
![]() Appreciate that DNA binding transcription factors, proteins that bind to specific DNA sequences that are often located near to transcriptional promoter elements, can either activate or repress gene transcription.
Appreciate that DNA binding transcription factors, proteins that bind to specific DNA sequences that are often located near to transcriptional promoter elements, can either activate or repress gene transcription.
![]() Recognize that DNA binding transcription factors are often modular proteins that are composed of structurally and functionally distinct domains, which can directly or indirectly control mRNA gene transcription, either through contacts with RNA polymerase and its cofactors, or through interactions with coregulators that modulate nucleosome structure via covalent modifications and/or displacement.
Recognize that DNA binding transcription factors are often modular proteins that are composed of structurally and functionally distinct domains, which can directly or indirectly control mRNA gene transcription, either through contacts with RNA polymerase and its cofactors, or through interactions with coregulators that modulate nucleosome structure via covalent modifications and/or displacement.
![]() Understand that nucleosome-directed regulatory events typically increase or decrease the accessibility of the underlying DNA such as enhancer or promoter sequences, although nucleosome modification can also create new binding sites for other coregulators.
Understand that nucleosome-directed regulatory events typically increase or decrease the accessibility of the underlying DNA such as enhancer or promoter sequences, although nucleosome modification can also create new binding sites for other coregulators.
![]() Understand that the processes of transcription, RNA processing, and nuclear export of RNA are all coupled.
Understand that the processes of transcription, RNA processing, and nuclear export of RNA are all coupled.
BIOMEDICAL IMPORTANCE
Organisms adapt to environmental changes by altering gene expression. The mechanisms controlling gene expression have been studied in detail and often involve modulation of gene transcription. Control of transcription ultimately results from changes in the mode of interaction of specific regulatory molecules, usually proteins, with various regions of DNA in the controlled gene. Such interactions can either have a positive or negative effect on transcription. Transcription control can result in tissue-specific gene expression, and gene regulation is influenced by hormones, heavy metals, and chemicals. In addition to transcription level controls, gene expression can also be modulated by gene amplification, gene rearrangement, posttranscriptional modifications, RNA stabilization, translational control, protein modification, and protein stabilization. Many of the mechanisms that control gene expression are used to respond to developmental cues, growth factors, hormones, environmental agents, and therapeutic drugs. Dysregulation of gene expression can lead to human disease. Thus, a molecular understanding of these processes will lead to development of agents that alter pathophysiologic mechanisms or inhibit the function or arrest the growth of pathogenic organisms.
REGULATED EXPRESSION OF GENES IS REQUIRED FOR DEVELOPMENT, DIFFERENTIATION, & ADAPTATION
The genetic information present in each normal somatic cell of a metazoan organism is practically identical. The exceptions are found in those few cells that have amplified or rearranged genes in order to perform specialized cellular functions or cells that have undergone oncogenic transformation. Expression of the genetic information must be regulated during ontogeny and differentiation of the organism and its cellular components. Furthermore, in order for the organism to adapt to its environment and to conserve energy and nutrients, the expression of genetic information must be cued to extrinsic signals and respond only when necessary. As organisms have evolved, more sophisticated regulatory mechanisms have appeared which provide the organism and its cells with the responsiveness necessary for survival in a complex environment. Mammalian cells possess about 1000 times more genetic information than does the bacterium Escherichia coli. Much of this additional genetic information is probably involved in regulation of gene expression during the differentiation of tissues and biologic processes in the multicellular organism and in ensuring that the organism can respond to complex environmental challenges.
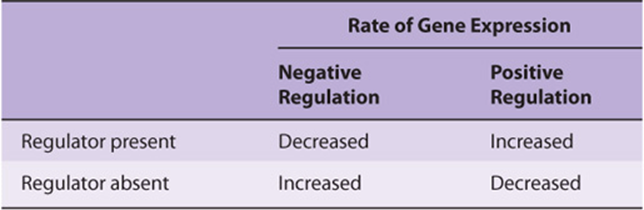
In simple terms, there are only two types of gene regulation: positive regulation and negative regulation (Table 38-1). When the expression of genetic information is quantitatively increased by the presence of a specific regulatory element, regulation is said to be positive; when the expression of genetic information is diminished by the presence of a specific regulatory element, regulation is said to be negative. The element or molecule mediating negative regulation is said to be a negative regulator, a silencer or repressor; that mediating positive regulation is a positive regulator, an enhancer or activator. However, a double negative has the effect of acting as a positive. Thus, an effector that inhibits the function of a negative regulator will appear to bring about a positive regulation. Many regulated systems that appear to be induced are in fact derepressed at the molecular level. (See Chapter 9 for explanation of these terms.)
TABLE 38–1 Effects of Positive and Negative Regulation on Gene Expression
BIOLOGIC SYSTEMS EXHIBIT THREE TYPES OF TEMPORAL RESPONSES TO A REGULATORY SIGNAL
Figure 38–1 depicts the extent or amount of gene expression in three types of temporal response to an inducing signal. A type A response is characterized by an increased extent of gene expression that is dependent upon the continued presence of the inducing signal. When the inducing signal is removed, the amount of gene expression diminishes to its basal level, but the amount repeatedly increases in response to the reappearance of the specific signal. This type of response is commonly observed in prokaryotes in response to sudden changes of the intracellular concentration of a nutrient. It is also observed in many higher organisms after exposure to inducers such as hormones, nutrients, or growth factors (Chapter 42).
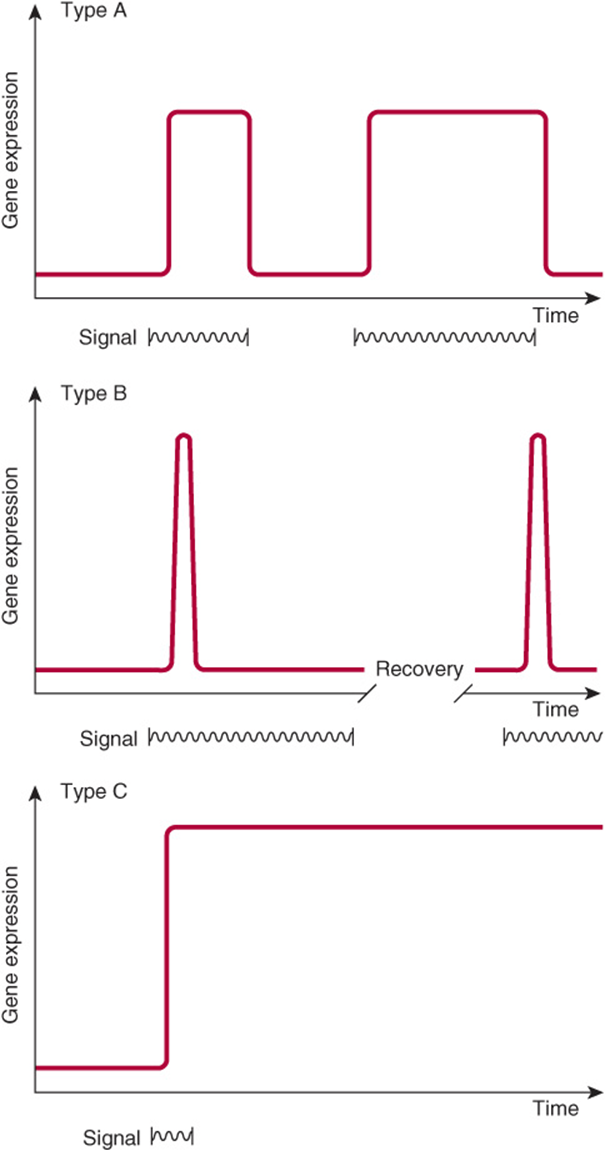
FIGURE 38–1 Diagrammatic representations of the responses of the extent of expression of a gene to specific regulatory signals (such as a hormone as a function of time).
A type B response exhibits an increased amount of gene expression that is transient even in the continued presence of the regulatory signal. After the regulatory signal has terminated and the cell has been allowed to recover, a second transient response to a subsequent regulatory signal may be observed. This phenomenon of response-desensitization recovery characterizes the action of many pharmacologic agents, but it is also a feature of many naturally occurring processes. This type of response commonly occurs during development of an organism, when only the transient appearance of a specific gene product is required although the signal persists.
The type C response pattern exhibits, in response to the regulatory signal, an increased extent of gene expression that persists indefinitely even after termination of the signal. The signal acts as a trigger in this pattern. Once expression of the gene is initiated in the cell, it cannot be terminated even in the daughter cells; it is therefore an irreversible and inherited alteration. This type of response typically occurs during the development of differentiated function in a tissue or organ.
Simple Unicellular and Multicellular Organisms Serve as Valuable Models for the Study of Gene Expression in Mammalian Cells
Analysis of the regulation of gene expression in prokaryotic cells helped establish the principle that information flows from the gene to a messenger RNA to a specific protein molecule. These studies were aided by the advanced genetic analyses that could be performed in prokaryotic and lower eukaryotic organisms such as baker’s yeast, Saccharomyces cerevisiae, and the fruit fly, Drosophila melanogaster, among others. In recent years, the principles established in these studies, coupled with a variety of molecular biology techniques, have led to remarkable progress in the analysis of gene regulation in higher eukaryotic organisms, including mammals. In this chapter, the initial discussion will center on prokaryotic systems. The impressive genetic studies will not be described, but the physiology of gene expression will be discussed. However, nearly all of the conclusions about this physiology have been derived from genetic studies and confirmed by molecular genetic and biochemical experiments.
Some Features of Prokaryotic Gene Expression Are Unique
Before the physiology of gene expression can be explained, a few specialized genetic and regulatory terms must be defined for prokaryotic systems. In prokaryotes, the genes involved in a metabolic pathway are often present in a linear array called an operon, for example, the lac operon. An operon can be regulated by a single promoter or regulatory region. The cistron is the smallest unit of genetic expression. As described in Chapter 9, some enzymes and other protein molecules are composed of two or more nonidentical subunits. Thus, the “one gene, one enzyme” concept is not necessarily valid. The cistron is the genetic unit coding for the structure of the subunit of a protein molecule, acting as it does as the smallest unit of genetic expression. Thus, the one gene, one enzyme idea might more accurately be regarded as a one cistron, one subunit concept. A single mRNA that encodes more than one separately translated protein is referred to as a polycistronic mRNA. For example, the polycistronic lac operon mRNA is translated into three separate proteins (see below). Operons and polycistronic mRNAs are common in bacteria but not in eukaryotes.
An inducible gene is one whose expression increases in response to an inducer or activator, a specific positive regulatory signal. In general, inducible genes have relatively low basal rates of transcription. By contrast, genes with high basal rates of transcription are often subject to downregulation by repressors.
The expression of some genes is constitutive, meaning that they are expressed at a reasonably constant rate and not known to be subject to regulation. These are often referred to as housekeeping genes. As a result of mutation, some inducible gene products become constitutively expressed. A mutation resulting in constitutive expression of what was formerly a regulated gene is called a constitutive mutation.
Analysis of Lactose Metabolism in E coli Led to the Operon Hypothesis
Jacob and Monod in 1961 described their operon model in a classic paper. Their hypothesis was to a large extent based on observations on the regulation of lactose metabolism by the intestinal bacterium E coli. The molecular mechanisms responsible for the regulation of the genes involved in the metabolism of lactose are now among the best-understood in any organism. β-Galactosidase hydrolyzes the β-galactoside lactose to galactose and glucose. The structural gene for β-galactosidase (lacZ) is clustered with the genes responsible for the permeation of lactose into the cell (lacY) and for thiogalactoside transacetylase (lacA). The structural genes for these three enzymes, along with the lac promoter and lac operator (a regulatory region), are physically associated to constitute the lac operon as depicted in Figure 38–2. This genetic arrangement of the structural genes and their regulatory genes allows for coordinate expression of the three enzymes concerned with lactose metabolism. Each of these linked genes is transcribed into one large polycistronic mRNA molecule that contains multiple independent translation start (AUG) and stop (UAA) codons for each of the three cistrons. Thus, each protein is translated separately, and they are not processed from a single large precursor protein.
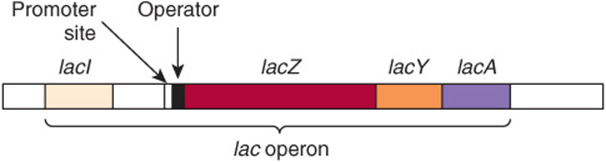
FIGURE 38–2 The positional relationships of the structural and regulatory genes of the lac operon. lacZ encodes β-galactosidase, lacY encodes a permease, and lacA encodes a thiogalactoside transacetylase. lacI encodes the lac operon repressor protein.
It is now conventional to consider that a gene includes regulatory sequences as well as the region that encodes the primary transcript. Although there are many historical exceptions, a gene is generally italicized in lower case and the encoded protein, when abbreviated, is expressed in roman type with the first letter capitalized. For example, the gene lacI encodes the repressor protein LacI. When E coli is presented with lactose or some specific lactose analogs under appropriate nonrepressing conditions (eg, high concentrations of lactose, no or very low glucose in media; see below), the expression of the activities of β-galactosidase, galactoside permease, and thiogalactoside transacetylase is increased 100-fold to 1000-fold. This is a type A response, as depicted in Figure 38–1. The kinetics of induction can be quite rapid; lac-specific mRNAs are fully induced within 5-6 min after addition of lactose to a culture; β-galactosidase protein is maximal within 10 min. Under fully induced conditions, there can be up to 5000 β-galactosidase molecules per cell, an amount about 1000 times greater than the basal, uninduced level. Upon removal of the signal, that is, the inducer, the synthesis of these three enzymes declines.
When E coli is exposed to both lactose and glucose as sources of carbon, the organisms first metabolize the glucose and then temporarily stop growing until the genes of the lac operon become induced to provide the ability to metabolize lactose as a usable energy source. Although lactose is present from the beginning of the bacterial growth phase, the cell does not induce those enzymes necessary for catabolism of lactose until the glucose has been exhausted. This phenomenon was first thought to be attributable to repression of the lac operon by some catabolite of glucose; hence, it was termed catabolite repression. It is now known that catabolite repression is in fact mediated by a catabolite gene activator protein (CAP) in conjunction with cAMP (Figure 17–5). This protein is also referred to as the cAMP regulatory protein (CRP). The expression of many inducible enzyme systems or operons in E coliand other prokaryotes is sensitive to catabolite repression, as discussed below.
The physiology of induction of the lac operon is well understood at the molecular level (Figure 38–3). Expression of the normal lacI gene of the lac operon is constitutive; it is expressed at a constant rate, resulting in formation of the subunits of the lac repressor. Four identical subunits with molecular weights of 38,000 assemble into a tetrameric Lac repressor molecule. The LacI repressor protein molecule, the product of lacI, has a high affinity (dissociation constant, Kd about 10-13 mol/L) for the operator locus. The operator locus is a region of double-stranded DNA that exhibits a twofold rotational symmetry and an inverted palindrome (indicated by arrows about the dotted axis) in a region that is 21 bp long, as shown below:
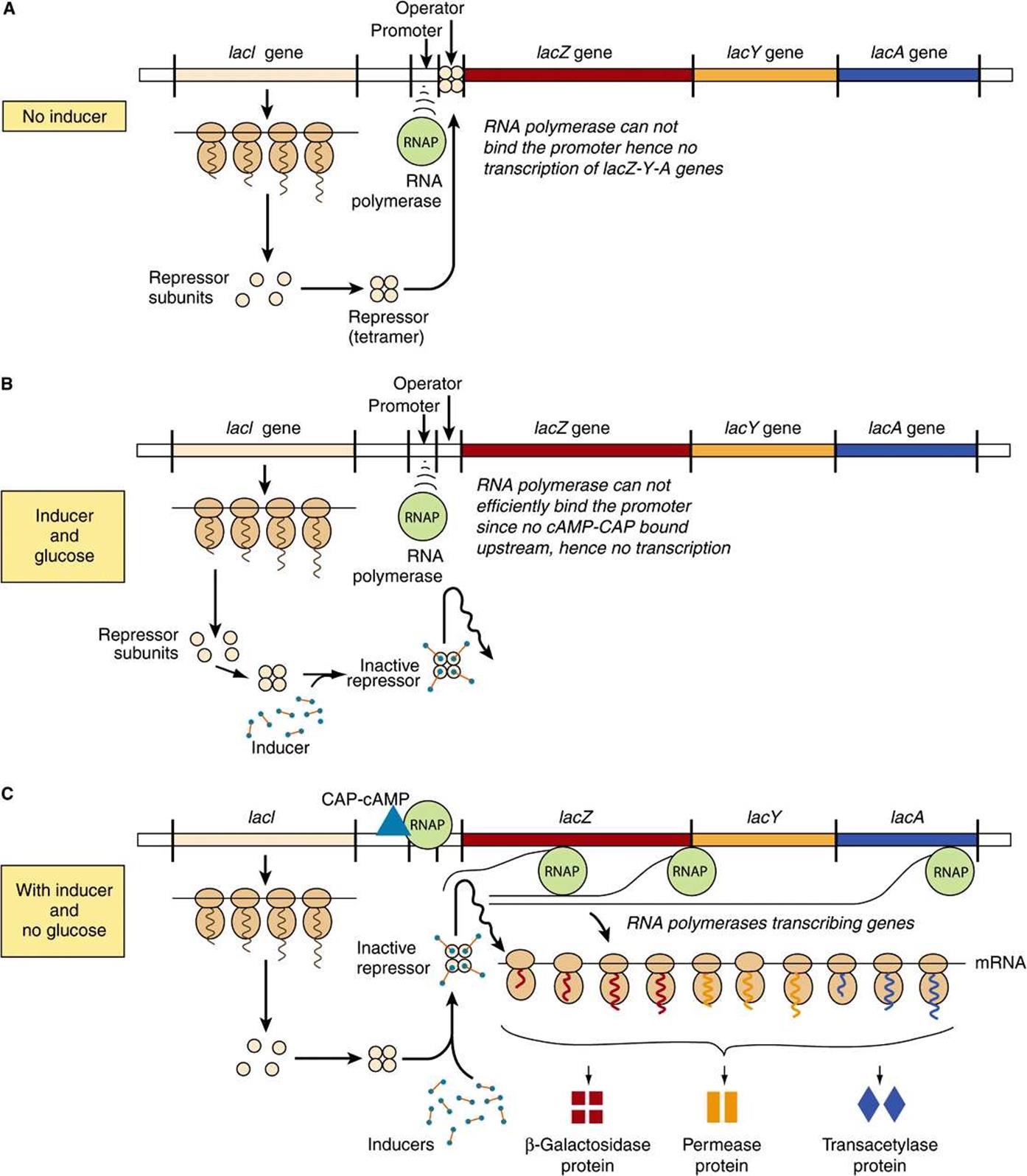
FIGURE 38–3 The mechanism of repression and derepression of the lac operon. When no inducer is present (A) the constitutively synthesized lacI gene products forms a repressor tetramer molecule that binds at the operator locus. Repressor-operator binding prevents the binding of RNA polymerase and consequently prevents transcription of the lacZ, lacY, and lacA structural genes into a polycistonic mRNA. When inducer is present, but glucose is also present in the culture medium (B), the tetrameric repressor molecules are conformationally altered by inducer, and cannot efficiently bind to the operator locus (affinity of binding reduced >1000-fold). However, RNA polymerase will not efficiently bind the promoter and initiate transcription, therefore the operon is not transcribed. However, when inducer is present and glucose is depleted from the medium (C) adenyl cylase is activated and cAMP is produced. This cAMP binds with high affinity to its binding protein the Cyclic AMP Activator Protein, or CRP. The camp-CAP complex binds to its recognition sequence (CRE, the cAMP Response Element) located ~15 bp upstream of the promoter. Direct protein-protein contacts between the CRE-bound CAP and the RNA polymerase increases promoter binding >20-fold; hence RNAP will efficiently transcribe the structural genes lacZ, lacY, and lacA, and the polycistronic mRNA molecule formed can be translated into the corresponding protein molecules β-galactosidase, permease, and transacetylase as shown, whick allows for the catabolism of lactose as the sole carbon source for growth.

At any one time, only two of the four subunits of the repressor appear to bind to the operator, and within the 21-base-pair region nearly every base of each base pair is involved in LacI recognition and binding. The binding occurs mostly in the major groove without interrupting the base-paired, doublehelical nature of the operator DNA. The operator locus is between the promoter site, at which the DNA-dependent RNA polymerase attaches to commence transcription, and the transcription initiation site of the lacZ gene. the structural gene for β-galactosidase (Figure 38–2). When attached to the operator locus, the LacI repressor molecule prevents transcription of the distal structural genes, lacZ, lacY, and lacA by interfering with the binding of RNA polymerase to the promoter; RNA polymerase and LacI repressor cannot be effectively bound to the lac operon at the same time. Thus, the LacI repressor molecule is a negative regulator; in its presence (and in the absence of inducer; see below), expression from the lacZ, lacY, and lacA genes is very, very low. There are normally 20-40 repressor tetramer molecules in the cell, a concentration of tetramer sufficient to effect, at any given time, >95% occupancy of the one lac operator element in a bacterium, thus ensuring low (but not zero) basal lac operon gene transcription in the absence of inducing signals.
A lactose analog that is capable of inducing the lac operon while not itself serving as a substrate for β-galactosidase is an example of a gratuitous inducer. An example is isopropylthiogalactoside (IPTG). The addition of lactose or of a gratuitous inducer such as IPTG to bacteria growing on a poorly utilized carbon source (such as succinate) results in prompt induction of the lac operon enzymes. Small amounts of the gratuitous inducer or of lactose are able to enter the cell even in the absence of permease. The LacI repressor molecules—both those attached to the operator loci and those free in the cytosol—have a high affinity for the inducer. Binding of the inducer to repressor molecule induces a conformational change in the structure of the repressor and causes it to dissociate from operator DNA because its affinity for the operator is now 104 times lower (Kd about 10-9 mol/L) than that of LacI in the absence of IPTG. DNA-dependent RNA polymerase can now bind to the promoter (ie, Figures 36-3 and 36-8), and transcription will begin, although this process is relatively inefficient (see below). In such a manner, an inducer derepresses the lac operon and allows transcription of the structural genes for β-galactosidase, galactoside permease, and thiogalactoside transacetylase. Translation of the polycistronic mRNA can occur even before transcription is completed. Derepression of the lac operon allows the cell to synthesize the enzymes necessary to catabolize lactose as an energy source. Based on the physiology just described, IPTG-induced expression of transfected plasmids bearing the lac operator-promoter ligated to appropriate bioengineered constructs is commonly used to express mammalian recombinant proteins in E coli.
In order for the RNA polymerase to form a PIC at the promoter site most efficiently, there must also be present the CAP to which cAMP is bound. By an independent mechanism, the bacterium accumulates cAMP only when it is starved for a source of carbon. In the presence of glucose—or of glycerol in concentrations sufficient for growth—the bacteria will lack sufficient cAMP to bind to CAP because the glucose inhibits adenylyl cyclase, the enzyme that converts ATP to cAMP (see Chapter 41). Thus, in the presence of glucose or glycerol, cAMP-saturated CAP is lacking, so that the DNA-dependent RNA polymerase cannot initiate transcription of the lac operon at the maximal rate. However, in the presence of the CAP-cAMP complex, which binds to DNA just upstream of the promoter site, transcription occurs at maximal levels (Figure 38–3). Studies indicate that a region of CAP directly contacts the RNA polymerase α-subunit, and these protein-protein interactions facilitate the binding of RNAP to the promoter. Thus, the CAP-cAMP regulator is acting as a positive regulator because its presence is required for optimal gene expression. The lac operon is therefore controlled by two distinct, ligand-modulated DNA binding trans-factors; one that acts positively (cAMP-CRP complex) to facilitate productive binding of RNA polymerase to the promoter and one that acts negatively (LacI repressor) that antagonizes RNA polymerase promoter binding. Maximal activity of the lac operon occurs when glucose levels are low (high cAMP with CAP activation) and lactose is present (LacI is prevented from binding to the operator).
When the lacI gene has been mutated so that its product, LacI, is not capable of binding to operator DNA, the organism will exhibit constitutive expression of the lac operon. In a contrary manner, an organism with a lacI gene mutation that produces a LacI protein which prevents the binding of an inducer to the repressor will remain repressed even in the presence of the inducer molecule, because the inducer cannot bind to the repressor on the operator locus in order to derepress the operon. Similarly, bacteria harboring mutations in their lac operator locus such that the operator sequence will not bind a normal repressor molecule constitutively express the lac operon genes. Mechanisms of positive and negative regulation comparable to those described here for the lac system have been observed in eukaryotic cells (see below).
The Genetic Switch of Bacteriophage Lambda (λ) Provides Another Paradigm for Protein-DNA Interactions and Transcriptional Regulation in Eukaryotic Cells
Like some eukaryotic viruses (eg, herpes simplex virus and HIV), some bacterial viruses can either reside in a dormant state within the host chromosomes or can replicate within the bacterium and eventually lead to lysis and killing of the bacterial host. Some E. coli harbor such a “temperate” virus, bacteriophage lambda (λ). When lambda infects an organism of that species, it injects its 45,000-bp, double-stranded, linear DNA genome into the cell (Figure 38–4). Depending upon the nutritional state of the cell, the lambda DNA will either integrate into the host genome (lysogenic pathway) and remain dormant until activated (see below), or it will commence replicating until it has made about 100 copies of complete, protein-packaged virus, at which point it causes lysis of its host (lytic pathway). The newly generated virus particles can then infect other susceptible hosts. Poor growth conditions favor lysogeny while good growth conditions promote the lytic pathway of lambda growth.
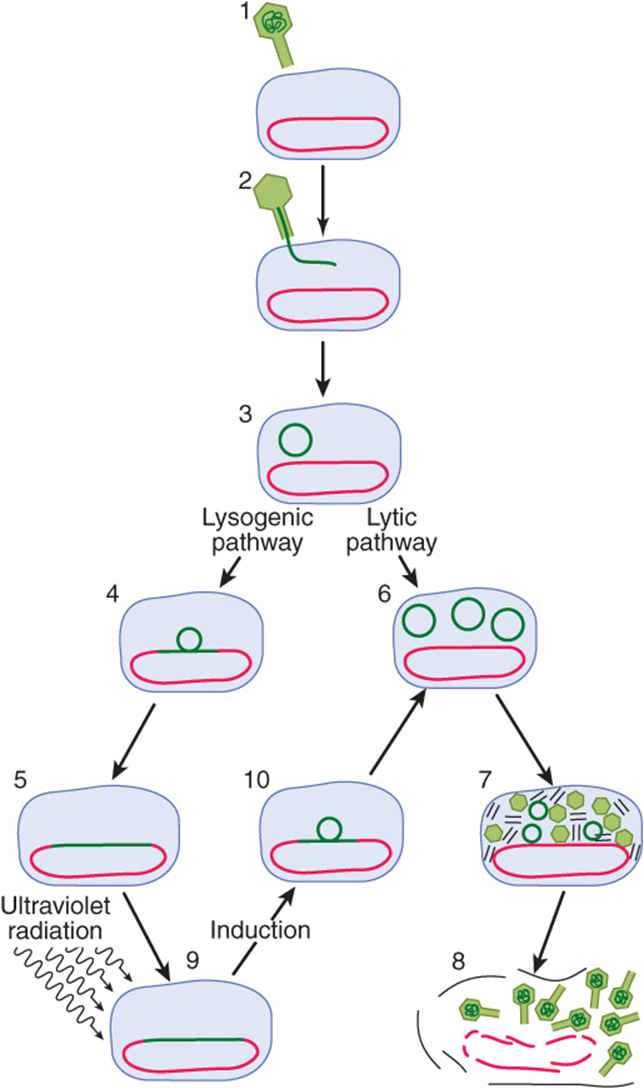
FIGURE 38–4 Infection of the bacterium E. coli by phage lambda begins when a virus particle attaches itself to specific receptors on the bacterial cell (1) and injects its DNA (dark green line) into the cell (2, 3). Infection can take either of two courses depending on which of two sets of viral genes is turned on. In the lysogenic pathway, the viral DNA becomes integrated into the bacterial chromosome (red) (4,5), where it replicates passively as the bacterial DNA and cell divides. This dormant genomically integrated virus is called a prophage, and the cell that harbors it is called a lysogen. In the alternative lytic mode of infection, the viral DNA replicates itself (6) and directs the synthesis of viral proteins (7). About 100 new virus particles are formed. The proliferating viruses induce lysis of the cell (8). A prophage can be “induced” by a DNA damaging agent such as ultraviolet radiation (9). The inducing agent throws a switch, so that a different set of genes is turned on. Viral DNA loops out of the chromosome (10) and replicates; the virus proceeds along the lytic pathway. (Reproduced, with permission, from Ptashne M, Johnson AD, Pabo CO: A genetic switch in a bacterial virus. Sci Am [Nov] 1982;247:128.)
When integrated into the host genome in its dormant state, lambda will remain in that state until activated by exposure of its bacterial host to DNA-damaging agents. In response to such a noxious stimulus, the dormant bacteriophage becomes “induced” and begins to transcribe and subsequently translate those genes of its own genome that are necessary for its excision from the host chromosome, its DNA replication, and the synthesis of its protein coat and lysis enzymes. This event acts like a trigger or type C (Figure 38–1) response; that is, once dormant lambda has committed itself to induction, there is no turning back until the cell is lysed and the replicated bacteriophage released. This switch from a dormant or prophage state to a lytic infection is well understood at the genetic and molecular levels and will be described in detail here; though less well understood at the molecular level, HIV and herpes viruses can behave similarly.
The lytic/lysogenic genetic switching event in lambda is centered around an 80-bp region in its double-stranded DNA genome referred to as the “right operator” (OR) (Figure 38–5A). The right operator is flanked on its left side by the structural gene for the lambda repressor protein, cI, and on its right side by the structural gene encoding another regulatory protein called cro. When lambda is in its prophage state—that is, integrated into the host genome—the cI repressor gene is the only lambda gene that is expressed. When the bacteriophage is undergoing lytic growth, the cI repressor gene is not expressed, but the cro gene—as well as many other lambda genes—is expressed. That is, when the repressor gene is on, the cro gene is off, and when the cro gene is on, the cI repressor gene is off. As we shall see, these two genes regulate each other’s expression and thus, ultimately, the decision between lytic and lysogenic growth of lambda. This decision between repressor gene transcription and cro gene transcription is a paradigmatic example of a molecular transcriptional switch.
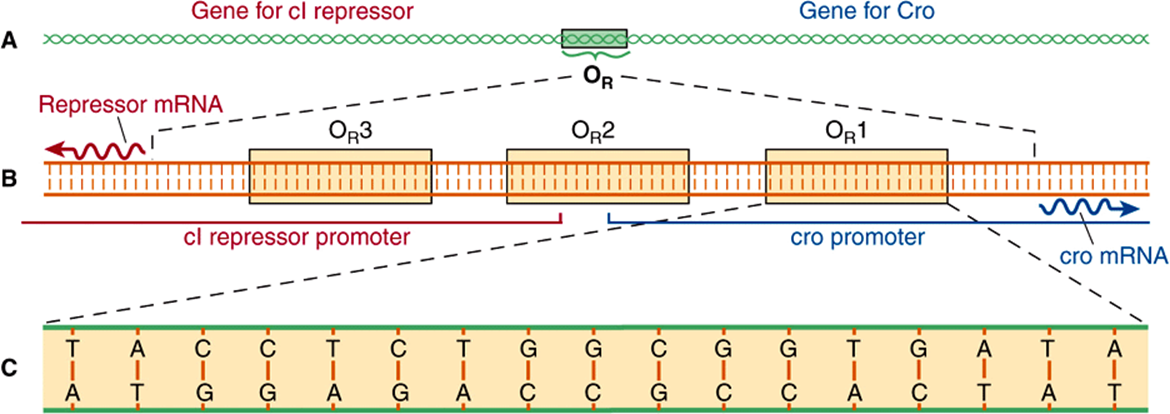
FIGURE 38–5 Right operator (OR) is shown in increasing detail in this series of drawings. The operator is a region of the viral DNA some 80 bp long (A). To its left lies the gene encoding lambda repressor (cI), to its right the gene (cro) encoding the regulator protein Cro. When the operator region is enlarged (B), it is seen to include three subregions, OR1, OR2, and OR3, each 17 bp long. They are recognition sites to which both repressor and Cro can bind. The recognition sites overlap two promoters—sequences of bases to which RNA polymerase binds in order to transcribe these genes into mRNA (wavy lines), that are translated into protein. Site OR1 is enlarged (C) to show its base sequence. Note that in the OR region of the lambda chromosome, both strands of DNA act as a template for transcription. (Reproduced, with permission, from Ptashne M, Johnson AD, Pabo CO: A genetic switch in a bacterial virus. Sci Am [Nov] 1982;247:128.)
The 80-bp lambda right operator, OR, can be subdivided into three discrete, evenly spaced, 17-bp cis-active DNA elements that represent the binding sites for either of two bacteriophage lambda regulatory proteins. Importantly, the nucleotide sequences of these three tandemly arranged sites are similar but not identical (Figure 38–5B). The three related cis-elements, termed operators OR1, OR2, and OR3, can be bound by either cI or Cro proteins. However, the relative affinities of cI and Cro for each of the sites vary, and this differential binding affinity is central to the appropriate operation of the lambda phage lytic or lysogenic “molecular switch.” The DNA region between the croand repressor genes also contains two promoter sequences that direct the binding of RNA polymerase in a specified orientation, where it commences transcribing adjacent genes. One promoter directs RNA polymerase to transcribe in the rightward direction and, thus, to transcribe cro and other distal genes, while the other promoter directs the transcription of the cI repressor gene in the leftward direction (Figure 38–5B).
The product of the repressor gene, the 236-amino-acid, 27 kDa cI repressor protein, exists as a two-domain molecule in which the amino terminal domain binds to operator DNA and the carboxyl terminal domain promotes the association of one repressor protein with another to form a dimer. A dimer of repressor molecules binds to operator DNA much more tightly than does the monomeric form (Figure 38–6A to 38-6C).
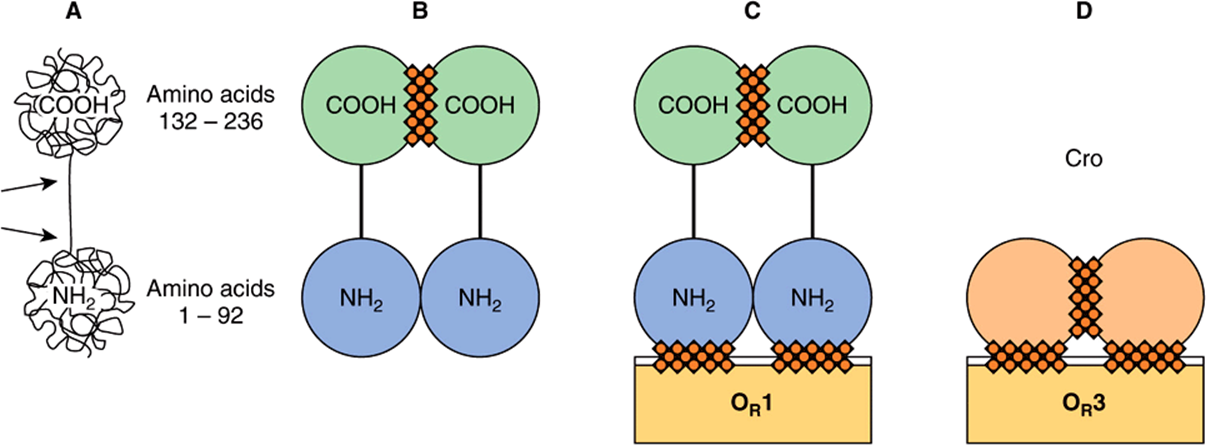
FIGURE 38–6 Schematic molecular structures of cI (lambda repressor, shown in A, B, and C) and Cro (D). The lambda repressor protein is a polypeptide chain 236 amino acids long. The chain folds itself into a dumbbell shape with two substructures: an amino terminal (NH2) domain and a carboxyl terminal (COOH) domain. The two domains are linked by a region of the chain that is less structured and susceptible to cleavage by proteases (indicated by the two arrows in A). Single repressor molecules (monomers) tend to reversibly associate to form dimers. (B) A dimer is held together mainly by contact between the carboxyl terminal domains (hatching). Repressor dimers bind to (and can dissociate from) the recognition sites in the operator region; they display differential affinites for the three operator sites, ![]() (C). It is the DBD of the repressor molecule that makes contact with the DNA (hatching). Cro (D) has a single domain with sites that promote dimerization and other sites that promote binding of dimers to operator, cro exhibits the highest affinity for OR3, opposite the sequence binding preference of the cI protein. (Reproduced, with permission, from Ptashne M, Johnson AD, Pabo CO: A genetic switch in a bacterial virus. Sci Am [Nov] 1982;247:128.)
(C). It is the DBD of the repressor molecule that makes contact with the DNA (hatching). Cro (D) has a single domain with sites that promote dimerization and other sites that promote binding of dimers to operator, cro exhibits the highest affinity for OR3, opposite the sequence binding preference of the cI protein. (Reproduced, with permission, from Ptashne M, Johnson AD, Pabo CO: A genetic switch in a bacterial virus. Sci Am [Nov] 1982;247:128.)
The product of the cro gene, the 66-amino-acid, 9-kDa Cro protein, has a single domain but also binds the operator DNA more tightly as a dimer (Figure 38–6D). The Cro protein’s single domain mediates both operator binding and dimerization.
In a lysogenic bacterium—that is, a bacterium containing an integrated dormant lambda prophage—the lambda repressor dimer binds preferentially to OR1 but in so doing, by a cooperative interaction, enhances the binding (by a factor of 10) of another repressor dimer to OR2 (Figure 38–7). The affinity of repressor for OR3 is the least of the three operator subregions. The binding of repressor to OR1 has two major effects. The occupation of OR1 by repressor blocks the binding of RNA polymerase to the rightward promoter and in that way prevents expression of cro. Second, as mentioned above, repressor dimer bound to OR1 enhances the binding of repressor dimer to OR2. The binding of repressor to OR2 has the important added effect of enhancing the binding of RNA polymerase to the leftward promoter that overlaps OR3 and thereby enhances transcription and subsequent expression of the repressor gene. This enhancement of transcription is mediated through direct protein-protein interactions between promoter-bound RNA polymerase and OR2-bound repressor, much as described above for CAP protein and RNA polymerase on the lac operon. Thus, the lambda repressor is both a negative regulator, by preventing transcription of cro, and a positive regulator, by enhancing transcription of its own gene, cI. This dual effect of repressor is responsible for the stable state of the dormant lambda bacteriophage; not only does the repressor prevent expression of the genes necessary for lysis, but it also promotes expression of itself to stabilize this state of differentiation. In the event that intracellular repressor protein concentration becomes very high, this excess repressor will bind to OR3 and by so doing diminish transcription of the repressor gene from the leftward promoter, by blocking RNAP binding to the cI promoter, until the repressor concentration drops and repressor dissociates itself from OR3. Interestingly, similar examples of repressor proteins also having the ability to activate transcription have been observed in eukaryotes.
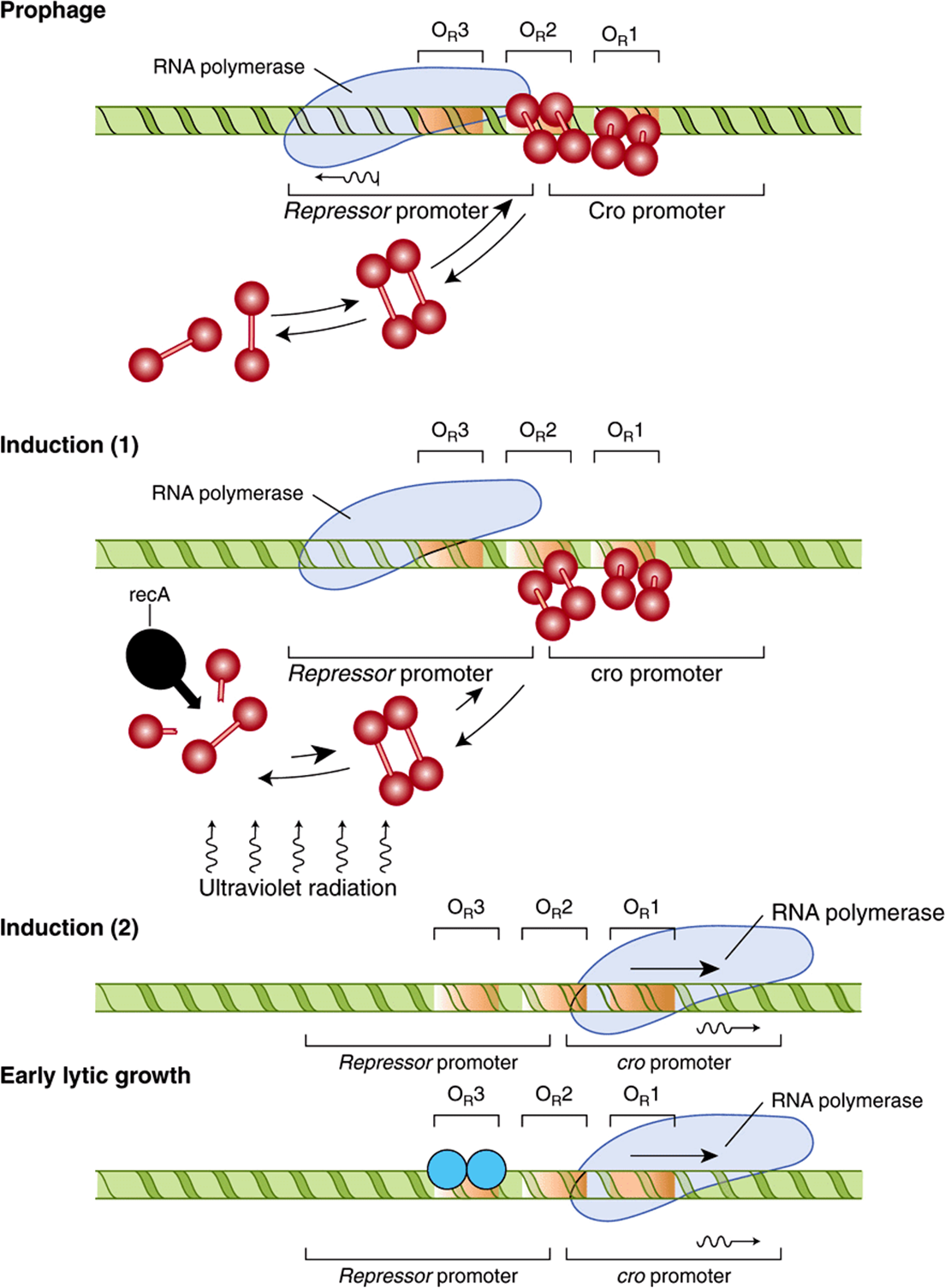
FIGURE 38–7 Configuration of the lytic/lysogenic switch is shown at four stages of the lambda life cycle. The lysogenic pathway (in which the virus remains dormant as a prophage) is selected when a repressor dimer binds to OR1, thereby making it likely that OR2 will be filled immediately by another dimer. In the prophage (top), the repressor dimers bound at OR1 and OR2 prevent RNA polymerase from binding to the rightward promoter and so block the synthesis of Cro (negative control). The repressors also enhance the binding of polymerase to the leftward promoter (positive control), with the result that the repressor gene is transcribed into RNA (wavy line) and more repressor is synthesized, maintaining the lysogenic state. The prophage is induced (middle) when ultraviolet radiation activates the protease recA, which cleaves repressor monomers. The equilibrium of free monomers, free dimers, and bound dimers is thereby shifted, and dimers leave the operator sites. RNA polymerase is no longer encouraged to bind to the leftward promoter, so that repressor is no longer synthesized. As induction proceeds, all the operator sites become vacant, thus polymerase can bind to the rightward promoter and Cro is synthesized. During early lytic growth, a single Cro dimer binds to OR3 (light blue shaded circles), the site for which it has the highest affinity. Consequently, RNA polymerase cannot bind to the leftward promoter, but the rightward promoter remains accessible. Polymerase continues to bind there, transcribing cro and other early lytic genes. Lytic growth ensues (bottom).(Reproduced, with permission, from Ptashne M, Johnson AD, Pabo CO: A genetic switch in a bacterial virus. Sci Am [Nov] 1982;247:128.)
With such a stable, repressive, cI-mediated, lysogenic state, one might wonder how the lytic cycle could ever be entered. However, this process does occur quite efficiently. When a DNA-damaging signal, such as ultraviolet light, strikes the lysogenic host bacterium, fragments of single-stranded DNA are generated that activate a specific co-protease coded by a bacterial gene and referred to as recA (Figure 38–7). The activated recA protease hydrolyzes the portion of the repressor protein that connects the amino terminal and carboxyl terminal domains of that molecule (see Figure 38–6A). Such cleavage of the repressor domains causes the repressor dimers to dissociate, which in turn causes dissociation of the repressor molecules from OR2 and eventually from OR1. The effects of removal of repressor from OR1 and OR2 are predictable. RNA polymerase immediately has access to the rightward promoter and commences transcribing the cro gene, and the enhancement effect of the repressor at OR2 on leftward transcription is lost (Figure 38–7).
The resulting newly synthesized Cro protein also binds to the operator region as a dimer, but its order of preference is opposite to that of repressor (Figure 38–7). That is, Cro binds most tightly to OR3, but there is no cooperative effect of Cro at OR3 on the binding of Cro to OR2. At increasingly higher concentrations of Cro, the protein will bind to OR2 and eventually to OR1.
Occupancy of OR3 by Cro immediately turns off transcription from the leftward cI promoter and in that way prevents any further expression of the repressor gene. The molecular switch is thus completely “thrown” in the lytic direction. The cro gene is now expressed, and the repressor gene is fully turned off. This event is irreversible, and the expression of other lambda genes begins as part of the lytic cycle. When Cro repressor concentration becomes quite high, it will eventually occupy OR1 and in so doing reduce the expression of its own gene, a process that is necessary in order to effect the final stages of the lytic cycle.
The three-dimensional structures of Cro and of the lambda repressor protein have been determined by X-ray crystallography, and models for their binding and effecting the above-described molecular and genetic events have been proposed and tested. Both bind to DNA using helix-turn-helix DNA-binding domain (DBD) motifs (see below). To date, this system provides arguably the best understanding of the molecular events involved in gene activation and repression.
Detailed analysis of the lambda repressor led to the important concept that transcription regulatory proteins have several functional domains. For example, lambda repressor binds to DNA with high affinity. Repressor monomers form dimers, cooperatively interact with each other, and repressor interacts with RNA polymerase, to enhance or block promoter binding or RNAP open complex formation (see Figure 36–3). The protein-DNA interface and the three protein-protein interfaces all involve separate and distinct domains of the repressor molecule. As will be noted below (see Figure 38–19), this is a characteristic shared by most (perhaps all) molecules that regulate transcription.
SPECIAL FEATURES ARE INVOLVED IN REGULATION OF EUKARYOTIC GENE TRANSCRIPTION
Most of the DNA in prokaryotic cells is organized into genes, and the templates always have the potential to be transcribed if appropriate positive and negative trans-factors are activated. A very different situation exists in mammalian cells, in which relatively little of the total DNA is organized into mRNA encoding genes and their associated regulatory regions. The function of the extra DNA is being actively investigated (ie, Chapter 39; the ENCODE Projects). More importantly, as described in Chapter 35, the DNA in eukaryotic cells is extensively folded and packed into the protein-DNA complex called chromatin. Histones are an important part of this complex since they both form the structures known as nucleosomes (see Chapter 35) and also factor significantly into gene regulatory mechanisms as outlined below.
The Chromatin Template Contributes Importantly to Eukaryotic Gene Transcription Control
Chromatin structure provides an additional level of control of gene transcription. As discussed in Chapter 35, large regions of chromatin are transcriptionally inactive while others are either active or potentially active. With few exceptions, each cell contains the same complement of genes. The development of specialized organs, tissues, and cells and their function in the intact organism depend upon the differential expression of genes.
Some of this differential expression is achieved by having different regions of chromatin available for transcription in cells from various tissues. For example, the DNA containing the β-globin gene cluster is in “active” chromatin in the reticulocyte but in “inactive” chromatin in muscle cells. All the factors involved in the determination of active chromatin have not been elucidated. The presence of nucleosomes and of complexes of histones and DNA (see Chapter 35) certainly provides a barrier against the ready association of transcription factors with specific DNA regions. The dynamics of the formation and disruption of nucleosome structure are therefore an important part of eukaryotic gene regulation.
Histone covalent modification, also dubbed the histone code, is an important determinant of gene activity. Histones are subjected to a wide range of specific posttranslational modifications (Table 35-1). These modifications are dynamic and reversible. Histone acetylation and deacetylation are best understood. The surprising discovery that histone acetylase and other enzymatic activities are associated with the coregulators involved in regulation of gene transcription (see Chapter 42) has provided a new concept of gene regulation. Acetylation is known to occur on lysine residues in the amino terminal tails of histone molecules, and has been consistently correlated with transcription, or alternatively transcriptional potential. Histone acetylation reduces the positive charge of these tails and likely contributes to a decrease in the binding affinity of histone for the negatively charged DNA. Such covalent modification of the histones creates new binding sites for additional proteins such as ATP-dependent chromatin remodeling complexes, which contain subunits that carry structural domains that specifically bind to histones that have been subjected to coregulator-deposited PTMs. These complexes can increase accessibility of adjacent DNA sequences by removing nucleosomal histones. Together then coregulators (chromatin modifiers and chromatin remodellers), working in conjunction, can open up gene promoters and regulatory regions, facilitating binding of other trans-factors and RNA polymerase II and GTFs (see Figures 36-10 and 36-11). Histone deacetylation catalyzed by transcriptional corepressors would have the opposite effect. Different proteins with specific acetylase and deacetylase activities are associated with various components of the transcription apparatus. The proteins that catalyze the histone PTMs are sometimes referred to as “code writers” while the proteins that recognize, bind and interpret these histone PTMs are termed “code readers” and the enzymes that remove histone PTMs are called “code erasers”Collectively then, there histone PTMs represent a very dynamic, potentially information-rich source of regulatory information. The exact rules and mechanisms defining the specificity of these various processes are under investigation. Some specific examples are illustrated in Chapter 42. A variety of commercial enterprises are working to develop drugs that specifically alter the ability of the proteins that modulate the histone code.
There is evidence that the methylation of deoxycytidine residues, 5MeC, (in the sequence 5′-meCpG-3’) in DNA may effect changes in chromatin so as to preclude its active transcription, as described in Chapter 35. For example, in mouse liver, only the unmethylated ribosomal genes can be expressed, and there is evidence that many animal viruses are not transcribed when their DNA is methylated. Acute demethylation of 5MeC residues in specific regions of steroid hormone inducible genes has been associated with an increased rate of transcription of the gene. However, it is not yet possible to generalize that methylated DNA is transcriptionally inactive, that all inactive chromatin is methylated, or that active DNA is not methylated.
Finally, the binding of specific transcription factors to cognate DNA elements may result in disruption of nucleosomal structure. Many eukaryotic genes have multiple protein-binding DNA elements. The serial binding of transcription factors to these elements—in a combinatorial fashion—may either directly disrupt the structure of the nucleosome, prevent its re-formation, or recruit, via protein-protein interactions, multiprotein coregulator complexes that have the ability to covalently modify and/or remodel nucleosomes. These reactions result in chromatin-level structural changes that in the end increase DNA accessibility to other factors and the transcription machinery (cf. above).
Eukaryotic DNA that is in an “active” region of chromatin can be transcribed. As in prokaryotic cells, a promoter dictates where the RNA polymerase will initiate transcription, but the promoter in mammalian cells (Chapter 36) is more complex. In addition, the trans-acting factors generally come from other chromosomes (and so act in trans), whereas this consideration is moot in the case of the single chromosome-containing prokaryotic cells. Additional complexity is added by elements or factors that enhance or repress transcription, define tissue-specific expression, and modulate the actions of many effector molecules. Finally, recent results suggest that gene activation and repression might occur when particular genes move into or out of different subnuclear compartments or locations.
Epigenetic Mechanisms Contribute Importantly to the Control of Gene Transcription
The molecules and regulatory biology described above contributes importantly to transcriptional regulation. Indeed, in recent years the role of covalent modification of DNA and histone and nonhistone proteins and the newly discovered ncRNAs has received tremendous attention in the field of gene regulation research, particularly through investigation into how such chemical modifications and/or molecules stably alter gene expression patterns without altering the underlying DNA gene sequence. This field of study has been termed epigenetics. As mentioned in Chapter 35, one aspect of these mechanisms, PTMs of histones has been dubbed the histone code or histone epigenetic code. The term “epigenetics” means “above genetics” and refers to the fact that these regulatory mechanisms do not change the underlying, regulated DNA sequence, but rather simply the expression patterns of this DNA. Epigenetic mechanisms play key roles in the establishment, maintenance, and reversibility of transcriptional states. A key feature of epigenetic mechanisms is that the controlled transcriptional on/off states can be maintained through multiple rounds of cell division. This observation indicates that there must be robust mechanisms to maintain and stably propagate these epigenetic states.
Two forms of epigenetic signals, cis- and trans-epigenetic signals, can be described; these are schematically illustrated in Figure 38–8. A simple trans-signaling event composed of positive transcriptional feedback mediated by an abundant, diffusible transactivator that partitions between mother and daughter cell at each division is depicted in Figure 38–8A. As long as the indicated, transcription factor is expressed at a sufficient level to allow all subsequent daughter cells to inherit the trans-epigenetic signal (transcription factor), such cells will have the cellular or molecular phenotype dictated by the other target genes of this transcriptional activator. Shown in Figure 38–8panel B is an example of how a cis-epigenetic signal (such as a specific 5MeCpG methylation mark) can be stably propagated to the two daughter cells following cell division. The hemi-methylated (ie, only one of the two DNA strands is 5MeC modified) DNA mark generated during DNA replication directs the methylation of the newly replicated strand through the action of ubiquitous maintenance DNA methylases. This 5MeC methylation results in both DNA daughter strands having the complete cis-epigenetic mark.
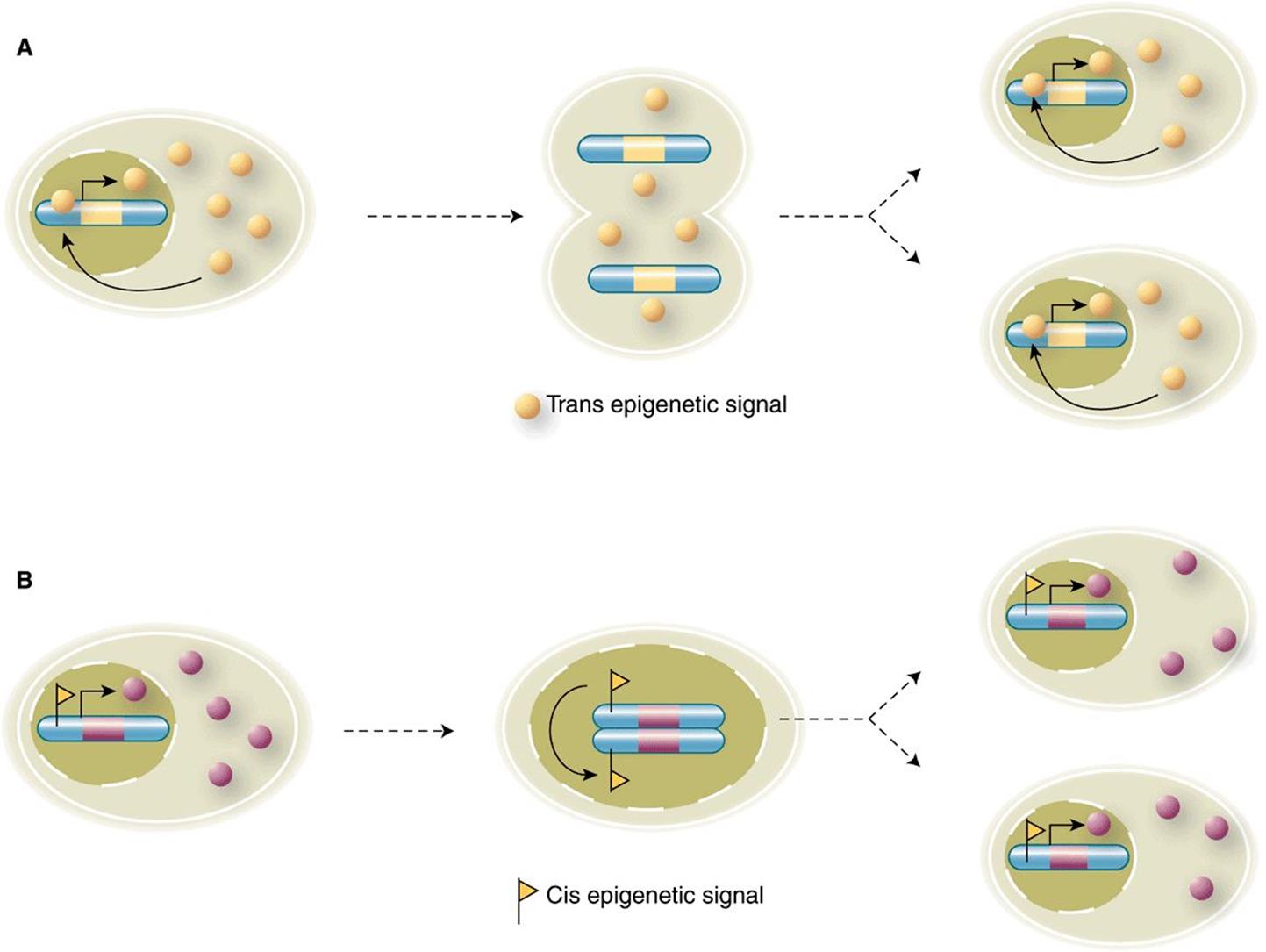
FIGURE 38–8 cis- and trans-epigenetic signals. (A) An example of an epigenetic signal that acts in trans. A DNA binding transactivator protein (yellow circle) is transcribed from its cognate gene (yellow bar) located on a particular chromosome (blue). The expressed protein is freely diffusible between nuclear and cytoplasmic compartments. Note that excess transactivator re-enters the nucleus following cell division, binds to its own gene and activates transcription in both daughter cells. This cycle re-establishes the positive feedback loop in effect prior to cell division, and thereby enforces stable expression of this transcriptional activator protein in both cells. (B) A cis-epigenetic signal; a gene (pink) located on a particular chromosome (blue) carries a cis-epigenetic signal (small yellow flag) within the regulatory region upstream of the pink gene transcription unit. In this case, the epigenetic signal is associated with active gene transcription and subsequent gene product production (pink circles). During DNA replication, the newly replicated chromatid serves as a template that elicits and templates the introduction of the same epigenetic signal, or mark, on the newly synthesized, unmarked chromatid. Consequently, both daughter cells contain the pink gene in a similarly cis-epigenetically marked state, which ensures expression in an identical fashion in both cells. See text for more detail. (Image Taken from: Roberto Bonasio, R, Tu, S, Reinberg D (2010), “Molecular Signals of Epigenetic States”. Science 330:612-616. Reprinted with permission from AAAS.)
Both cis- and trans-epigenetic signals result in stable and hereditable expression states, and therefore represent type C gene expression responses (ie, Figure 38–1). However, it is important to note that both states can be reversed if either the trans- or cis-epigenetic signals are removed by, for example, extinguishing the expression of the enforcing transcription factor (trans-signal) or by removing a DNA cis-epigenetic signal (via DNA demethylation). Enzymes have been described that, at least in vitro, can remove both protein PTMs and 5MeC modifications.
Stable transmission of epigenetic on/off states can be effected by multiple molecular mechanisms. Shown in Figure 38–9 are three ways by which cis-epigenetic marks can be propagated through a round of DNA replication. The first example of epigenetic mark transmission involves the propagation of DNA 5MeC marks, and occurs as described above in Figure 38–8. The second example of epigenetic state transmission illustrates how a nucleosomal histone PTM (in this example, Lysine K-27 trimethylated histone H3; H3K27me3) can be propagated. In this example immediately following DNA replication, both H3K27me3-marked and H3-unmarked nucleosomes randomly reform on both daughter DNA strands. The polycomb repressive complex 2 (PRC2), composed of EED-SUZ12-EZH2 and RbAP subunits, binds to the nucleosome containing the preexisting H3K27me3 mark via the EED subunit. Binding of PRC2 to this Histone mark stimulates the methylase activity of the PRC2 subunit EZH2, which results in the local methylation of nucleosomal H3. Histone H3 methylation thus causes the full, stable transmission of the H3K27me3 epigenetic mark to both chromatids. Finally, locus/sequence-specific targeting of nucleosomal histone epigenetic cis-signals can be attained through the action of ncRNAs as depicted in Figure 38–9, panel C. Here a specific ncRNA interacts with target DNA sequences and the resulting RNA-DNA complex is recognized by RBP, an RNA-binding protein. Then, likely through a specific adaptor protein (A), the RNA-DNA-RBP complex recruits a chromatin modifying complex (CMC) that locally modifies nucleosomal histones. Again, this mechanism leads to the transmission of a stable epigenetic mark.
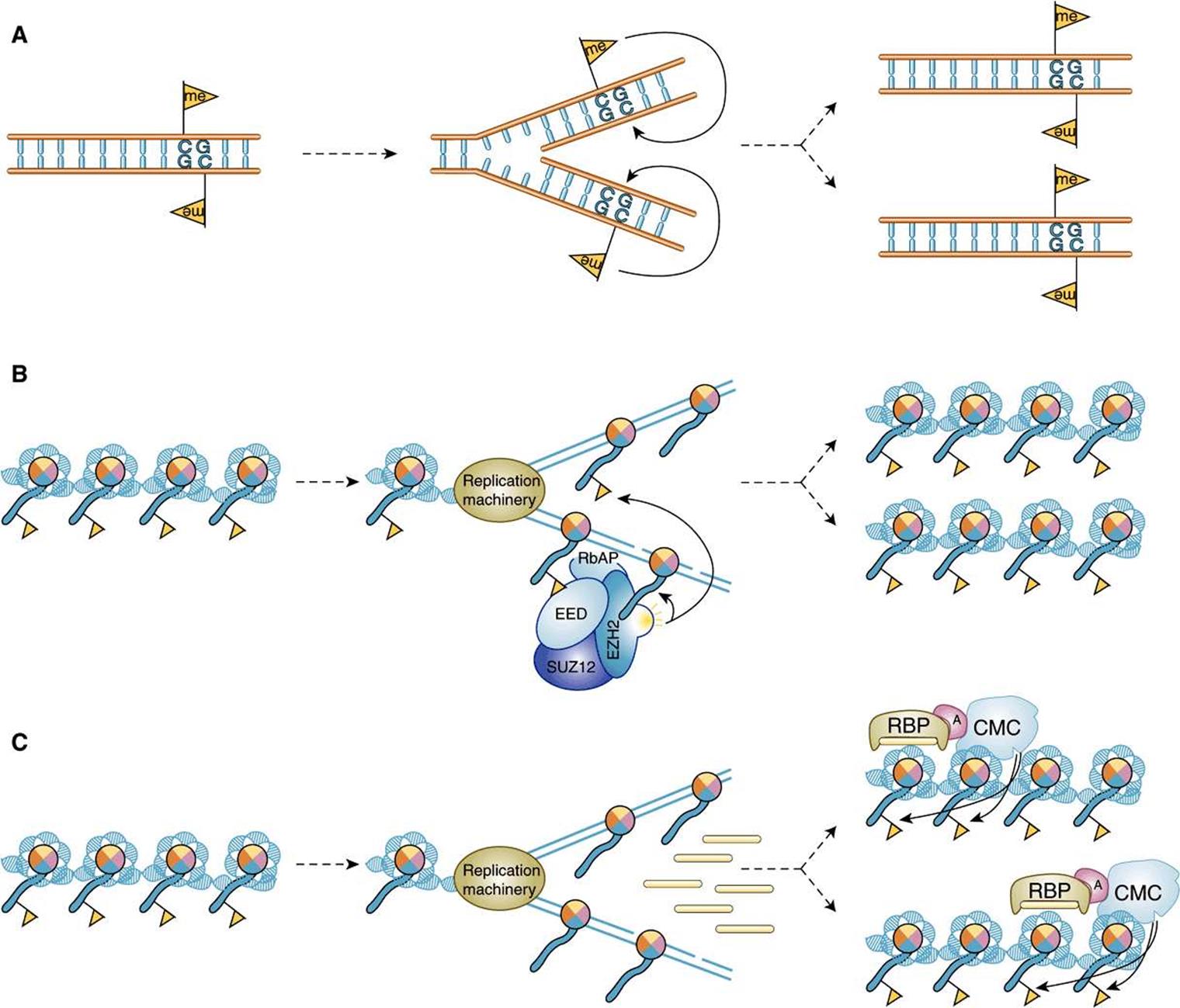
FIGURE 38–9 Mechanisms for the transmission and propagation of epigenetic signals following a round of DNA replication. (A) Propagation of a 5MeC signal (yellow flag; see Figure 38–8B). (B) Propagation of a histone PTM mark epigenetic signal (H3K27me) that is mediated through the action of the PRC2 CMC, a four subunit protein composed of EED, EZH2 histone methylase, RbAP and SUZ12. Note that in this context PRC2 is a both a histone code reader (via the methylated histone binding domain in EED) and histone code writer (via the SET domain histone methylase within EZH2). Location-specific deposition of the histone PTM cis-epigenetic signal is targeted by the recognition of the H3K27me marks in preexisting nucleosomal histones (yellow flag). (C) Another example of the transmission of a histone epigenetic signal (yellow flag) except here signal-targeting is mediated through the action of small ncRNAs, which work in concert with an RNA-binding protein (RBP), an Adaptor (A) protein, and a CMC. See text for more detail. (Image Taken from: Roberto Bonasio, R, Tu, S, Reinberg D (2010), “Molecular Signals of Epigenetic States”. Science 330:612-616. Reprinted with permission from AAAS.)
Additional work will be required to establish the complete molecular details of these epigenetic processes, determine how ubiquitously these mechanisms operate, identify the full complement of molecules involved, and genes controlled. Epigenetic signals are critically important to gene regulation as evidenced by the fact that mutations and/or overexpression of many of the molecules that contribute to epigenetic control lead to human disease.
Certain DNA Elements Enhance or Repress Transcription of Eukaryotic Genes
In addition to gross changes in chromatin affecting transcriptional activity, certain DNA elements facilitate or enhance initiation at the promoter and hence are termed enhancers. Enhancer elements, which typically contain multiple binding sites for transactivator proteins, differ from the promoter in notable ways. They can exert their positive influence on transcription even when separated by tens of thousands of base pairs from a promoter; they work when oriented in either direction; and they can work upstream (5’) or downstream (3’) from the promoter. Enhancers are promiscuous; they can stimulate any promoter in the vicinity and may act on more than one promoter. The viral SV40 enhancer can exert an influence on, for example, the transcription of β-globin by increasing its transcription 200-fold in cells containing both the SV40 enhancer and the β-globin gene on the same plasmid (see below and Figure 38–10); in this case the SV40 enhancer β-globin gene was constructed using recombinant DNA technology—see Chapter 39. The enhancer element does not produce a product that in turn acts on the promoter, since it is active only when it exists within the same DNA molecule as (ie, cis to) the promoter. Enhancer-binding proteins are responsible for this effect. The exact mechanisms by which these transcription activators work are subject to intensive investigation. Certainly, enhancer-binding trans-factors have been shown to interact with a plethora of other transcription proteins. These interactions include chromatin-modifying coactivators, mediator, as well as the individual components of the basal RNA polymerase II transcription machinery. Ultimately, transfactor-enhancer DNA-binding events result in an increase in the binding of the basal transcription machinery to the promoter. Enhancer elements and associated binding proteins often convey nuclease hypersensitivity to those regions where they reside (Chapter 35). A summary of the properties of enhancers is presented in Table 38-2.
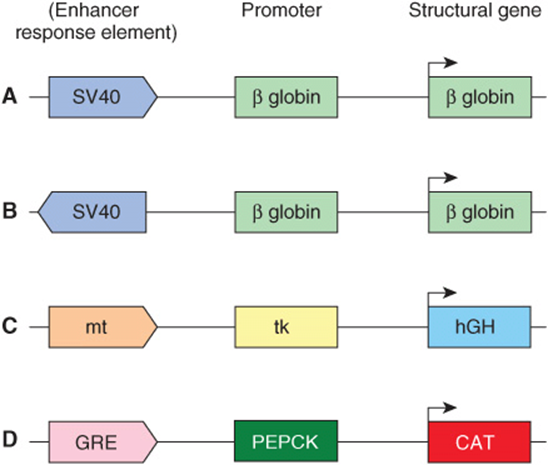
FIGURE 38–10 A schematic illustrating the action of enhancers and other cis-acting regulatory elements. These model chimeric genes, all constructed by recombinant DNA techniques (Chapter 39) in vitro, consist of a reporter (structural) gene that encodes a protein that can be readily assayed, and that is not normally produced in the cells to be studied, a promoter that ensures accurate initiation of transcription, and the indicated regulatory elements. In all cases, high-level transcription from the indicated chimeras depends upon the presence of enhancers, which stimulate transcription ≥ 100-fold over basal transcriptional levels (ie, transcription of the same chimeric genes containing just promoters fused to the structural genes). Examples (A) and (B) illustrate the fact that enhancers (eg, SV40) work in either orientation and upon a heterologous promoter. Example (C) illustrates that the metallothionein (mt) regulatory element (which under the influence of cadmium or zinc induces transcription of the endogenous mt gene and hence the metal-binding mt protein) will work through the thymidine kinase (tk) promoter to enhance transcription of the human growth hormone (hGH) gene. The engineered genetic constructions were introduced into the male pronuclei of single-cell mouse embryos and the embryos placed into the uterus of a surrogate mother to develop as transgenic animals. Offspring have been generated under these conditions, and in some the addition of zinc ions to their drinking water effects an increase in growth hormone expression in liver. In this case, these transgenic animals have responded to the high levels of growth hormone by becoming twice as large as their normal litter mates. Example (D) illustrates that a glucocorticoid response element (GRE) will work through homologous (PEPCK gene) or heterologous promoters (not shown; ie, tk) promoter, SV40 promoter, β-globin promoter, etc) to drive expression of the chloramphenicol acetyl transferase (CAT) reporter gene.
TABLE 38–2 Summary of the Properties of Enhancers

One of the best-understood mammalian enhancer systems is that of the β-interferon gene. This gene is induced upon viral infection of mammalian cells. One goal of the cell, once virally infected, is to attempt to mount an antiviral response—if not to save the infected cell, then to help to save the entire organism from viral infection. Interferon production is one mechanism by which this is accomplished. This family of proteins is secreted by virally infected cells. Secreted interferon interacts with neighboring cells to cause an inhibition of viral replication by a variety of mechanisms, thereby limiting the extent of viral infection. The enhancer element controlling induction of the β-interferon gene, which is located between nucleotides –110 and –45 relative to the transcription start site (+1), is well characterized. This enhancer is composed of four distinct clustered cis-elements, each of which is bound by unique trans-factors. One cis-element is bound by the transacting factor NF-KB, one by a member of the IRF (interferon regulatory factor) family of trans-factors, and a third by the heterodimeric leucine zipper factor ATF-2/c-Jun (see below). The fourth factor is the ubiquitous, abundant architectural transcription factor known as HMG I(Y). Upon binding to its A+T-rich binding sites, HMG I(Y) induces a significant bend in the DNA. There are four such HMG I(Y) binding sites interspersed throughout the enhancer. These sites play a critical role in forming a particular 3D structure, along with the aforementioned three trans-factors, by inducing a series of critically spaced DNA bends. Consequently, HMG I(Y) induces the cooperative formation of a unique, stereospecific, 3D structure within which all four factors are active when viral infection signals are sensed by the cell. The structure formed by the cooperative assembly of these four factors is termed the β-interferon enhanceosome (see Figure 38–11), so named because of its obvious structural similarity to the nucleosome, also a unique three-dimensional protein-DNA structure that wraps DNA about an assembly of proteins (see Figures 35-1 and 35-2). The enhanceosome, once formed, induces a large increase in β-interferon gene transcription upon virus infection. It is not simply the protein occupancy of the linearly apposed cis-element sites that induces β-interferon gene transcription—rather, it is the formation of the enhanceosome proper that provides appropriate surfaces for the recruitment of coactivators that results in the enhanced formation of the PIC on the cis-linked promoter and thus transcription activation.
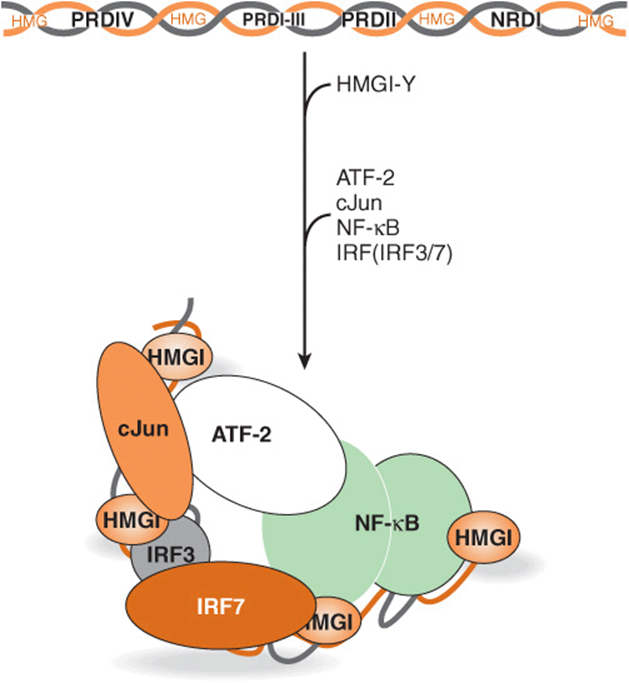
FIGURE 38–11 Formation and putative structure of the enhanceosome formed on the human β-interferon gene enhancer. Diagrammatically represented at the top is the distribution of the multiple cis-elements (HMG, PRDIV, PRDI-III, PRDII, NRDI) composing the β-interferon gene enhancer. The intact enhancer mediates transcriptional induction of the β-interferon gene (over 100-fold) upon virus infection of human cells. The cis-elements of this modular enhancer represent the binding sites for the trans-factors HMG I(Y), cJun-ATF-2, IRF3-IRF7, and NF-KB, respectively. The factors interact with these DNA elements in an obligatory, ordered, and highly cooperative fashion as indicated by the arrow. Initial binding of four HMG I(Y) proteins induces sharp DNA bends in the enhancer, causing the entire 70-80 bp region to assume a high level of curvature. This curvature is integral to the subsequent highly cooperative binding of the other trans-factors since this enables the DNA-bound factors to make important, direct protein-protein interactions that both contribute to the formation and stability of the enhanceosome and generate a unique 3D surface that serves to recruit chromatin-modifying coregulators that carry enzymatic activities (eg, Swi/Snf: ATPase, chromatin remodeler and P/CAF: histone acetyltransferase) as well as the general transcription machinery (RNA polymerase II and GTFs). Although four of the five cis-elements (PRDIV, PRDI-III, PRDII, NRDI) independently can modestly stimulate (~10-fold) transcription of a reporter gene in transfected cells (see Figures 38-10 and 38-12), all five cis-elements, in appropriate order, are required to form an enhancer that can appropriately stimulate mRNA gene transcription (ie, ≥100-fold) in response to viral infection of a human cell. This distinction indicates the strict requirement for appropriate enhanceosome architecture for efficient trans-activation. Similar enhanceosomes, involving distinct cis- and trans-factors and coregulators, are proposed to form on many other mammalian genes.
The cis-acting elements that decrease or repress the expression of specific genes have also been identified. Because fewer of these elements have been studied, it is not possible to formulate generalizations about their mechanism of action—though again, as for gene activation, chromatin level covalent modifications of histones and other proteins by (repressor)-recruited multisubunit corepressors have been implicated.
Tissue-Specific Expression May Result From Either the Action of Enhancers or Repressors or a Combination of Both Cis-Acting Regulatory Elements
Many genes are now recognized to harbor enhancer or activator elements in various locations relative to their coding regions. In addition to being able to enhance gene transcription, some of these enhancer elements clearly possess the ability to do so in a tissue-specific manner. Thus, the enhancer element associated with the immunoglobulin genes between the J and C regions enhances the expression of those genes preferentially in lymphoid cells. Similarly by fusing known or suspected tissue-specific enhancers to reporter genes (see below) and introducing these chimeric enhancer-reporter constructs microsurgically into single-cell embryo, one can create a transgenic animal (see Chapter 39), and rigorously test whether a given test enhancer truly drives expression in a cell-or tissue-specific fashion. This transgenic animal approach has proved useful in studying tissue-specific gene expression.
Reporter Genes Are Used to Define Enhancers & Other Regulatory Elements
By ligating regions of DNA suspected of harboring regulatory sequences to various reporter genes (the reporter or chimeric gene approach) (Figures 38-10, 38-12, & 38-13), one can determine which regions in the vicinity of structural genes have an influence on their expression. Pieces of DNA thought to harbor regulatory elements are ligated to a suitable reporter gene and introduced into a host cell (Figure 38–12). Basal expression of the reporter gene will be increased if the DNA contains an enhancer. Addition of a hormone or heavy metal to the culture medium will increase expression of the reporter gene if the DNA contains a hormone or metal response element (Figure 38–13). The location of the element can be pinpointed by using progressively shorter pieces of DNA, deletions, or point mutations (Figure 38–13).
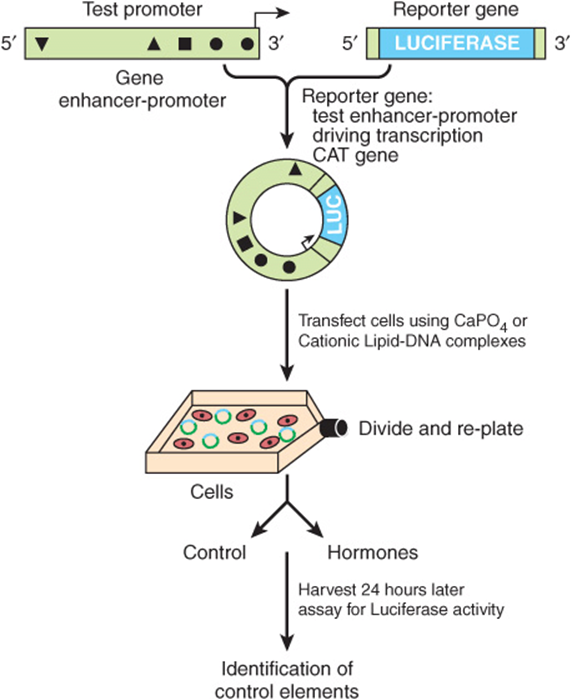
FIGURE 38–12 The use of reporter genes to define DNA regulatory elements. A DNA fragment bearing regulatory cis-elements (triangles, square, circles in diagram) from the gene in question—in this example, approximately 2 kb of 5′-flanking DNA and cognate promoter—is ligated into a plasmid vector that contains a suitable reporter gene—in this case, the enzyme firefly luciferase, abbreviated LUC. Whatever reporter gene is utilized in these experiments, the reporter can not be present in the transfected cells. Consequently, any detection of these activities in a cell extract means that the cell was successfully transfected by the plasmid. Not shown here, but typically one cotransfects an additional reporter such as Renilla luciferase to serve as a transfection efficiency control. Assay conditions for the firefly and Renilla luciferases are different, hence the two activities can be sequentially assayed using the same cell extract. An increase of firefly luciferase activity over the basal level, for example, after addition of one or more hormones, means that the region of DNA inserted into the reporter gene plasmid contains functional hormone response elements (HRE). Progressively shorter pieces of DNA, regions with internal deletions, or regions with point mutations can be constructed and inserted to pinpoint the response element (see Figure 38–13 for deletion mapping of the relevant HREs).
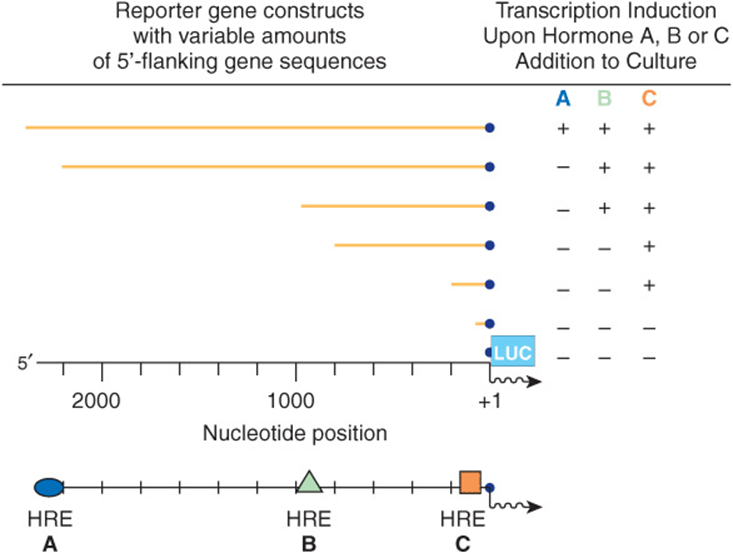
FIGURE 38–13 Mapping hormone response elements (HREs) (A), (B), and (C) using the reporter gene-transfection approach. A family of reporter genes, constructed as described in Figure 38–10, can be transfected individually into a recipient cell. By analyzing when certain hormone responses are lost in comparison to the 5′ deletion end point, specific hormone-responsive elements can be located.
This strategy, typically using transfected cells in culture (ie, cells induced to take up exogenous DNAs), has led to the identification of hundreds of enhancers, repressors, tissue-specific elements, and hormone, heavy metal, and drug-response elements. The activity of a gene at any moment reflects the interaction of these numerous cis-acting DNA elements with their respective trans-acting factors. Overall, transcriptional output is determined by the balance of positive and negative signaling to the transcription machinery. The challenge now is to figure out how this occurs at the molecular level.
Combinations of DNA Elements & Associated Proteins Provide Diversity in Responses
Prokaryotic genes are often regulated in an on-off manner in response to simple environmental cues. Some eukaryotic genes are regulated in the simple on-off manner, but the process in most genes, especially in mammals, is much more complicated. Signals representing a number of complex environmental stimuli may converge on a single gene. The response of the gene to these signals can have several physiologic characteristics. First, the response may extend over a considerable range. This is accomplished by having additive and synergistic positive responses counterbalanced by negative or repressing effects. In some cases, either the positive or the negative response can be dominant. Also required is a mechanism whereby an effector such as a hormone can activate some genes in a cell while repressing others and leaving still others unaffected. When all of these processes are coupled with tissue-specific element factors, considerable flexibility is afforded. These physiologic variables obviously require an arrangement much more complicated than an on-off switch. The array of DNA elements in a promoter specifies—with associated factors—how a given gene will respond and how long a particular response is maintained. Some simple examples are illustrated in Figure 38–14.
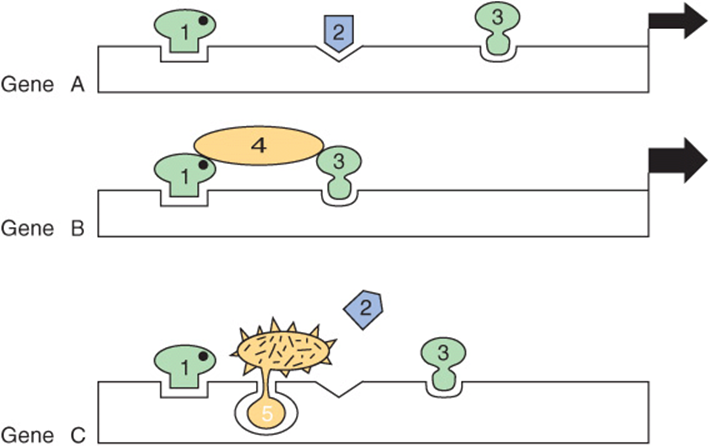
FIGURE 38–14 Combinations of DNA elements and proteins provide diversity in the response of a gene. Gene A is activated (the width of the arrow indicates the extent) by the combination of transcripitonal activator proteins 1, 2, and 3 (probably with coactivators, as shown in Figure 36–10). Gene B is activated, in this case more effectively, by the combination of one, three, and four; note that transcription factor 4 does not contact DNA directly in this example. The activators could form a linear bridge that links the basal machinery to the promoter, or this could be accomplished by looping out of the DNA. In either case, the purpose is to direct the basal transcription machinery to the promoter. Gene C is inactivated by the combination of transcription factors 1, 5, and 3; in this case, factor 5 is shown to preclude the essential binding of factor 2 to DNA, as occurs in example A. If activator 1 helps repressor 5 bind and if activator 1 binding requires a ligand (solid dot), it can be seen how the ligand could activate one gene in a cell (gene A) and repress another (gene C) in the same cell.
Transcription Domains Can Be Defined by Locus Control Regions & Insulators
The large number of genes in eukaryotic cells and the complex arrays of transcription regulatory factors present an organizational problem. Why are some genes available for transcription in a given cell whereas others are not? If enhancers can regulate several genes from tens of kilobase distances and are not position- and orientation-dependent, how are they prevented from triggering transcription of all cis-linked genes in the vicinity? Part of the solution to these problems is arrived at by having the chromatin arranged in functional units that restrict patterns of gene expression. This may be achieved by having the chromatin form a structure with the nuclear matrix or other physical entity, or compartment within the nucleus. Alternatively, some regions are controlled by complex DNA elements called locus control regions (LCRs). An LCR—with associated bound proteins—controls the expression of a cluster of genes. The best-defined LCR regulates expression of the globin gene family over a large region of DNA. Another mechanism is provided by insulators. These DNA elements, also in association with one or more proteins, prevent an enhancer from acting on a promoter on the other side of an insulator in another transcription domain. Insulators thus serve as transcriptional boundary elements.
SEVERAL MOTIFS COMPOSE THE DNA BINDING DOMAINS OF REGULATORY TRANSCRIPTION FACTOR PROTEINS
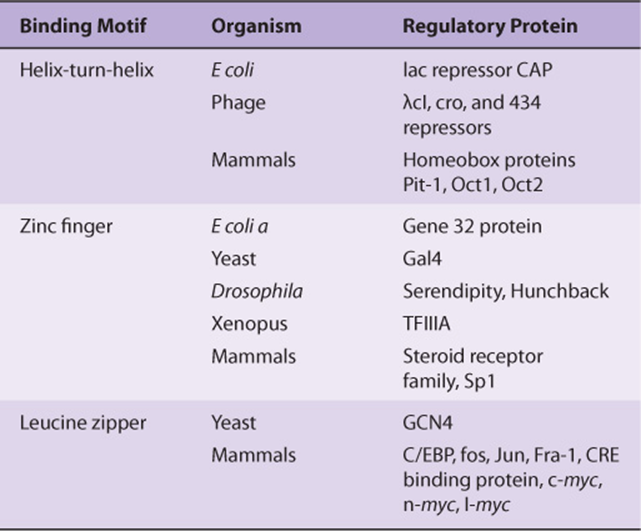
The specificity involved in the control of transcription requires that regulatory proteins bind with high affinity and specificity to the correct region of DNA. Three unique motifs—the helix-turn-helix, the zinc finger, and the leucine zipper— account for many of these specific protein-DNA interactions. Examples of proteins containing these motifs are given in Table 38-3.
TABLE 38–3 Examples of Transcription Factors That Contain Various DNA Binding Motifs
Comparison of the binding activities of the proteins that contain these motifs leads to several important generalizations.
1. Binding must be of high affinity to the specific site and of low affinity to other DNA.
2. Small regions of the protein make direct contact with DNA; the rest of the protein, in addition to providing the trans-activation domains, may be involved in the dimerization of monomers of the binding protein, may provide a contact surface for the formation of heterodimers, may provide one or more ligand-binding sites, or may provide surfaces for interaction with coactivators or corepressors.
3. The protein-DNA interactions are maintained by hydrogen bonds, ionic interactions and van der Waals forces.
4. The motifs found in these proteins are unique; their presence in a protein of unknown function suggests that the protein may bind to DNA.
5. Proteins with the helix-turn-helix or leucine zipper motifs form dimers, and their respective DNA-binding sites are symmetric palindromes. In proteins with the zinc finger motif, the binding site is repeated two to nine times. These features allow for cooperative interactions between binding sites and enhance the degree and affinity of binding.
The Helix-Turn-Helix Motif
The first motif described was the helix-turn-helix. Analysis of the 3D structure of the lambda Cro transcription regulator has revealed that each monomer consists of three antiparallel β sheets and three α helices (Figure 38–15).The dimer forms by association of the antiparallel β3 sheets. The α3 helices form the DNA recognition surface, and the rest of the molecule appears to be involved in stabilizing these structures. The average diameter of an α helix is 1.2 nm, which is the approximate width of the major groove in the B form of DNA.
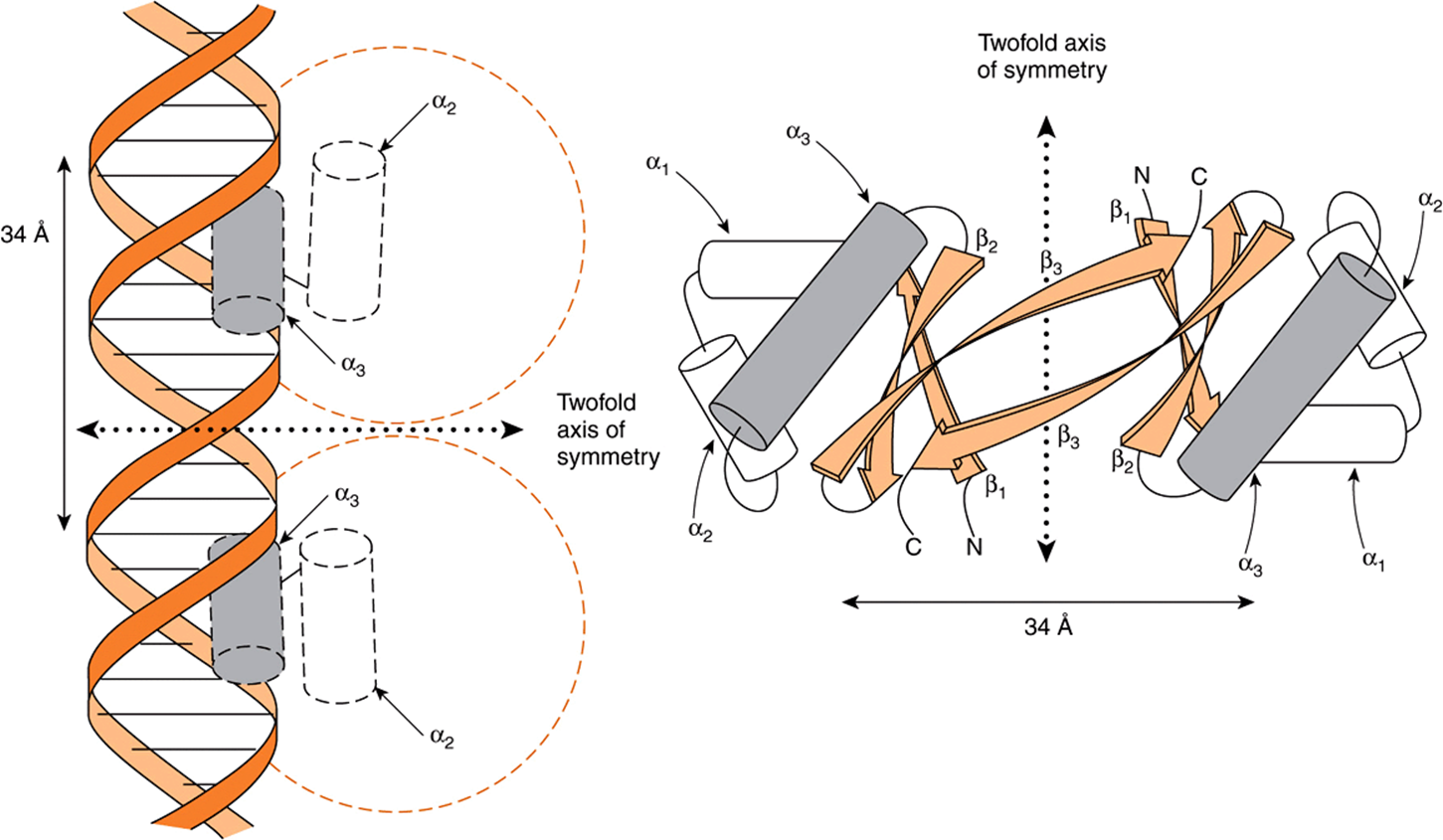
FIGURE 38–15 A schematic representation of the 3D structure of Cro protein and its binding to DNA by its helix-turn-helix motif (left). The Cro monomer consists of three antiparallel β sheets (β1-β3) and three α-helices (α1-α3). The helix-turn-helix motif is formed because the α3 and α2 helices are held at about 90 degrees to each other by a turn of four amino acids. The α3 helix of Cro is the DNA recognition surface (shaded). Two monomers associate through the antiparallel β3 sheets to form a dimer that has a twofold axis of symmetry (right). A Cro dimer binds to DNA through its α3 helices, each of which contacts about 5 bp on the same surface of the major groove (see Figure 38–6). The distance between comparable points on the two DNA α-helices is 34 Å, which is the distance required for one complete turn of the double helix. (Courtesy of B Mathews.)
The DNA recognition domain of each Cro monomer interacts with 5 bp and the dimer binding sites span 3.4 nm, allowing fit into successive half turns of the major groove on the same surface (Figure 38–15). X-ray analyses of the λ cI repressor, CAP (the cAMP receptor protein of E. coli), tryptophan repressor, and phage 434 repressor, all also display this dimeric helix-turn-helix structure that is present in eukaryotic DNA-binding proteins as well (see Table 38-3).
The Zinc Finger Motif
The zinc finger was the second DNA binding motif whose atomic structure was elucidated. It was known that the protein TFIIIA, a positive regulator of 5S RNA gene transcription, required zinc for activity. Structural and biophysical analyses revealed that each TFIIIA molecule contains nine zinc ions in a repeating coordination complex formed by closely spaced cysteine-cysteine residues followed 12-13 amino acids later by a histidine-histidine pair (Figure 38–16). In some instances—notably the steroid-thyroid nuclear hormone receptor family—the His-His doublet is replaced by a second Cys-Cys pair. The protein containing zinc fingers appears to lie on one face of the DNA helix, with successive fingers alternatively positioned in one turn in the major groove. As is the case with the recognition domain in the helix-turn-helix protein, each TFIIIA zinc finger contacts about 5 bp of DNA. The importance of this motif in the action of steroid hormones is underscored by an “experiment of nature.” A single amino acid mutation in either of the two zinc fingers of the 1,25(OH)2-D3 receptor protein results in resistance to the action of this hormone and the clinical syndrome of rickets.
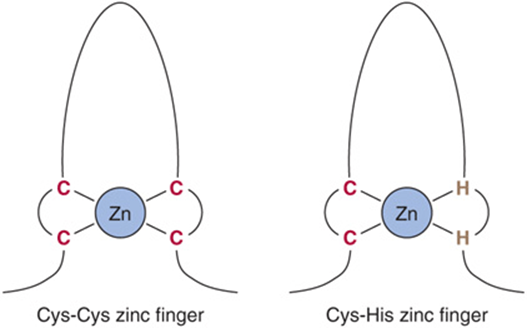
FIGURE 38–16 Zinc fingers are a series of repeated domains (two to nine) in which each is centered on a tetrahedral coordination with zinc. In the case of TFIIIA, the coordination is provided by a pair of cysteine residues (C) separated by 12-13 amino acids from a pair of histidine (H) residues. In other zinc finger proteins, the second pair also consists of C residues. Zinc fingers bind in the major groove, with adjacent fingers making contact with 5 bp along the same face of the helix.
The Leucine Zipper Motif
Careful analysis of a 30-amino-acid sequence in the carboxyl terminal region of the enhancer binding protein C/EBP revealed a novel structure, the leucine zipper motif. As illustrated in Figure 38–17, this region of the protein forms an α helix in which there is a periodic repeat of leucine residues at every seventh position. This occurs for eight helical turns and four leucine repeats. Similar structures have been found in a number of other proteins associated with the regulation of transcription in mammalian and yeast cells. This structure allows two identical or nonidentical monomers (eg, Jun-Jun or Fos-Jun) to “zip together” in a coiled coil and form a tight dimeric complex (Figure 38–17). This protein-protein interaction may serve to enhance the association of the separate DBDs with their target (Figure 38–17).
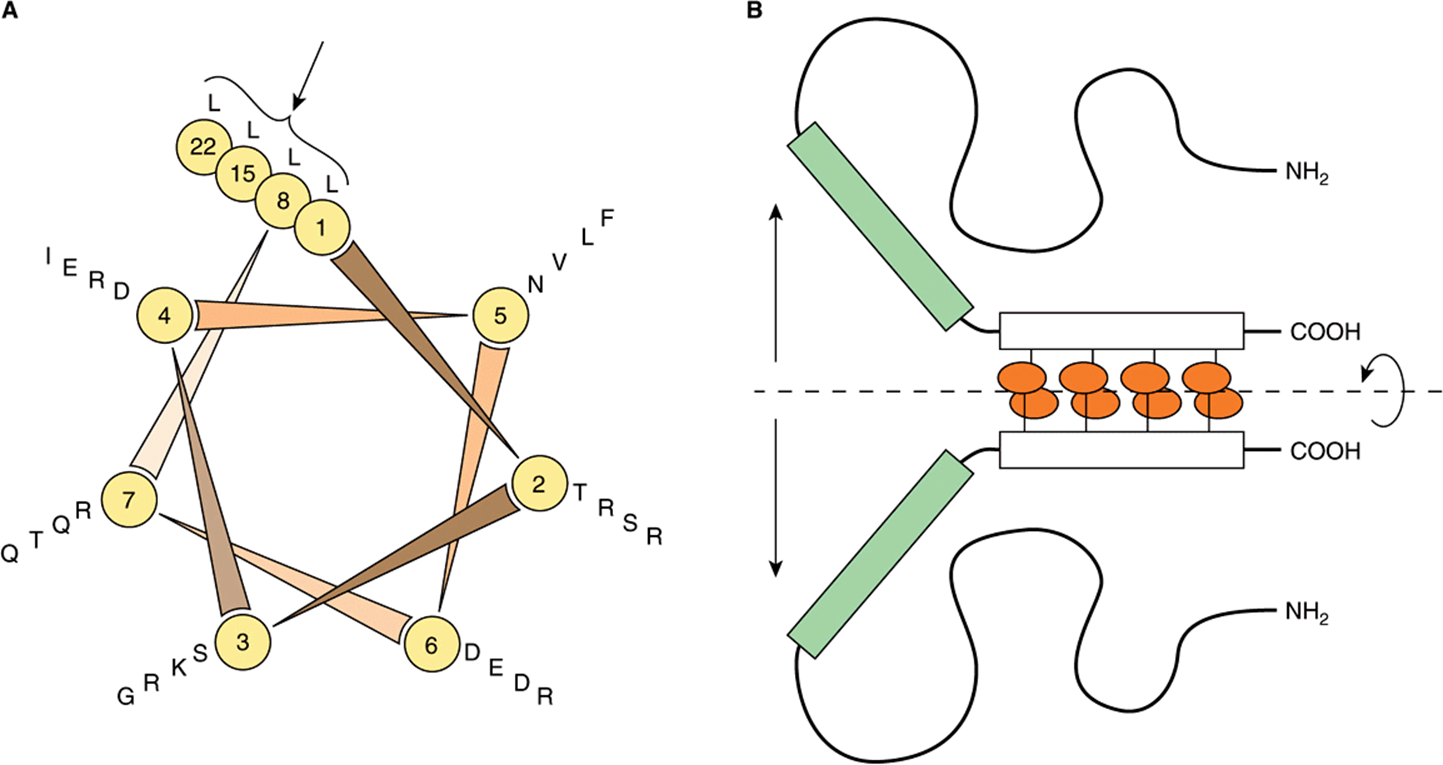
FIGURE 38–17 The leucine zipper motif. (A) It shows a helical wheel analysis of a carboxyl terminal portion of the DNA binding protein C/EBP. The amino acid sequence is displayed end-to-end down the axis of a schematic α-helix. The helical wheel consists of seven spokes that correspond to the seven amino acids that comprise every two turns of the α-helix. Note that leucine residues (L) occur at every seventh position (in this schematic C/EBP amino acid residues 1, 8, 15, 22; see arrow). Other proteins with “leucine zippers” have a similar helical wheel pattern. (B) It is a schematic model of the DNA-binding domain of C/EBP. Two identical C/EBP polypeptide chains are held in dimer formation by the leucine zipper domain of each polypeptide (denoted by the rectangles and attached ovals). This association is required to hold the DNA binding domains of each polypeptide (the shaded rectangles) in the proper conformation for DNA binding. (Courtesy of S McKnight.)
THE DNA BINDING & TRANSACTIVATION DOMAINS OF MOST REGULATORY PROTEINS ARE SEPARATE
DNA binding could result in a general conformational change that allows the bound protein to activate transcription, or these two functions could be served by separate and independent domains. Domain swap experiments suggest that the latter is typically the case.
The GAL1 gene product is involved in galactose metabolism in yeast. Transcription of this gene is positively regulated by the GAL4 protein, which binds to an upstream activator sequence (UAS), or enhancer, through an amino terminal domain. The amino terminal 73-amino-acid DBD of GAL4 was removed and replaced with the DBD of LexA, an E coli DNA-binding protein. This domain swap resulted in a molecule that did not bind to the GAL1 UAS and, of course, did not activate the GAL1 gene (Figure 38–18). If, however, the lexA operator—the DNA sequence normally bound by the lexADBD— was inserted into the promoter region of the GAL gene thereby replacing the normal GAL1 enhancer, the hybrid protein bound to this promoter (at the lexA operator) and it activated transcription of GAL1. This experiment, which has been repeated a number of times, affords solid evidence that the carboxyl terminal region of GAL4 causes transcriptional activation. These data also demonstrate that the DBD and transactivation domains (ADs) are independent and noninteractive. The hierarchy involved in assembling gene transcription-activating complexes includes proteins that bind DNA and transactivate; others that form protein-protein complexes which bridge DNA-binding proteins to transactivating proteins; and others that form protein-protein complexes with components of coregulators or the basal transcription apparatus. A given protein may thus have several modular surfaces or domains that serve different functions (see Figure 38–19). As described in Chapter 36, the primary purpose of these complex assemblies is to facilitate the assembly and/or activity of the basal transcription apparatus on the cis-linked promoter.
FIGURE 38–18 Domain-swap experiments demonstrate the independent nature of DNA binding and transcription activation domains. The GAL1 gene promoter contains an upstream activating sequence (UAS) or enhancer that is bound by the regulatory transcription factor GAL4 (A). GAL4, like the lambda cI protein is modular, and contains an N-terminal DBD and an C-terminal activation domain, or AD. When the GAL4 transcription factor binds the GAL1 enhancer/UAS, activation of GAL1 gene transcription ensues (Active). A chimeric protein, in which the amino terminal DNA-binding domain (DBD) of GAL4 is removed and replaced with the DBD of the E. coli protein LexA (LexA DBD-GAL4 AD), fails to stimulate GAL1 transcription because the LexA DBD cannot bind to the GAL1 enhancer/UAS (B). By contrast, the LexA DBD-GAL4 AD fusion protein does increase GAL1 transcription when the lexA operator (the natural target for the LexA DBD) is inserted into the GAL1 promoter region (C), replacing the normal GAL1 UAS.
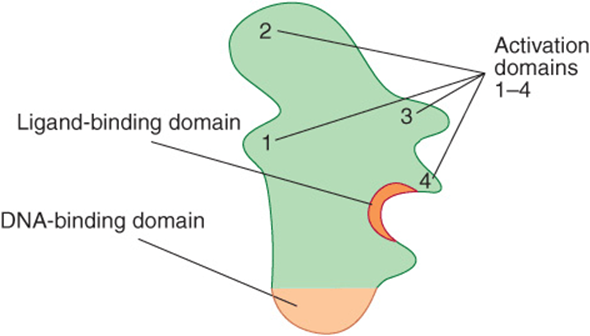
FIGURE 38–19 Proteins that regulate transcription have several domains. This hypothetical transcription factor has a DBD that is distinct from a ligand-binding domain (LBD) and several activation domains (ADs) (1-4). Other proteins may lack the DBD or LBD and all may have variable numbers of domains that contact other proteins, including coregulators and those of the basal transcription complex (see also Chapters 41 & 42).
GENE REGULATION IN PROKARYOTES & EUKARYOTES DIFFERS IN IMPORTANT RESPECTS
In addition to transcription, eukaryotic cells employ a variety of mechanisms to regulate gene expression (Table 38-4). The nuclear membrane of eukaryotic cells physically segregates gene transcription from translation, since ribosomes exist only in the cytoplasm. Many more steps, especially in RNA processing, are involved in the expression of eukaryotic genes than of prokaryotic genes, and these steps provide additional sites for regulatory influences that cannot exist in prokaryotes. These RNA processing steps in eukaryotes, described in detail in Chapter 36, include capping of the 5′ ends of the primary transcripts, addition of a polyadenylate tail to the 3′ ends of transcripts, and excision of intron regions to generate spliced exons in the mature mRNA molecule. To date, analyses of eukaryotic gene expression provide evidence that regulation occurs at the level of transcription, nuclear RNA processing, mRNA stability, and translation. In addition, gene amplification and rearrangement influence gene expression.
TABLE 38–4 Gene Expression is Regulated by Transcription and in Numerous Other Ways at the RNA Level in Eukaryotic Cells
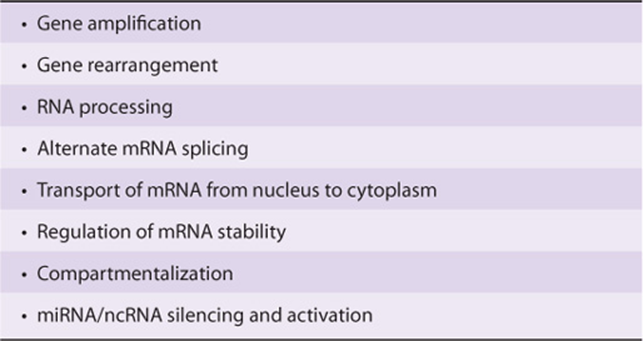
Owing to the advent of recombinant DNA technology, much progress has been made in recent years in the understanding of eukaryotic gene expression. However, because most eukaryotic organisms contain so much more genetic information than do prokaryotes and because manipulation of their genes is so much more difficult, molecular aspects of eukaryotic gene regulation are less well understood than the examples discussed earlier in this chapter. This section briefly describes a few different types of eukaryotic gene regulation.
miRNAs Modulate Gene Expression by Altering mRNA Function
As noted in Chapter 35 the recently discovered class of eukaryotic small RNAs, termed miRNAs, contribute importantly to the control of gene expression. These ~22 nucleotide RNAs regulate the translatability of specific mRNAs by either inhibiting translation or inducing mRNA degradation, though in a few cases miRNAs have been shown to stimulate mRNA function (translation). At least a portion of the miRNA-driven modulation of mRNA activity is thought to occur in the P body (Figure 37–11). miRNA action can result in dramatic changes in protein production and hence gene expression. miRNAs have been implicated in numerous human diseases such as heart disease, cancer, muscle wasting, viral infection and diabetes.
miRNAs, like the DNA-binding transcription factors described in detail above, are transactive, and once synthesized and appropriately processed, interact with specific proteins and bind target mRNAs, typically in 3′ untranslated mRNA regions (Figure 36–17). Binding of miRNAs to mRNA targets is directed by normal base-pairing rules. In general, if miRNA-mRNA base pairing has one or more mismatches, translation of the cognate “target” mRNA is inhibited, whereas if miRNA-mRNA base pairing is perfect over all 22 nucleotides, the corresponding mRNA is degraded.
Given the tremendous and ever growing import of miRNAs, many scientists and biotechnology companies are actively studying miRNA biogenesis, transport, and function in hopes of curing human disease. Time will tell the magnitude and universality of miRNA-mediated gene regulation. It is likely that in the near future scientists will unveil the medical significance of these intriguing small RNAs.
Eukaryotic Genes Can Be Amplified or Rearranged During Development or in Response to Drugs
During early development of metazoans, there is an abrupt increase in the need for specific molecules such as ribosomal RNA and messenger RNA molecules for proteins that make up such organs as the eggshell. One way to increase the rate at which such molecules can be formed is to increase the number of genes available for transcription of these specific molecules. Among the repetitive DNA sequences within the genome are hundreds of copies of ribosomal RNA genes. These genes preexist repetitively in the DNA of the gametes and thus are transmitted in high copy numbers from generation to generation. In some specific organisms such as the fruit fly (drosophila), there occurs during oogenesis an amplification of a few preexisting genes such as those for the chorion (eggshell) proteins. Subsequently, these amplified genes, presumably generated by a process of repeated initiations during DNA synthesis, provide multiple sites for gene transcription (Figures 36-4 and 38-20).
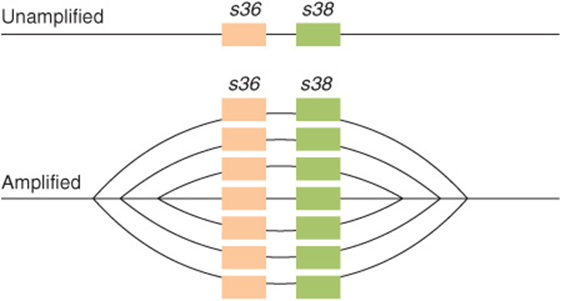
FIGURE 38–20 Schematic representation of the amplification of chorion protein genes s36 and s38. (Reproduced, with permission, from Chisholm R: Gene amplification during development. Trends Biochem Sci 1982;7:161. Copyright © 1982. Reprinted, with permission, from Elsevier.)
As noted in Chapter 36, the coding sequences responsible for the generation of specific protein molecules are frequently not contiguous in the mammalian genome. In the case of antibody encoding genes, this is particularly true. As described in detail in Chapter 50, immunoglobulins are composed of two polypeptides, the so-called heavy (about 50 kDa) and light (about 25 kDa) chains. The mRNAs encoding these two protein subunits are encoded by gene sequences that are subjected to extensive DNA sequence-coding changes. These DNA coding changes are integral to generating the requisite recognition diversity central to appropriate immune function.
IgG heavy and light chain mRNAs are encoded by several different segments that are tandemly repeated in the germline. Thus, for example, the IgG light chain is composed of variable (VL), joining (JL), and constant (CL) domains or segments. For particular subsets of IgG light chains, there are roughly 300 tandemly repeated VL gene coding segments, 5 tandemly arranged JL coding sequences, and roughly 10 CL gene coding segments. All of these multiple, distinct coding regions are located in the same region of the same chromosome, and each type of coding segment (VL, JL, and CL) is tandemly repeated in head-to-tail fashion within the segment repeat region. By having multiple VL, JL, and CL segments to choose from, an immune cell has a greater repertoire of sequences to work with to develop both immunologic flexibility and specificity. However, a given functional IgG light chain transcription unit—like all other “normal” mammalian transcription units—contains only the coding sequences for a single protein. Thus, before a particular IgG light chain can be expressed, single VL, JL, and CL coding sequences must be recombined to generate a single, contiguous transcription unit excluding the multiple nonutilized segments (ie, the other approximately 300 unused VL segments, the other 4 unused JL segments, and the other 9 unused CL segments). This deletion of unused genetic information is accomplished by selective DNA recombination that removes the unwanted coding DNA while retaining the required coding sequences: one VL, one JL, and one CL sequence. (VLsequences are subjected to additional point mutagenesis to generate even more variability—hence the name.) The newly recombined sequences thus form a single transcription unit that is competent for RNA polymerase II-mediated transcription into a single monocistronic mRNA. Although the IgG genes represent one of the best-studied instances of directed DNA rearrangement modulating gene expression, other cases of gene regulatory DNA rearrangement have been described in the literature. Indeed, as detailed below, drug-induced gene amplification is an important complication of cancer chemotherapy.
In recent years, it has been possible to promote the amplification of specific genetic regions in cultured mammalian cells. In some cases, a several 1000-fold increase in the copy number of specific genes can be achieved over a period of time involving increasing doses of selective drugs. In fact, it has been demonstrated in patients receiving methotrexate for cancer that malignant cells can develop drug resistance by increasing the number of genes for dihydrofolate reductase, the target of methotrexate. Gene amplification and deletion events involving 10 to 1,000,000 of bp of DNA such as these occur spontaneously in vivo—ie, in the absence of exogenously supplied selective agents—and these unscheduled extra rounds of replication can become stabilized in the genome under appropriate selective pressures.
Alternative RNA Processing Is Another Control Mechanism
In addition to affecting the efficiency of promoter utilization, eukaryotic cells employ alternative RNA processing to control gene expression. This can result when alternative promoters, intron-exon splice sites, or polyadenylation sites are used. Occasionally, heterogeneity within a cell results, but more commonly the same primary transcript is processed differently in different tissues. A few examples of each of these types of regulation are presented below.
The use of alternative transcription start sites results in a different 5′ exon on mRNAs encoding mouse amylase and myosin light chain, rat glucokinase, and drosophila alcohol dehydrogenase and actin. Alternative polyadenylation sites in the μ immunoglobulin heavy chain primary transcript result in mRNAs that are either 2700 bases long (μm) or 2400 bases long (μs). This results in a different carboxyl terminal region of the encoded proteins such that the μm protein remains attached to the membrane of the B lymphocyte and the μs immunoglobulin is secreted. Alternative splicing and processing results in the formation of seven unique α-tropomyosin mRNAs in seven different tissues. It is not clear how these processing-splicing decisions are made or whether these steps can be regulated.
Regulation of Messenger RNA Stability Provides Another Control Mechanism
Although most mRNAs in mammalian cells are very stable (half-lives measured in hours), some turn over very rapidly (half-lives of 10-30 minutes). In certain instances, mRNA stability is subject to regulation. This has important implications since there is usually a direct relationship between mRNA amount and the translation of that mRNA into its cognate protein. Changes in the stability of a specific mRNA can therefore have major effects on biologic processes.
Messenger RNAs exist in the cytoplasm as ribonucleoprotein particles (RNPs). Some of these proteins protect the mRNA from digestion by nucleases, while others may under certain conditions promote nuclease attack. It is thought that mRNAs are stabilized or destabilized by the interaction of proteins with these various structures or sequences. Certain effectors, such as hormones, may regulate mRNA stability by increasing or decreasing the amount of these proteins.
It appears that the ends of mRNA molecules are involved in mRNA stability (Figure 38–21). The 5′ cap structure in eukaryotic mRNA prevents attack by 5′ exonucleases, and the poly(A) tail prohibits the action of 3′ exonucleases. In mRNA molecules with those structures, it is presumed that a single endonucleolytic cut allows exonucleases to attack and digest the entire molecule. Other structures (sequences) in the 5′ untranslated region (5’ UTR), the coding region, and the 3′ UTR are thought to promote or prevent this initial endonucleolytic action (Figure 38–21). A few illustrative examples will be cited.
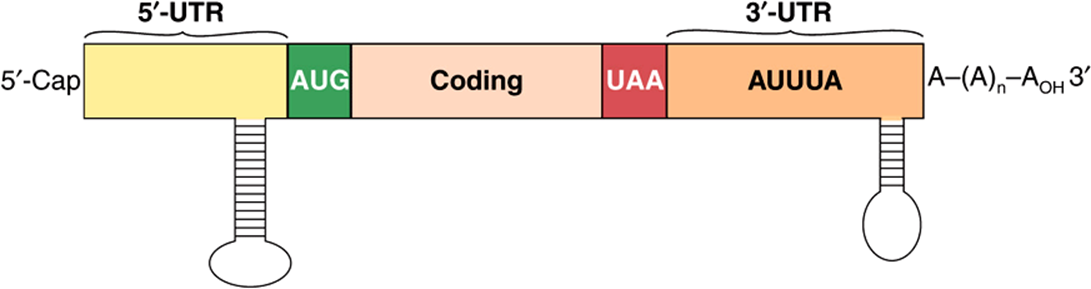
FIGURE 38–21 Structure of a typical eukaryotic mRNA showing elements that are involved in regulating mRNA stability. The typical eukaryotic mRNA has a 5′ noncoding sequence, or untranslated region (5’ UTR), a coding region, and a 3′ untranslated region (3’ UTR). Essentially all mRNAs are capped at the 5′ end, and most have a polyadenylate sequence, 100-200 nucleotides long at their 3′ end. The 5′ cap and 3′ poly(A) tail protect the mRNA against exonuclease attack and are bound by specific proteins that interact to facilitate translation (see Figure 37–7). Stem-loop structures in the 5′ and 3′ NCS, and the AU-rich region in the 3′ NCS are thought to represent the binding sites for specific proteins that modulate mRNA stability. miRNAs typically target sequences in the 3′ UTR.
Deletion of the 5′ UTR results in a threefold to fivefold prolongation of the half-life of c-myc mRNA. Shortening the coding region of histone mRNA results in a prolonged half-life. A form of autoregulation of mRNA stability indirectly involves the coding region. Free tubulin binds to the first four amino acids of a nascent chain of tubulin as it emerges from the ribosome. This appears to activate an RNase associated with the ribosome which then digests the tubulin mRNA.
Structures at the 3′ end, including the poly(A) tail, enhance or diminish the stability of specific mRNAs. The absence of a poly(A) tail is associated with rapid degradation of mRNA, and the removal of poly(A) from some RNAs results in their destabilization. Histone mRNAs lack a poly(A) tail but have a sequence near the 3′ terminal that can form a stem-loop structure, and this appears to provide resistance to exonucleolytic attack. Histone H4 mRNA, for example, is degraded in the 3′-5’ direction but only after a single endonucleolytic cut occurs about nine nucleotides from the 3′ end in the region of the putative stem-loop structure. Stem-loop structures in the 3′ noncoding sequence are also critical for the regulation, by iron, of the mRNA encoding the transferrin receptor. Stem-loop structures are also associated with mRNA stability in bacteria, suggesting that this mechanism may be commonly employed.
Other sequences in the 3′ ends of certain eukaryotic mRNAs appear to be involved in the destabilization of these molecules. Some of this is mediated through the action of specific miRNAs as discussed above. In addition, of particular interest are AU-rich regions, many of which contain the sequence AUUUA. This sequence appears in mRNAs that have a very short half-life, including some encoding oncogene proteins and cytokines. The importance of this region is underscored by an experiment in which a sequence corresponding to the 3′ UTR of the short-half-life colony-stimulating factor (CSF) mRNA, which contains the AUUUA motif, was added to the 3′ end of the β-globin mRNA. Instead of becoming very stable, this hybrid β-globin mRNA now had the short-half-life characteristic of CSF mRNA. Much of this mRNA metabolism likely occurs in cytoplasmic P bodies.
From the few examples cited, it is clear that a number of mechanisms are used to regulate mRNA stability and hence function—just as several mechanisms are used to regulate the synthesis of mRNA. Coordinate regulation of these two processes confers on the cell remarkable adaptability.
SUMMARY
![]() The genetic constitutions of metazoan somatic cells are nearly all identical.
The genetic constitutions of metazoan somatic cells are nearly all identical.
![]() Phenotype (tissue or cell specificity) is dictated by differences in gene expression of this complement of genes.
Phenotype (tissue or cell specificity) is dictated by differences in gene expression of this complement of genes.
![]() Alterations in gene expression allow a cell to adapt to environmental changes, developmental cues, and physiological signals.
Alterations in gene expression allow a cell to adapt to environmental changes, developmental cues, and physiological signals.
![]() Gene expression can be controlled at multiple levels by changes in transcription, RNA processing, localization, and stability or utilization. Gene amplification and rearrangements also influence gene expression.
Gene expression can be controlled at multiple levels by changes in transcription, RNA processing, localization, and stability or utilization. Gene amplification and rearrangements also influence gene expression.
![]() Transcription controls operate at the level of protein-DNA and protein-protein interactions. These interactions display protein domain modularity and high specificity.
Transcription controls operate at the level of protein-DNA and protein-protein interactions. These interactions display protein domain modularity and high specificity.
![]() Several different classes of DNA-binding domains have been identified in transcription factors.
Several different classes of DNA-binding domains have been identified in transcription factors.
![]() Chromatin and DNA modifications contribute importantly in eukaryotic transcription control by modulating DNA accessibility and specifying recruitment of specific coactivators and corepressors to target genes.
Chromatin and DNA modifications contribute importantly in eukaryotic transcription control by modulating DNA accessibility and specifying recruitment of specific coactivators and corepressors to target genes.
![]() Several epigenetic mechanisms for gene control have been described and the molecular mechanisms through which these processes operate are beginning to be elucidated at the molecular level.
Several epigenetic mechanisms for gene control have been described and the molecular mechanisms through which these processes operate are beginning to be elucidated at the molecular level.
![]() miRNA and siRNAs modulate mRNA translation and stability; these mechanisms complement transcription controls to regulate gene expression.
miRNA and siRNAs modulate mRNA translation and stability; these mechanisms complement transcription controls to regulate gene expression.
REFERENCES
Bhaumik SR, Smith E, Shilatifard A, et al: Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol 2007;14;1008.
Bird AP, Wolffe AP: Methylation-induced repression—belts, braces and chromatin. Cell 1999;99:451.
Bonasio R, Tu S, Reinberg D: Molecular signals of epigenetic states. Science 2010;330:612-616.
Busby S, Ebright RH: Promoter structure, promoter recognition, and transcription activation in prokaryotes. Cell 1994;79:743.
Gerstein MB, Lu ZJ, Van Nostrand EL, et al: Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 2010;330:1775-1787.
Jacob F, Monod J: Genetic regulatory mechanisms in protein synthesis. J Mol Biol 1961;3:318.
Klug A: The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem 2010;79:213-231.
Lemon B, Tjian R: Orchestrated response: a symphony of transcription factors for gene control. Genes Dev 2000;14:2551.
Letchman DS: Transcription factor mutations and disease. N Engl J Med 1996;334:28.
Margueron R, Reinberg D: The polycomb complex PRC2 and its mark in life. Nature 2011;469:343-349.
Näär AM, Lemon BD, Tjian R: Transcriptional coactivator complexes. Annu Rev Biochem 2001;70:475. Nabel CS, Kohli RM: Demystifying DNA Demethylation Science 2011;333:1229-1230.
Oltz EM: Regulation of antigen receptor gene assembly in lymphocytes. Immunol Res 2001;23:121.
Ørom UA, Derrien T, Beringer M, et al: Long noncoding RNAs with enhancer-like function in human cells. Cell 2010;143: 46-58.
Ptashne M: A Genetic Switch, 2nd ed. Cell Press and Blackwell Scientific Publications, 1992.
Roeder RG: Transcriptional regulation and the role of diverse coactivators in animal cells. FEBS Lett 2005;579:909.
Ruthenburg AJ, Li H, Patel DJ, et al: Multivalent engagement of chromatin modifications by linked binding molecules. Nature Rev Mol Cell Bio 2007;8:983.
Segal E, Fondufe-Mittendorf Y, Chen L, et al: A genomic code for nucleosome positioning. Nature 2006;442:772.
Small EM, Olson EN: Pervasive roles of microRNAs in cardiovascular biology. Nature 2011;469:336-342.
The modENCODE Consortium, Roy S, Ernst J, Kharchenko PV, et al: Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 2010;330,1787-1797.
Valencia-Sanchez MA, Liu J, Hannon GJ, et al: Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev 2006;20:515.
Yang XJ, Seto E: HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007;26:5310.
Weake VM, Workman JL: Inducible gene expression: diverse regulatory mechanisms. Nat Rev Genet 2010;11:426-437.
Wu R, Bahl CP, Narang SA: Lactose operator-repressor interaction. Curr Top Cell Regul 1978;13:137.
Zhang Z, Pugh BF: High-resolution genome-wide mapping of the primary structure of chromatin. Cell 2011;144:175-186.