Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION VI. Special Topics
Chapter 55. Cancer: An Overview
Robert K. Murray, MD, PhD, Molly Jacob, MB BS, MD, PhD, & Joe Varghese, MB BS, MD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Present an overview of important aspects of the biochemical and genetic features of cancer cells.
Present an overview of important aspects of the biochemical and genetic features of cancer cells.
![]() Describe important properties of oncogenes and tumor suppressor genes.
Describe important properties of oncogenes and tumor suppressor genes.
![]() Briefly describe the concepts of genomic instability, aneuploidy, and angiogenesis in tumors.
Briefly describe the concepts of genomic instability, aneuploidy, and angiogenesis in tumors.
![]() Discuss the use of tumor markers for following responses to treatments and to detect recurrences.
Discuss the use of tumor markers for following responses to treatments and to detect recurrences.
![]() Appreciate that recent understanding of the biology of cancer has led to the development of various new therapies.
Appreciate that recent understanding of the biology of cancer has led to the development of various new therapies.
BIOMEDICAL IMPORTANCE
Cancers constitute the second most common cause of death, after cardiovascular disease, in the USA and many other countries. Approximately 6-7 million people around the world die from cancer each year, and this figure is projected to increase. Humans of all ages develop cancer, and a wide variety of organs are affected. Worldwide, the main types of cancer accounting for mortality are those involving the lung, stomach, colon, rectum, liver and breast. Other types of cancers that lead to death include cervical, esophageal, and prostate cancers. Skin cancers are very common, but apart from melanomas, are generally not as aggressive as those mentioned above. The incidence of many cancers increases with age. Hence, as people live longer many more will develop the disease. Hereditary factors play a role in some types of tumors. Apart from great individual suffering caused by the disease, the economic burden to society is immense.
SOME GENERAL COMMENTS ON NEOPLASMS
A neoplasm refers to any abnormal new growth of tissue. It may be benign or malignant in nature. The term “cancer” is usually associated with malignant tumors. Tumors can arise in any organ in the body and result in different clinical features, depending on the location of the growth.
Cancer cells are characterized by certain key properties: (1) they proliferate rapidly and display diminished growth control, (2) they display loss of contact inhibition in vitro, and (3) they invade local tissues and spread(metastasize) to other parts of the body. These properties are characteristic of cells of malignant tumors. It is the last property that is generally responsible for the deaths of patients who have cancer. Cells of benign tumors also show diminished control of growth, but do not invade local tissue or spread to other parts of the body. Other important properties of cancer cells are as follows: (1) they are self-sufficient in growth signals, (2) they are insensitive to anti-growth signals, (3) they stimulate local angio-genesis, and (4) they are often able to evade apoptosis. These points are summarized in Figure 55–1.
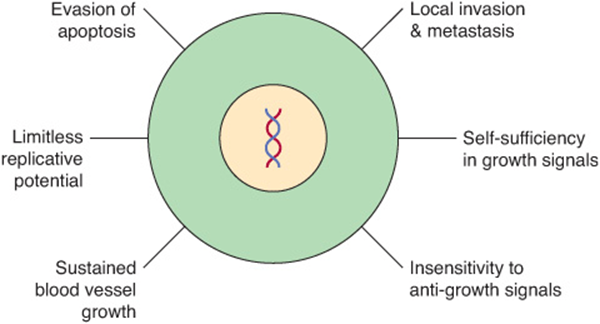
FIGURE 55–1 Six major features of cancer cells. Other important properties of cancer cells are shown in Figure 55–2. (With permission, After Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell 2000;100:57).
Figure 55–2 shows a number of other important properties associated with cancer cells. These various points will be discussed below.
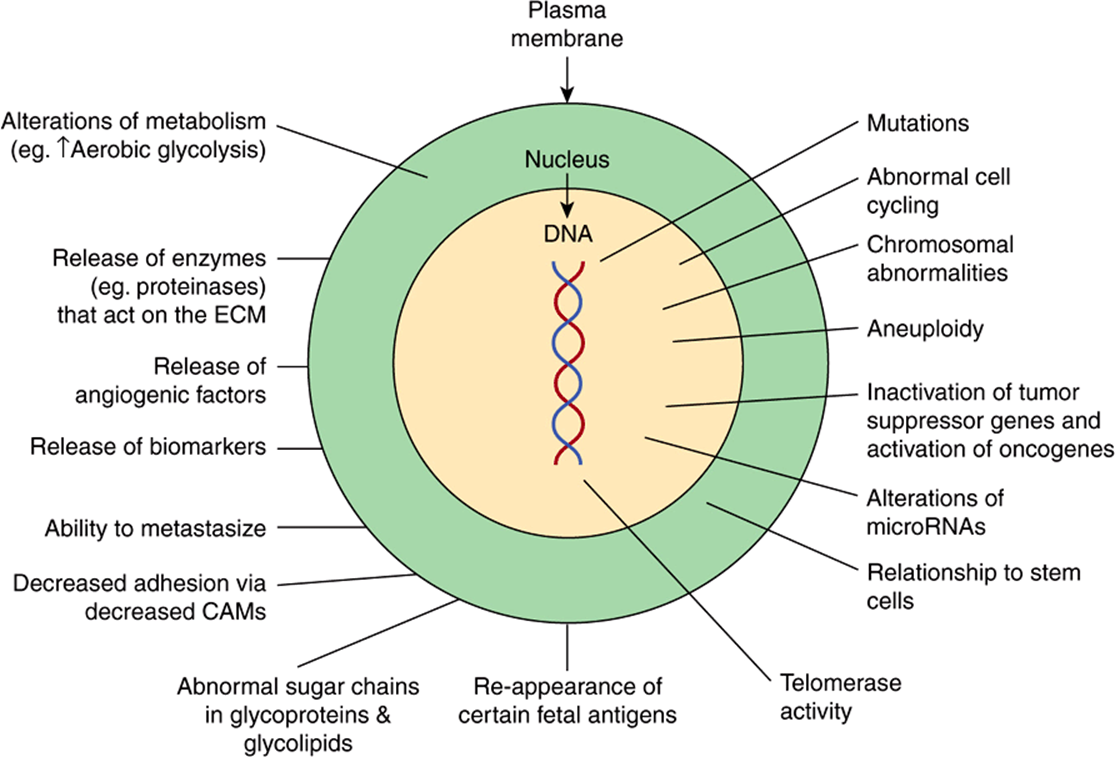
FIGURE 55–2 Some biochemical and genetic changes occurring in human cancer cells. Many changes, in addition to those indicated in Figure 55–1, are observed in cancer cells, only some of which are shown here. The roles of mutations in activating oncogenes and inactivating tumor suppressor genes are discussed in the text. Abnormalities of cell cycling and of chromosome structure, including aneuploidy, are common. Alterations of microRNA molecules that regulate gene activities have been reported, and the relationship of stem cells to cancer cells is a very active area of research. Telomerase activity is often detectable in cancer cells. Tumors sometimes synthesize certain fetal antigens, which may be measurable in the blood. Changes in plasma membrane constituents (eg, alteration of the sugar chains of various glycoproteins—some of which are cell adhesion molecules—and glycolipids) have been detected in many studies, and may be of importance in relation to decreased cell adhesion and metastasis. Various molecules can pass out of cancer cells and can be detected in the blood as tumor biomarkers. Angiogenic factors and various proteinases are also released by some tumors. Many changes in metabolism have been observed; for example, cancer cells often exhibit a high rate of aerobic glycolysis. (CAM, cell adhesion molecule; ECM, extracellular matrix.)
The central issues in cancer are to elucidate the biochemical and genetic mechanisms that underlie the uncontrolled growth of cancer cells, their ability to invade and metastasize and to develop successful treatments that destroy cancer cells, while causing minimal damage to normal cells. Considerable progress has been made in understanding the basic nature of cancer cells, with a central finding being that cancer is a disease due to abnormalities in key genes. However, many aspects of the behavior of cancer cells, in particular their ability to spread, have yet to be fully explained. In addition, despite improvements in treatment of certain types of cancers, therapies are still often unsuccessful. The study of cancer (oncology) is a huge area, so this short chapter can only introduce the reader to some key concepts.
A Glossary at the end of this chapter summarizes the meanings of many of the terms used.
FUNDAMENTAL FEATURES OF CARCINOGENESIS
Nonlethal genetic damage is the initiating event in carcinogenesis. There are principally four classes of genes, which when affected by such damage, can result in the development of a tumor. These are proto-oncogenes, tumor suppressor genes, genes involved in DNA repair, and those that are involved in apoptosis. Cancer is of clonal origin, with a single abnormal cell multiplying to become a mass of cells forming a tumor. Carcinogenesis is thus a multistep process, with multiple genetic alterations occurring in cells, transforming normal cells into malignant ones. Hence, a tumor often takes several years to develop.
CAUSES OF GENETIC DAMAGE
Genetic damage can be due to acquired or inherited mutations. The former occur due to exposure to environmental carcinogens while the latter are hereditary. Such hereditary abnormalities result in a number of familial conditionsthat predispose to hereditary cancer. These mutations are found in specific genes (eg, tumor suppressor genes) present in the germ cells and are discussed later.
Spontaneous mutations, some of which may predispose to cancer, occur at a frequency of approximately 10-7 to 10-6 per cell per generation. This rate will increase in tissues subject to a high rate of proliferation, increasing the generation of cancer cells from affected parent cells. Oxidative stress (see Chapter 45), by producing increased numbers of reactive oxygen species, may be a factor in increasing the mutation rate.
RADIANT ENERGY, CHEMICALS, AND CERTAIN VIRUSES ARE THE MAJOR KNOWN CAUSES OF CANCER
In general, there are three classes of carcinogens, exposure to which result in tumor formation. These are radiant energy, chemicals, and certain oncogenic viruses (see Figure 55–3). The first two cause mutations in DNA, and the third class generally acts by introducing novel genes into normal cells.
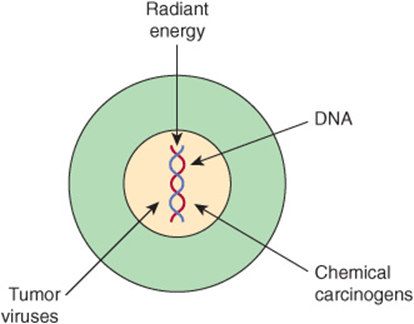
FIGURE 55–3 Radiant energy, chemical carcinogens and certain viruses can cause cancer.
We shall only describe briefly how radiant energy, chemicals, and oncogenic viruses cause cancer.
Radiant Energy can be Carcinogenic
Ultraviolet rays, x-rays, and γ-rays are mutagenic and carcinogenic. Extensive studies have shown that these agents can damage DNA in a number of ways, including the lesions listed in Table 55-1. Mutations in DNA, due to such damage, are thought to be the basic mechanism of carcinogenicity caused by radiant energy although the exact pathways are still under investigation. X-rays and γ-rays can cause formation of reactive oxygen species (ROS), which can also be mutagenic and probably contribute to the carcinogenic effects of radiant energy.
TABLE 55–1 Some Types of DNA Damage Caused by Radiant Energy

Exposure to ultraviolet radiation is common due to exposure to sunlight, which is its main source. Ample evidence exists to show that such radiation is linked to cancers of the skin. The risk of developing a skin cancer due to ultraviolet radiation increases with increasing frequency and intensity of exposure and decreasing melanin content of skin.
DNA damage produced by environmental agents is usually removed by DNA repair mechanisms. Individuals who have an inherited inability to repair DNA, as is seen in xeroderma pigmentosa (see Chapter 57) and ataxia telangiectasia, have increased risk of developing a malignancy.
Many Chemicals are Carcinogenic
A wide variety of chemical compounds are carcinogenic (see Table 55-2 and Figure 55–4). It is estimated that perhaps 80% of human cancers are caused by environmental factors, principally chemicals.
TABLE 55–2 Some Chemical Carcinogens
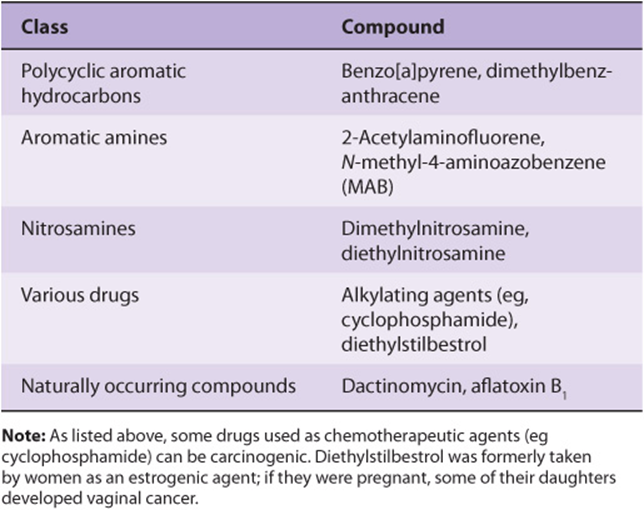
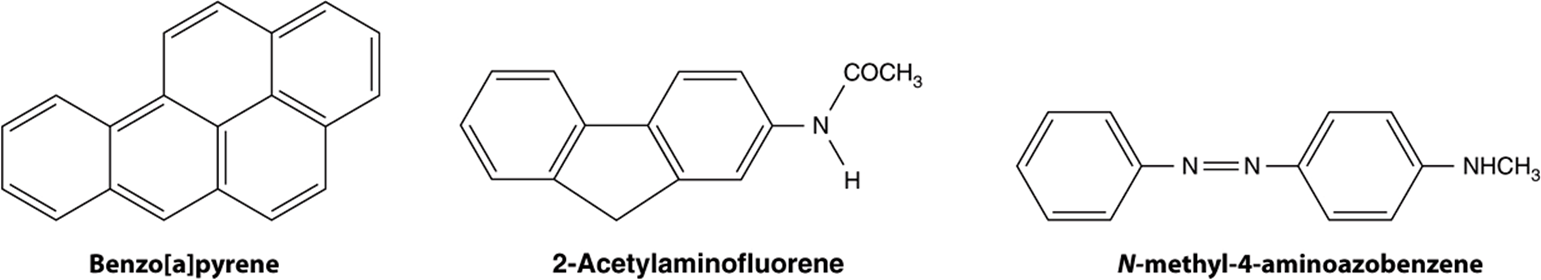
FIGURE 55–4 Structures of three experimentally widely used chemical carcinogens.
Extensive studies have been performed in the field of chemical carcinogenesis. Overall, most chemical carcinogens are thought to interact covalently with DNA, forming a wide variety of adducts. Depending on the extent of damage to DNA and its repair by DNA repair systems (see Chapter 35), a variety of mutations in DNA can result from exposure of an animal or human to chemical carcinogens, some of which contribute to the development of cancer.
Some chemicals interact directly with DNA (eg, methchlorethamine and β-propiolactone), but others (procarcinogens) require conversion by enzyme action to become ultimate carcinogens (Figure 55–5). Most ultimate carcinogens are electrophiles (molecules deficient in electrons) and readily attack nucleophilic (electron-rich) groups in DNA. Conversion of chemicals to ultimate carcinogens is principally due to the actions of various species of cytochrome P450 located in the endoplasmic reticulum (ER) (see Chapter 53). This fact is used in the Ames assay (see below), in which an aliquot of post-mitochondrial supernatant (containing ER) is added to the assay system as a source of cytochrome P450 enzymes.
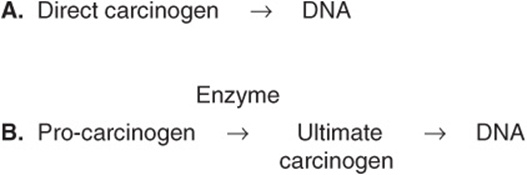
FIGURE 55–5 (A) Direct and (B) indirect carcinogens. Direct carcinogens can interact with DNA without prior enzyme activation. Indirect carcinogens are activated by an enzyme (eg a cytochrome P450 species) to the ultimate carcinogen and then interact with DNA.
Chemical carcinogenesis comprises two stages—initiation and promotion. Initiation is the stage where exposure to a chemical causes irreversible DNA damage and is a necessary initial event for a cell to become cancerous. Promotion comprises the stage at which an initiated cell begins to grow and proliferate. The cumulative effect of these stages is a neoplasm.
Chemical carcinogens can be identified by screening for their mutagenicity. A simple way to do this is by using the Ames assay (Figure 55–6). This relatively simple test, which detects mutations in Salmonella typhimuriumcaused by chemicals, has proven very valuable for screening purposes. A refinement of the Ames test is to add an aliquot of endoplasmic reticulum (ER) to the assay, to make it possible to indentify procarcino-gens. Very few, if any, compounds that have tested negative in the Ames test have been shown to cause tumors in animals. However, animal testing is required to show unambiguously that a chemical is carcinogenic.
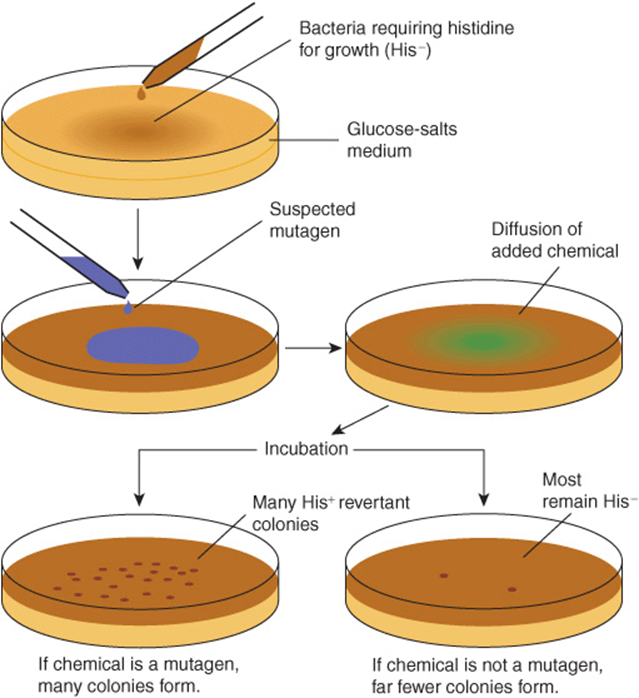
FIGURE 55–6 The Ames assay to screen for mutagens. The chemical tested will increase the frequency of reversion of His–to His+ cells if it is a mutagen and, therefore, a potential carcinogen. A control plate (not shown) contains the liquid in which the suspected mutagen is dissolved. Reproduced, with permission, from Nester EW et al: Microbiology: A Human Perspective. 5th ed. McGraw-Hill, 2007.
It should be noted that compounds that alter epigenetic factors (eg, stilbestrol), thus perhaps leading to cancer, would not test positive in the Ames test, as they are not mutagenic.
Approximately 15% of Human Cancers may be Caused by Viruses
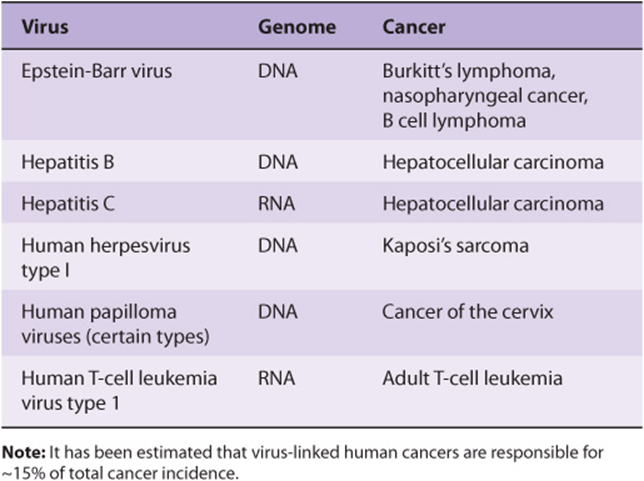
The study of tumor viruses has contributed very significantly to the understanding of cancer. For example, discovery of both oncogenes and tumor suppressor genes (see below) emerged from studies of oncogenic viruses. Both DNA and RNA viruses have been identified as being able to cause cancers in humans (Table 55-3). The details of how each of these viruses causes cancer will not be described here. In general, the genetic material of viruses is incorporated into the genome of the host cell. In the case of RNA viruses, this would occur after reverse transcription of the viral RNA to viral DNA. Such integration of viral DNA (called the provirus) with the host DNA results in various events such as deregulation of the cell cycle, inhibition of apoptosis, and abnormalities of cell signaling pathways. All these events are discussed later in this chapter. The DNA viruses often act by down-regulating the tumor suppressor genes P53 and RB (see below). RNA viruses often carry oncogenes in their genomes; how oncogenes act to cause malignancy is discussed below. It has been estimated that about 15% of human tumors may be caused by viruses.
TABLE 55–3 Some Viruses That Cause or Are Associated With Human Cancers
ONCOGENES AND TUMOR SUPPRESSOR GENES PLAY KEY ROLES IN CAUSING CANCER
Over the past 30 years or so, major advances have been made in understanding how cancer cells develop and grow. Two key findings were the discoveries of oncogenes and tumor suppressor genes. These discoveries pointed to specific mechanisms by which cell growth and division could be disturbed, resulting in abnormal growth. The overall effects of oncogenes and loss of activity of tumor suppressor genes are summarized in Figure 55–7.

FIGURE 55–7 Oncogenes and loss of activity of tumor suppressor genes drive cell growth towards cancer. Oncogenes encode various proteins that can drive the growth of cancer cells. Oncogenes are derived from proto-oncogenes. Tumor suppressor genes encode proteins that normally suppress cell growth, but which are inactivated when altered by mutations. MicroRNA molecules (not shown here) are also affected by mutations, and this can affect their normal regulatory functions. In addition, epigenetic changes (also not shown) affect gene expression, and hence growth of cancer cells.
Oncogenes are Derived from Proto-oncogenes and Encode a Wide Variety of Proteins that Affect Cell Growth and Cell Death
An oncogene can be defined as an altered gene whose product acts in a dominant manner to accelerate cell growth or cell division. It is derived by “activation” of normal cellular proto-oncogenes (which encode growth stimulating proteins). The mechanisms involved in such activation are listed in Table 55-4.
TABLE 55–4 Mechanisms of Activating Oncogenes
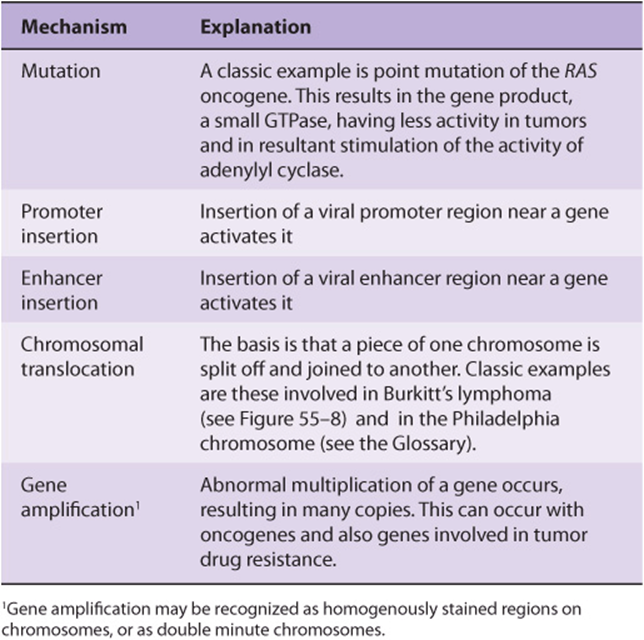
The Table lists an example of a point mutation occurring in the RAS oncogene, which encodes a small GTPase. Loss of the activity of this G protein (see Chapter 42) results in chronic stimulation of the activity of adenylyl cyclase, leading to cell proliferation. Another way an oncogene can be activated is via insertion of a promoter (see Figure 55–8(A)), in which integration of a retroviral provirus (ie, a DNA copy of the RNA genome of a tumor virus such as Rous sarcoma virus, made by reverse transcriptase) activates MYC, a neighboring host gene. Overproduction of the protein encoded by MYC (a transcription factor) stimulates cell proliferation. As the legend to Figure 55–8(A) indicates, a similar type of effect results from enhancer insertion. Chromosomal translocations are found quite frequently in cancer cells, with about a hundred different examples having been documented. The translocation found in cases of Burkitt’s lymphoma is illustrated in Figure 55–8(B). The overall effect of this translocation is also to activate MYC, resulting in cell proliferation. Yet another mechanism of oncogene activation is via gene amplification, which occurs quite commonly in various cancers. In this case, multiple copies of an oncogene are formed resulting in increased production of a growth-promoting protein.
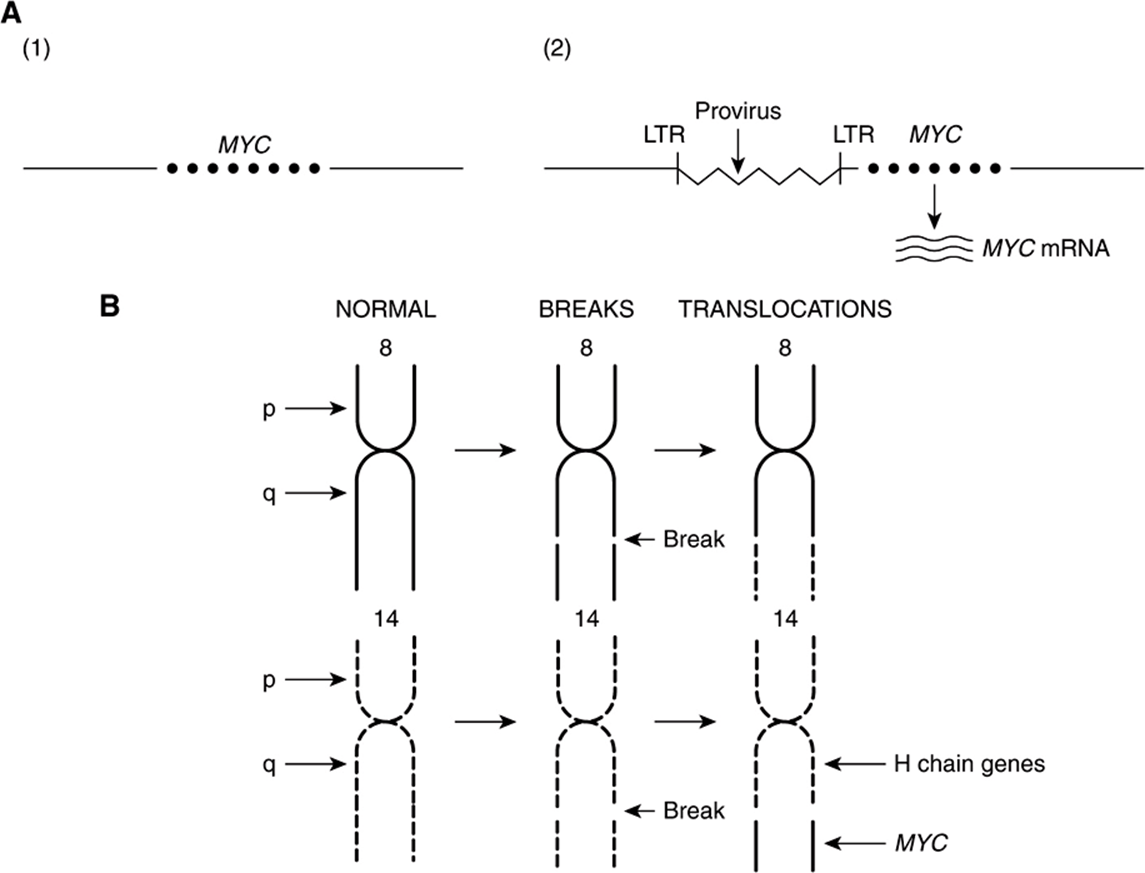
FIGURE 55–8 (A) Schematic representation of how promoter insertion may activate a proto-oncogene. (1). Normal chicken chromosome showing an inactive MYC gene. (2) An avian leukemia virus has integrated in the chromosome in its proviral form (a DNA copy of its RNA genome) adjacent to the MYC gene. Its right-hand long terminal repeat (LTR), containing a strong promoter (see Chapter 36), lies just upstream of the MYC gene and activates that gene, resulting in transcription of MYC mRNA. For simplicity, only one strand of DNA is depicted and other details have been omitted. Enhancer insertion acts similarly, except that the site of integration may be downstream or considerably upstream, and it cannot act as a promoter. Instead, a specific proviral sequence acts as an enhancer element (see Chapter 36), leading to activation of the MYC gene and its transcription. (B) Schematic representation of the reciprocal translocation involved in Burkitt’s lymphoma. The chromosomes involved are 8 and 14. A segment from the end of the q arm of chromosome 8 breaks off and moves to chromosome 14. The reverse process moves a small segment from the q arm of chromosome 14 to chromosome 8. The MYC gene is contained in the small piece of chromosome 8 that was transferred to chromosome 14; it is thus placed next to genes transcribing the heavy chains of immunoglobulin molecules, and itself becomes activated. Many other translocations have been identified, with perhaps the best known being that involved in formation of the Philadelphia chromosome (see the Glossary).
Once oncogenes are activated, how do their protein products act to promote development of cancer? Figure 55–9 shows certain of the ways in which they operate. Some affect cell signaling pathways (eg, the product of an oncogene may act as growth factor, a growth factor receptor, a G-protein or as a downstream signaling molecule). Others act to alter transcription or to deregulate the cell cycle. Yet others can affect cell-cell interactions or the process of apoptosis. These mechanisms help to explain many of the major features of cancer cells shown in Figure 55–1, such as their limitless replicative potential, their signaling defects, their ability to invade and spread, and their evasion of apoptosis.
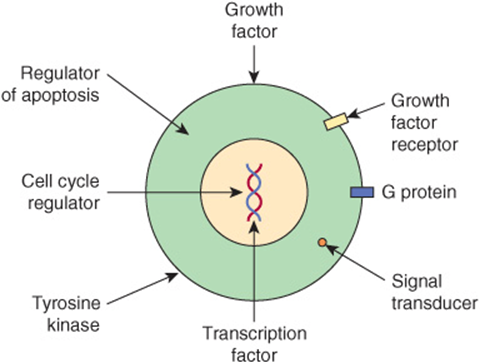
FIGURE 55–9 Some ways in which proteins encoded by oncogenes work. The Figure shows examples of various proteins encoded by oncogenes. The proteins are listed below with the corresponding oncogene given in parentheses along with its OMIM number. A growth factor, fibroblast growth factor 3 (INT2,164950); a growth factor receptor, epidermal growth factor receptor [EGFR] (HER1, 131550); a G protein (H-RAS-1, 190020); a signal transducer (BRAF, 164757); a transcription factor (MYC,190080); a tyrosine kinase and involved in cell-cell adhesion (SRC, 190090); a cell cycle regulator (PRAD, 168461); a regulator of apoptosis (BCL2, 151430).
Certain tumor viruses (eg, retroviruses) contain oncogenes. It was the study of such tumor viruses (eg, Rous sarcoma virus [RSV], a retrovirus) that first revealed the presence of oncogenes. Further study showed that viral oncogenes were derived from cellular proto-oncogenes that the tumor viruses had picked up during their passage through host cells.
Tumor Suppressor Genes Act to Inhibit Cell Growth and Cell Division
A tumor suppressor gene produces a protein product that normally suppresses cell growth or cell division. When such a gene is altered by mutation, the inhibitory effect of its product is lost or diminished, leading to increased cell growth or cell division. As first suggested by AG Knudson, based on studies of the inheritance of retinoblastomas, both copies of a tumor suppressor gene must be affected for it to lose its inhibitory effects on growth.
A useful distinction has been made between gatekeeper and caretaker functions of tumor suppressor genes. The former control cell proliferation, and include mainly genes that act to regulate the cell cycle and apoptosis, The latter are concerned with preserving the integrity of the genome, and include genes whose products are involved in recognizing and correcting DNA damage and maintaining chromosomal integrity during cell division.
Many oncogenes and tumor suppressor genes have now been identified. Only a few are mentioned here. Some differences between oncogenes and tumor suppressor genes are listed in Table 55-5.
TABLE 55–5 Some Differences Between Oncogenes and Tumor Suppressor Genes
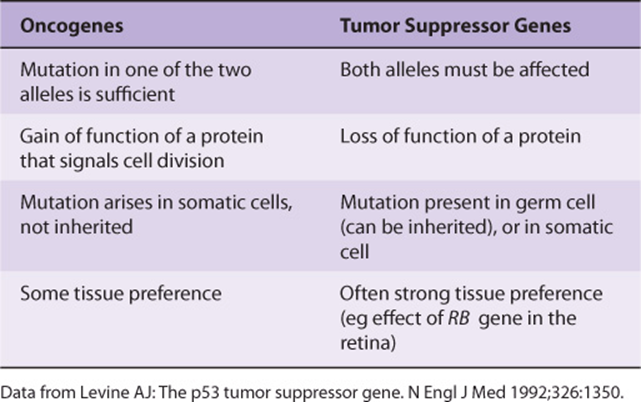
Table 55-6 lists some of the properties of two of the most studied oncogenes (MYC and RAS), and two of the most studied tumor suppressor genes (P53 and RB).
TABLE 55–6 Some Properties of a Few Important Oncogenes and Tumor Suppressor Genes


Studies of the Development of Colorectal Cancers Have Illuminated the Involvements of Specific Oncogenes and Tumor Suppressor Genes
Many types of tumors have been analyzed for genetic changes. One of the most informative areas in this respect has been analyses of the development of colorectal cancers by Vogelstein and colleagues. Their work, and that of others, has shown the involvement of various oncogenes and tumor suppressor genes in human cancer. (Case 4 in Chapter 57 describes the history of a patient with colorectal cancer). These workers analyzed various oncogenes, tumor suppressor genes, and certain other relevant genes in samples of normal colonic epithelium, of dysplastic epithelium (a preneoplastic condition, characterized by abnormal development of epithelium), of various stages of adenomatous polyps, and of adenocarcinomas. Some of their major findings are summarized in Figure 55–10. It can be seen that certain genes were found to be mutated at relatively specific stages of the total sequence shown. Functions of the various genes identified are listed in Table 55-7. The overall sequence of changes can vary somewhat from that shown, and other genes may also be involved. Similar studies have been performed on a number of other human tumors revealing somewhat different patterns of activation of oncogenes and mutations of tumor suppressor genes. Further mutations in these and other genes are involved in tumor progression, a phenomenon whereby clones of tumor cells become selected for fast growth rate and ability to spread. Thus, a relatively large tumor may contain a variety of cells with different genotypes, making successful treatment more difficult.
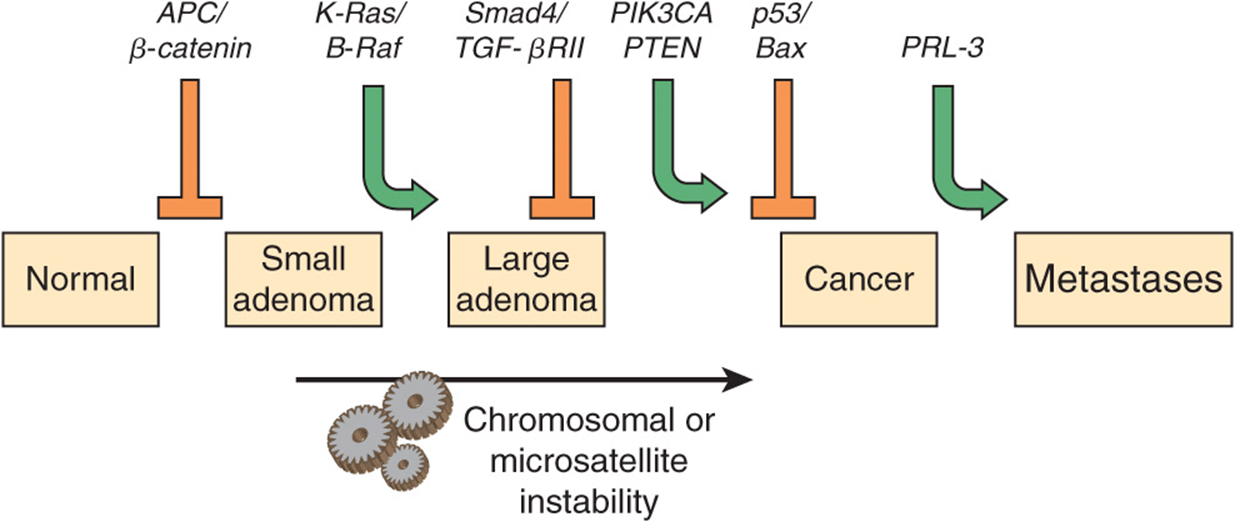
FIGURE 55–10 Multistep genetic changes associated with the development of colorectal cancers. Mutations in the APC gene initiate the formation of adenomas. One sequence of mutations in an oncogene and in various tumor suppressor genes that can result in further progression to large adenomas and cancer is indicated. Patients with familial adenomatous polyposis (OMIM 175100) inherit mutations in the APC gene and develop numerous dysplastic aberrant crypt foci (ACF), some of which progress as they acquire the other mutations indicated in the Figure. The tumors from patients with hereditary nonpolyposis colon cancer (OMIM 120435) go through a similar though not identical series of mutations; mutations in the mismatch repair system (see Chapter 35) speed up this process. K-RAS is an oncogene, and the other specific genes indicated are tumor suppressor genes. The chromosomal locations of the various genes shown here are known. The sequence of events shown here is not invariable in the development of all colorectal cancers. A variety of other genetic alterations have been described in a small fraction of advanced colorectal cancers. These may be responsible for the heterogeneity of biological and clinical properties observed among different cases. Instability of chromosomes and microsatellites (see Chapter 35) occurs in many tumors, and likely involves mutations in a considerable number of genes. (Reproduced, with permission, from Bunz F, Kinzler KW, Vogelstein B: Colorectal Tumors, Fig. 48-2, The Online Metabolic & Molecular Bases of Inherited Disease, www.ommbid.com)
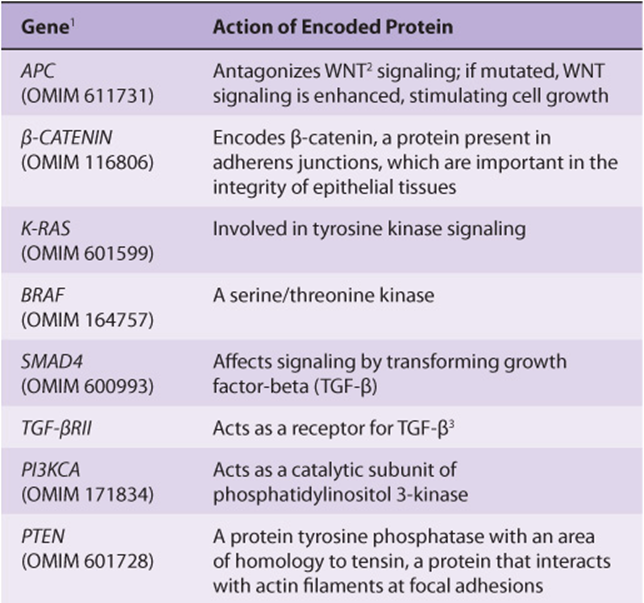
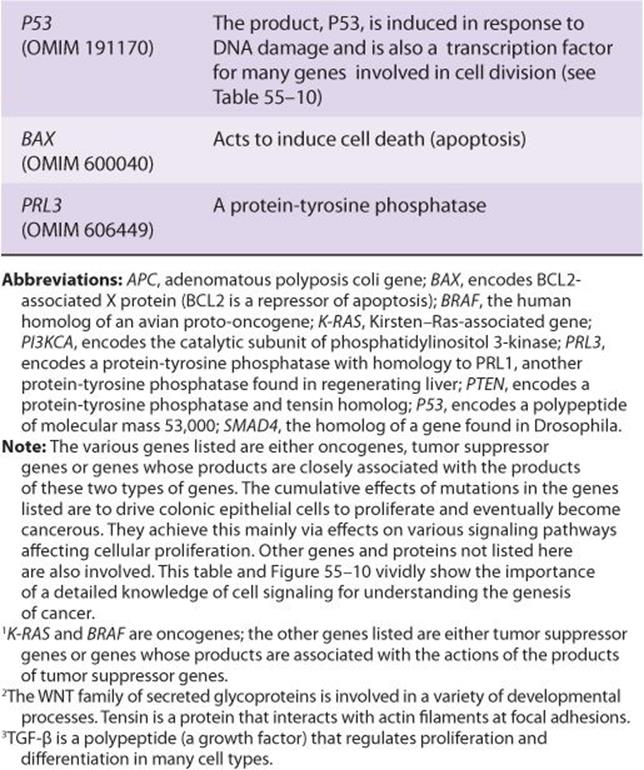
TABLE 55–7 Some Genes Associated with Colorectal Carcinogenesis
Several other inferences can be made from these results and those from other similar studies. The first of these is that cancer is truly a genetic disease, but in a somewhat different sense from the normal meaning of the phrase, insofar as many of the gene alterations are due to somatic mutations. Secondly, carcinogenesis is, as mentioned above, a multistep process. It is estimated that in most cases a minimum of five to six genes must be mutated for cancer to occur. Thirdly, additional subsequent mutations are thought to confer selective advantages on clones of cells, some of which acquire the ability to metastasize successfully (see below). Fourthly, many of the genes implicated in colorectal carcinogenesis and other types of cancers are involved in cell signaling events, showing the central role that alterations in signaling play in the development of cancer.
GROWTH FACTORS & ABNORMALITIES OF THEIR RECEPTORS AND SIGNALING PATHWAYS PLAY MAJOR ROLES IN CANCER DEVELOPMENT
There Are Many Growth Factors
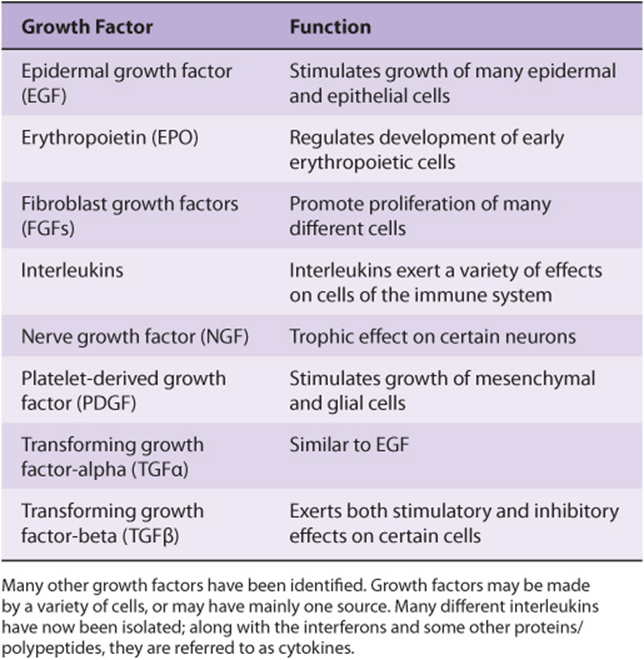
A large variety of polypeptide growth factors that work on human tissues and cells have been identified. Some are listed in Table 55-8. Here we focus mostly on their relationship with cancer.
TABLE 55–8 Some Polypeptide Growth Factors
Growth factors can act in an endocrine, paracrine, or autocrine manner and affect a wide variety of cells to produce a mitogenic response. As described earlier (Chapter 52), they play an important role in the differentiation of hematopoietic cells.
Growth inhibitory factors also exist. For example, transforming factor beta (TGF-β) exerts inhibitory effects on the growth of certain cells. Thus, chronic exposure to increased amounts of a growth factor or to decreased amounts of a growth inhibitory factor can alter the balance of cellular growth.
Growth Factors Work Via Specific Receptors and Transmembrane Signaling to Affect the Activities of Specific Genes
Growth factors produce their effects by interacting with specific receptors on cell surfaces, initiating various signaling events (Chapter 42). Many receptors for growth factors have been cloned. They generally have short membrane-spanning segments and external and cytoplasmic domains. A number (eg, those for epidermal growth factor [EGF], insulin and platelet-derived growth factor [PDGF]) have tyrosine kinase activities. The kinase activity, located in the cytoplasmic domains, causes autophosphorylation of the receptor protein and also phosphorylates certain other proteins.
Consideration of how PDGF acts illustrates how one particular growth factor brings about its effects. Interaction of PDGF with its receptor stimulates the activity of phospholipase C. This acts to split phosphatidylinositol bisphosphate (PIP2) into inositol trisphosphate (IP3) and diacylglycerol (DAG) (see Figure 42–6). Increased IP3 stimulates the release of intracellular Ca2+ and DAG increases the activity of protein kinase C (PKC). Hydrolysis of DAG may release arachidonic acid, which can stimulate production of prostaglandins and leukotrienes, each of which has various biologic effects. Exposure of target cells to PDGF can result in rapid (minutes to 1-2 h) activation of certain cellular proto-oncogenes (eg, MYC and FOS), which participate in stimulation of mitosis via effects on the cell cycle (see below). The bottom line is that growth factors interact with specific receptors, which stimulate specific signaling pathways to increase or decrease the activities of various genes that affect cell division.
MANY CANCERS CAN BE PREVENTED BY MODIFYING RISK FACTORS
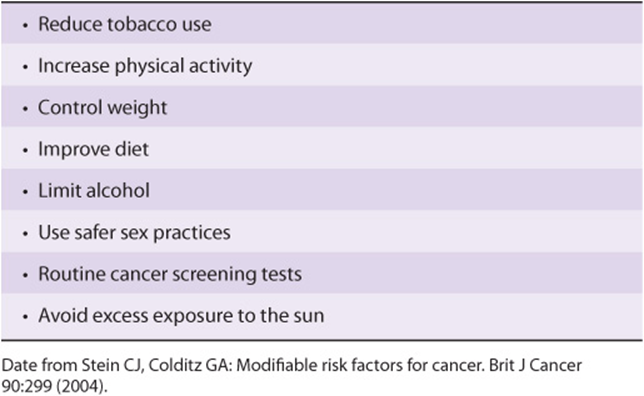
Modifiable risk factors have been linked to a wide variety of cancers. It has been estimated that over half of all cancers in developed countries could be prevented if the measures summarized in Table 55-9 were introduced on a population-wide basis. Smoking is still a major cause of cancer across the globe. It cannot be overemphasized that prevention and early detection of cancer are most critical if the disease is to be beaten.
TABLE 55–9 Measures That Might Prevent Approximately 50% of Cancers if Introduced on a Population-Wide Basis
ABNORMALITIES OF THE CELL CYCLE ARE UBIQUITOUS IN CANCER CELLS
Knowledge of the cell cycle is necessary for understanding many of the mechanisms involved in the development of cancer. It is also of importance because many anti-cancer drugs act only against cells that are dividing, or in a certain phase of the cycle.
Basic aspects of the cell cycle were described in Chapter 35. As shown in Figure 35–20, the cycle has four phases: G1, S, G2, and M. If cells are not cycling, they are said to be in G0 phase and are termed quiescent. Cells can be recruited into the cycle from G0, by various influences (eg, certain growth factors). Generation time is the time needed for a cell in G0 to enter the cycle and give rise to two daughter cells. The cells of a cancer usually have a shorter generation time than normal cells, and there are less of them in G0 phase.
The roles of various cyclins, cyclin-dependent kinases (CDKs), and a number of other important molecules that affect the cell cycle (eg, the genes RB and P53) were also described in Chapter 35. The points in the cycle at which some of these molecules act are indicated in Figure 35–21 and Table 35-7.
Because a major property of cancer cells is uncontrolled growth, many aspects of their cell cycle have been studied in considerable depth. Only a few results can be mentioned here. A variety of mutations affecting cyclins and CDKs have been reported. Many products of proto-oncogenes and tumor suppressor genes play important roles in regulating the normal cycle. A wide variety of mutations have been found in these types of genes, including RAS, MYC, RB, P53 (which are among the most studied, see below) and many others.
For example, as discussed in Chapter 35, the protein product of the RB gene is a cell cycle regulator. It acts via binding to the transcription factor E2F, blocking progression of the cell from G1 to S phase. Loss of the RB protein due to mutations thus removes this element of control of the cell cycle.
When damage to DNA occurs (by radiation or chemicals), the P53 protein increases in amount and activates transcription of genes that delay transit through the cycle. If the damage is too severe to repair, P53 activates genes that cause apoptosis (see below). If P53 is absent or inactive due to mutation, apoptosis does not occur and cells with damaged DNA persist, perhaps becoming progenitors of cancer cells.
GENOMIC INSTABILITY AND ANEUPLOIDY ARE IMPORTANT CHARACTERISTICS OF CANCER CELLS
As referred to above and also later in this Chapter, cancer cells have many mutations. One possible explanation for their genomic instability is that they have a mutator phenotype. This was originally postulated by Loeb and colleagues to be due to cancer cells having acquired mutations in genes involved in DNA replication and DNA repair, thus allowing mutations to accumulate. The concept was later expanded to include mutations that affect chromosomal segregation, DNA damage surveillance, and processes such as apoptosis.
The term genomic instability is frequently used to refer to two abnormalities shown by many cancer cells, microsatellite and chromosomal instability (CI). Microsatellite instability was described briefly in Chapter 35. It involves expansion or contraction of microsatellites, usually due to abnormalities of mismatch repair or to replication slippage. CI occurs more often than microsatellite instability, and the two are often mutually exclusive. It refers to gain or loss of chromosomes caused by abnormalities of chromosomal segregation during mitosis.
Another area of interest regarding CI is copy number variation (CNV) (see the Glossary). Associations of various CNVs with many cancers have been identified, and their precise roles in cancer are under investigation.
An important aspect of CI is aneuploidy, a very common feature of solid tumors. Aneuploidy exists when the chromosomal number of a cell is not a multiple of the haploid number. The degree of aneuploidy often correlates with a poor prognosis. This has suggested that abnormalities of chromosomal segregation may contribute to tumor progression by increasing genetic diversity. Some scientists believe that aneuploidy is a fundamental aspect of cancer.
Much research is proceeding on determining the basis of CI and aneuploidy. As shown in Figure 55–11, a number of different processes are involved in normal chromosomal segregation. Each of the processes shown is complex, involving various organelles and also many individual proteins. A textbook of Cell Biology should be consulted for details of these important processes. Studies are in progress to compare these processes in normal and tumor cells, and to determine which of the differences detected may be contributors to CI and aneuploidy. One hope of this line of research is that it might be possible to develop drugs that diminish or even prevent CI and aneuploidy.
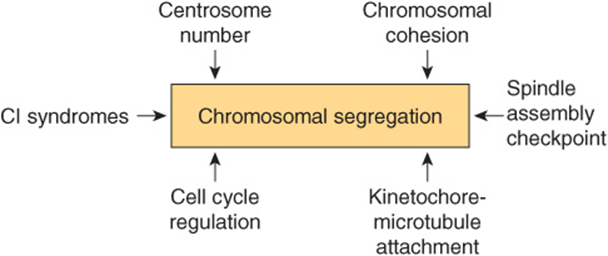
FIGURE 55–11 Some factors involved in chromosomal segregation which are relevant to understanding chromosomal instability (CI) and aneuploidy. CI syndromes include Bloom syndrome (OMIM 210900) and others. (Based on Thompson SL et al: Mechanisms of chromosomal instability. Curr Biol 2010;20(6):R285).
MANY CANCER CELLS DISPLAY ELEVATED LEVELS OF TELOMERASE ACTIVITY
There has been considerable interest in the involvement of telomeres (see Chapter 35) in a number of diseases and also in aging. With respect to cancer, when tumor cells divide rapidly their telomeres often shorten. Such telomeres (usually detected in leukocytes because of ease of obtaining them) have been implicated as a risk factor for many, but not all, solid tumors, (eg, breast cancer). Short telomeres appear to be of predictive value regarding the progression of chronic inflammatory diseases (such as ulcerative colitis and Barrett’s esophagus) to cancer. Abnormalities of telomere structure and function can contribute to CI (see above). The activity of telomerase, the main enzyme involved in synthesizing telomeres, is frequently elevated in cancer cells, providing one mechanism for overcoming telomere shortening. Selective inhibitors of telomerase have been considered as possible drugs for treating cancer, but have not as yet been translated into successful clinical use.
A NUMBER OF CANCERS HAVE A HEREDITARY PREDISPOSITION
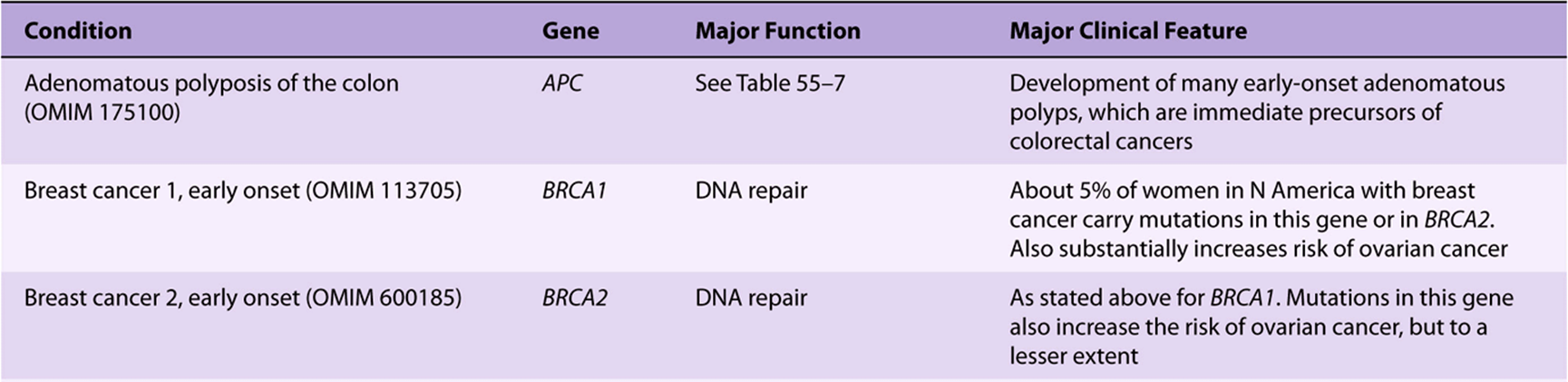
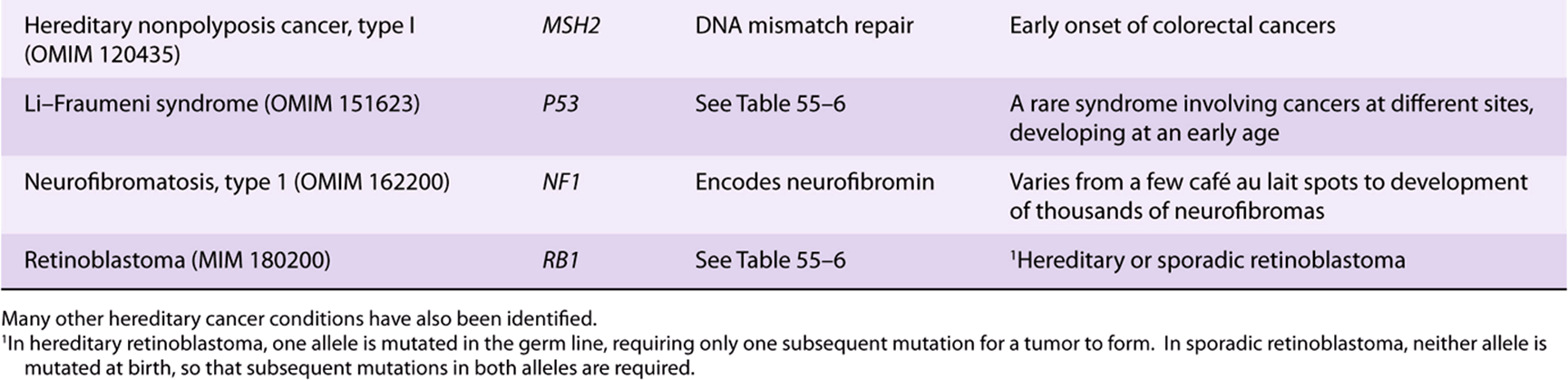
It has been known for many years that certain cancers have a hereditary basis. It is estimated that about 5% of cancers fall into this category. The discovery of oncogenes and tumor suppressor genes has allowed investigations of the basis of this phenomenon. Many hereditary types of cancer have now been recognized; only a few of these are listed in Table 55-10. In a number of cases, where a hereditary syndrome is suspected, appropriate genetic screening of families has allowed early interventions to be made. For example, some young women who have inherited either a mutated BRCA1 or BRCA2 gene have opted for prophylactic mastectomies to prevent cancer of the breast occurring in later life.
TABLE 55–10 Some Hereditary Cancer Conditions
WHOLE GENOME SEQUENCING OF TUMOR CELLS IS PROVIDING NEW INSIGHTS REGARDING CANCER
Since the completion of the Human Genome Project, the technology of large-scale sequencing and the analysis and interpretation of sequence data have advanced considerably. Sequencing has become much cheaper. This has led to studies, the aim of which is to completely sequence the genomes of a large number of different types of tumors. In this way, a comprehensive catalog of the types and numbers of mutations found in various types of cancers will eventually become available. It is hoped that such studies will lead to results that will also impact diagnostic testing and point to useful, new prognostic biomarkers. It should also reveal the involvement of many new genes in carcinogenesis, some of which may provide leads to new therapeutic approaches. To date, about 350 genes involved in cancer have been identified; it has been predicted that as many as 2000 may exist. It is of particular interest to identify mutations in genes that cause and accelerate cancers; these are known as driver mutations, whereas other mutations are called passenger mutations.
Studies on a number of types of cancer have already afforded interesting results. For example, in a study of DNA from a person suffering from acute myelocytic leukemia, about 750 somatic mutations were detected. Only 12 of these were in genes encoding proteins or regulatory RNAs; mutations in such genes are most likely to exert serious effects. (Coding areas of the genome only occupy about 2% of genomic DNA.) Mutations in other genomic sites may or may not have consequences. Whole genome sequencing on seven human primary prostate cancers has revealed the presence of surprisingly many genomic rearrangements.
A fascinating example of the information that can be provided by genome sequencing of cancers is provided by the results of a recent study of pancreatic carcinomas. These are among the most deadly cancers. However, an issue that had not been resolved is whether their lethality is due to their aggressiveness (ability to metastasize and grow), or to late diagnosis. In the study under consideration, the genomes of seven primary pancreatic cancers were sequenced, as were the genomes of metastases from them obtained at autopsy. Approximately 61 known cancer-related mutations were detected in each metastasis. Using a “molecular clock” technique borrowed from evolutionary biology, it was calculated how long it took the metastases to accumulate these mutations. Prior knowledge of the overall sequence of mutations made this possible. It was estimated that it took just over 10 years from the time of the initiating mutation for non-metastatic primary tumors to develop in the pancreas. Another 5 years was required for such tumors to gain metastatic potential. Thereafter, about 2 years elapsed before the tumors metastasized and death resulted. Thus, it was suggested that the evolution of many pancreatic cancers is a relatively slow process, and that pancreatic cancers are not highly aggressive. The problem is that they are difficult to diagnose. The hope is that methods such as detection of mutations in pancreatic cancer cells present in stool specimens, development of new blood biomarkers for pancreatic cancers, and perhaps new imaging techniques will allow early diagnosis, always a critical factor in the management of cancer. Within a relatively few years, complete sequence data on the genomes of many important types of tumors should be available, and, hopefully, will considerably advance understanding of the basic biology of cancer and provide information relevant to better drug design and less toxic treatment.
Distinct from gene sequencing studies, there have been many reports of involvements of microRNAs in cancer. Because of their importance in many aspects of gene control, the study of these molecules is already forming an important area of cancer research.
Several hundred types of cancers have been identified by classical histopathology. Thus, cancer is not one single disease. The work on genome sequencing of cancers, revealing the large number of mutations in cancer cells, suggests that there may well be not just hundreds of different cancers, but that each cancer may be genetically different.
CANCER CELLS HAVE ABNORMALITIES OF APOPTOSIS THAT PROLONG THEIR LIVES
Apoptosis is a genetically regulated program that, when activated, causes cell death. The main players are proteolytic enzymes named caspases, which normally exist as inactive procaspases. The name caspase reflects that they are cysteine proteases that split peptide bonds on the C-terminal end of aspartate residues. About 15 human caspases are known, although not all participate in apoptosis. When those involved in apoptosis are activated (mainly 2, 3, 6, 7, 8, 9 and 10), they participate in a cascade of events (compare with the coagulation cascade, Chapter 51) that ultimately kills cells by digesting various proteins and other molecules. The upstream caspases (eg, 2, 8, and 10) at the beginning of the cascade are often called initiators, and those downstream at the end of the pathway (eg, 3, 6, and 7) are called effectors or executioners. Caspase-activated DNase (CAD) fragments DNA, producing a characteristic laddering pattern detected by electrophoresis. Microscopic features of apoptosis include condensation of chromatin, changes of nuclear shape and membrane blebbing. The dead cells are rapidly disposed of by phagocytic activity, avoiding an inflammatory reaction.
Apoptosis differs from necrosis, a pathologic form of cell death that is not genetically programmed. Necrosis occurs on exposure to external agents, such as certain chemicals and extreme heat (eg, burns). Various hydrolytic enzymes (proteases, phospholipases, nucleases, etc) are involved in necrosis. Release of cell contents from dying cells can cause local inflammation, unlike apoptosis.
The overall process of apoptosis is complex and it is a matter of life and death (excuse the pun!) that it is tightly regulated. It includes proteins that act as receptors, adapters, procaspases and caspases, and pro-and antiapoptotic factors. There are extrinsic and intrinsic pathways, with mitochondria being important participants in the intrinsic pathway.
Figure 55–12 shows a much simplified diagram of some of the key events in apoptosis. Two major pathways are involved, the death receptor (extrinsic) pathway and the mitochondrial (intrinsic) pathway.
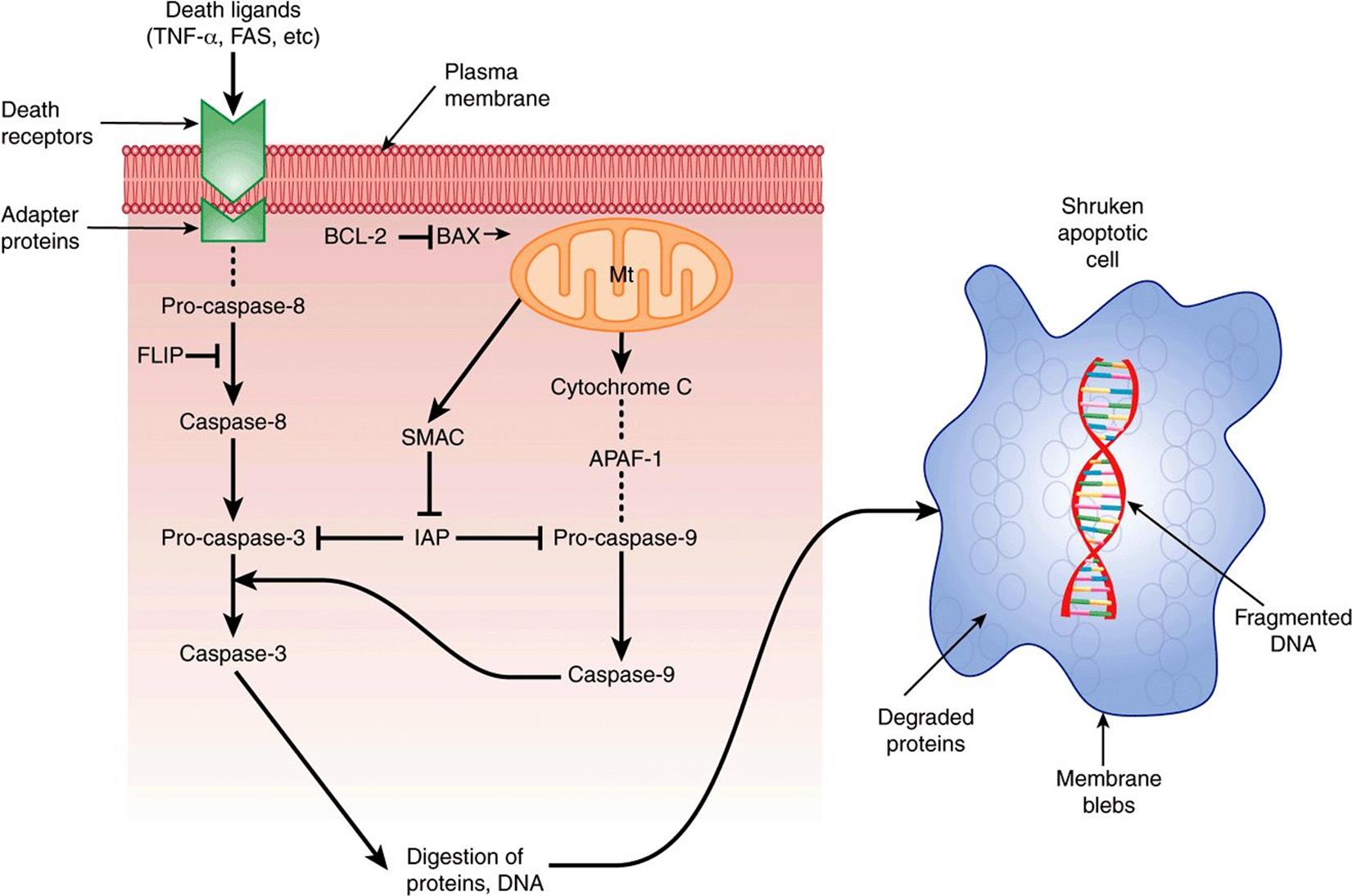
FIGURE 55–12 Scheme of apoptosis, much simplified. The left-hand side represents major events in the extrinsic pathway. Death signals include TNF-α and FAS (present on the surface of lymphocytes and some other cells). The signals (ligands) interact with specific death receptors (there are a number of them). The activated receptor then interacts with an adapter protein (FADD is one of a number of them), and then forms a complex with pro-caspase 8. (The complex is indicated by the … between the receptor and pro-caspase-8 in the Figure). Through a series of further steps, active caspase-3 is formed, which is a major effector (executioner) of cell damage. Regulation of the extrinsic pathway can occur due to the inhibitory effect of FLIP on the conversion of pro-caspase-8 to caspase-8, and also the inhibitory effect of IAP on pro-caspase-3. The right-hand side represents major events in the intrinsic (mt) pathway. Various cell stresses affect the permeability of the mt outer membrane, resulting in efflux of cytochrome c into the cytoplasm. This forms a multi-protein complex with APAF-1 and pro-caspase-9, called an apoptosome. Via these interactions, pro-caspase -9 is converted to caspase-9. This, in turn, can act on pro-caspase-3 to convert it to its active form. Regulation of the intrinsic pathway can occur at the level of BAX, which facilitates increasing mt permeability permitting efflux of cytochrome c, and is thus pro-apoptotic. BCL-2 opposes this effect of BAX and is thus anti-apoptotic. IAP also inhibits pro-caspase-9, and this effect of IAP can be overcome by SMAC. (APAF-1, apoptopic protease activating factor-1; BAX, BCL-2 associated X protein; BCL-2, B-cell CLL/lymphoma 2 (CLL represents chronic lymphatic leukemia); FADD, FAS-associated via death domain; FAS, FAS antigen; FLICE, FADD-like ICE; FLIP, FLICE inhibitory protein; IAP, inhibitor of apoptosis proteins; ICE, interleukin1-p convertase; SMAC, second mitochondria-derived activator of caspase.) ![]() signifies opposes the action of.
signifies opposes the action of.
Major features of the death receptor pathway are shown on the left-hand side of the Figure. External signals initiating apoptosis include the cytokine tumor necrosis factor-α (TNF-α) and FAS ligand. A number of death receptors have been identified. They are transmembrane proteins and some interact with adapter proteins (such as FADD). These complexes in turn interact with procaspase-8, resulting in its conversion to caspase-8 (an initiator). Caspase-3 (an effector) is activated via a series of further reactions. It digests important structural proteins such as lamin (this is associated with nuclear condensation), various cytoskeletal proteins, and enzymes involved in DNA repair, causing cell death.
Regulation of this pathway occurs at several levels. FLIP inhibits the conversion of pro-caspase-8 to its active form. Inhibitors of apoptosis (IAPs) inhibit the conversion of pro-caspase-3 to its active form. These effects can be overcome by the protein SMAC, which is released from mitochondria.
The mitochondrial pathway can be initiated by exposure to reactive oxygen species, DNA damage and other stimuli. This results in pores forming in the outer mitochondrial membrane, through which cytochrome c escapes into the cytoplasm. In the cytoplasm, cytochrome c interacts with APAF-1, procaspase-9 and ATP to form a multi-protein complex known as an apoptosome. As a result of this interaction, procaspase-9 is converted to its active form and, in turn, acts on procaspase-3 to produce caspase-3.
Regarding regulation, activation of the P53 gene up-regulates transcription of BAX. BAX is proapoptotic, in that it causes loss of mitochondrial membrane potential, helping initiate the mitochondrial apoptotic pathway. On the other hand, BCL-2 inhibits the loss of the membrane potential, and is thus antiapoptotic. IAPs inhibit conversion of procaspase 9 to caspase-9; SMAC can overcome this.
Note that the death pathway uses caspase-8 as an initiator, whereas the mitochondrial pathway uses caspase-9. These two pathways can interact. In addition, there are also other pathways of apoptosis not discussed here.
Cancer Cells Evade Apoptosis
Cancer cells have developed ways of evading apoptosis, and thus of continuing to grow and divide. In general, these result from mutations that cause loss of function of proteins that are proapoptotic, or from overexpression of anti-apoptotic genes. One such example concerns loss of function of the P53 gene, perhaps the most commonly mutated gene in cancers. Resultant loss of up-regulation of proapoptotic BAX (see above) shifts the balance in favor of antiapoptotic proteins. Overexpression of many anti-apoptotic genes is a frequent finding in cancers. The resulting evasion of apoptosis favors the continuing growth of cancers. Attempts are being made to develop drugs or other compounds that will specifically turn on apoptosis in cancer cells, terminating their lifespans.
As indicated above, apoptosis is a complex, multiregulated pathway with numerous participants, all of which are not mentioned here in this simplified account. It is also involved in various developmental and physiological processes. It may seem paradoxical, but regulated cell death is as important in maintaining health as is formation of new cells. In addition to cancer, apoptosis is implicated in other diseases, including certain autoimmune and chronic neurological disorders, such as Alzheimer disease and Parkinson disease, where excessive cell death (rather than excessive growth) is a feature.
Table 55-11 summarizes some of the principal features of apoptosis.
TABLE 55–11 Summary of Some Important Points Regarding Apoptosis

EPIGENETIC MECHANISMS ARE INVOLVED IN CANCER
There is growing evidence that epigenetic mechanisms (see Chapter 36) are involved in the causation of cancer. Methylation of specific cytosine bases in genes is implicated in turning off the activities of certain genes. Changes from normal in methylation/demethylation of cytosine residues in specific genes have been detected in cancer cells. Post-translational modifications of histones, such as acetylation, methylation, phosphorylation, and ubiquitination, also affect gene expression. Changes in acetylation of histones H3 and H4, affecting gene transcription, have been found in cancer cells. Mutations affecting the structures of protein complexes (eg, the SW1/SNF complexes) involved in chromatin remodeling can also affect gene transcription. Indeed, several of the components of the SW1/SNF complexes may act as tumor suppressor genes. Some of these points about epigenetics are summarized in Figure 55–13.
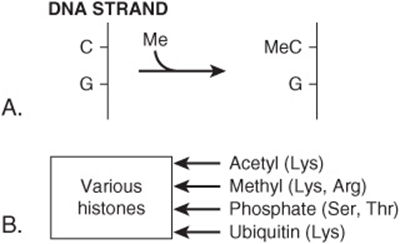
FIGURE 55–13 Some factors involved in epigenetics. (A) Methylation of cytosine to form 5′ methylcytosine. The cytosine is usually located next to a guanine residue, forming a CpG island. Methylation of cytosine by a methyltransferase is associated with silencing of the activities of certain genes. (B) Post-translational modifications of various histones. Specific residues in specific histones are modified by various enzymes, changing the conformations and activities of the modified histones. For example, acetylation of N-terminal lysines in certain histones is associated with opening up of chromatin and with increased transcription of certain genes. (C) Chromatin remodelling: see Figure 36–10.
A matter of particular interest regarding epigenetic changes is that many of them are potentially reversible. In this regard, 5’azadeoxycytidine is an inhibitor of methyltransferases, and suberoylanilide hydroxamic acid (SAHA, Vorinostat) acts to deacetylate histones. Both of these agents have been used to treat certain types of leukemias and lymphomas.
The increasing use of screening techniques for studying epigenetic changes (eg, analysis of the methylome [the sum total of methylation modifications in the genome]) in more types of cancers is likely to add considerably to knowledge in this area.
THERE IS MUCH INTEREST IN THE ROLE OF STEM CELLS IN CANCER
Stem cells were discussed briefly in Chapter 52. Many scientists are currently investigating the role of stem cells in cancer. Cancer stem cells are believed to harbor mutations that, either by themselves or in collaboration with further mutations, make these cells cancerous. They may be detected by the use of specific surface markers, or other techniques. It appears that surrounding tissues (eg, components of the extracellular matrix) can significantly influence the behavior of these cells. An important concept driving some of the research in this area is the belief that one of the reasons that cancer chemotherapy is often not successful is that a pool of cancer stem cells exists that is not susceptible to conventional chemotherapy. Reasons for this include the facts that many stem cells are relatively dormant, have active DNA repair systems, express drug transporters that can expel anticancer drugs, and are often resistant to apoptosis.
Evidence is accumulating that cancer stem cells do indeed play key roles in many types of neoplasia. If so, development of therapies with high specificity for killing these cells will prove of extreme value.
TUMORS OFTEN STIMULATE ANGIOGENESIS
Tumor cells need an adequate blood supply to provide nutrients for their survival. Both tumor cells and cells in tissues surrounding tumors have been found to secrete factors that stimulate the growth of new blood vessels (ie, angiogenesis). There has been much interest in tumor angiogenesis, partly because if it could be specifically inhibited, this could provide a selective method of killing tumor cells.
The growth of blood vessels supplying tumor cells can be stimulated by hypoxia and other factors. Hypoxia causes elevated levels of hypoxia-inducible factor-1 (HIF-1), which in turn increases levels of vascular endothelial growth factor (VEGF), a major stimulant of angiogenesis. Some five types of VEGF have been identified (A-E), with most interest focusing on VEGF A. They interact with specific tyrosine kinase receptors on endothelial and lymphatic cells. These receptors, via signaling pathways, cause up-regulation of the NF-κB pathway (see Chapter 50), resulting in proliferation of endothelial cells and formation of new blood vessels. Blood vessels supplying tumors are not normal; their structure is often disorganized and they are much leakier than normal blood vessels. Molecules other than VEGFs, such as angipoietin, beta-fibroblast growth factor, TGF-α, and placental growth factor, also stimulate angiogenesis. Certain other molecules also inhibit blood vessel growth (eg, angiogenin and endostatin).
Monoclonal antibodies to VEGF A have been developed (eg, bevacizumab or Avastatin) and have been used in the treatment of certain types of cancer (eg, colon and breast). They bind to VEGF and prevent it acting. They were found to increase the overall patient survival, but most patients eventually relapsed. It is felt that they are best used in combination with other anticancer therapies. Monoclonal antibodies to other growth factors that stimulate angiogenesis are also being developed and tried out therapeutically, as are small molecule inhibitors of angiogenesis. Inhibitors of angiogenesis are useful in other conditions, such as “wet” age-related macular degeneration and diabetic retinopathy, in which proliferation of blood vessels is a feature.
METASTASIS IS THE MOST SERIOUS ASPECT OF CANCER
It has been estimated that about 85% of the mortality associated with cancer results from metastasis. Spread of cancer is usually via lymphatics or blood vessels. Metastasis is a complex process, and its molecular bases are yet to be elucidated.
Figure 55–14 is a simplified scheme of metastasis. The earliest event is detachment of tumor cells from the primary tumor. The cells can then gain access to the circulation (or lymphatics), a process termed intravasation. Once in the circulation, they tend to arrest in the nearest small capillary bed. In that site, they extravasate and migrate through the neighboring ECM, before finding a site to settle. Thereafter, if they survive host defense mechanisms, they grow at variable rates. To ensure growth, they need an adequate blood supply, as discussed above. Some aspects of these events are now discussed in more detail.
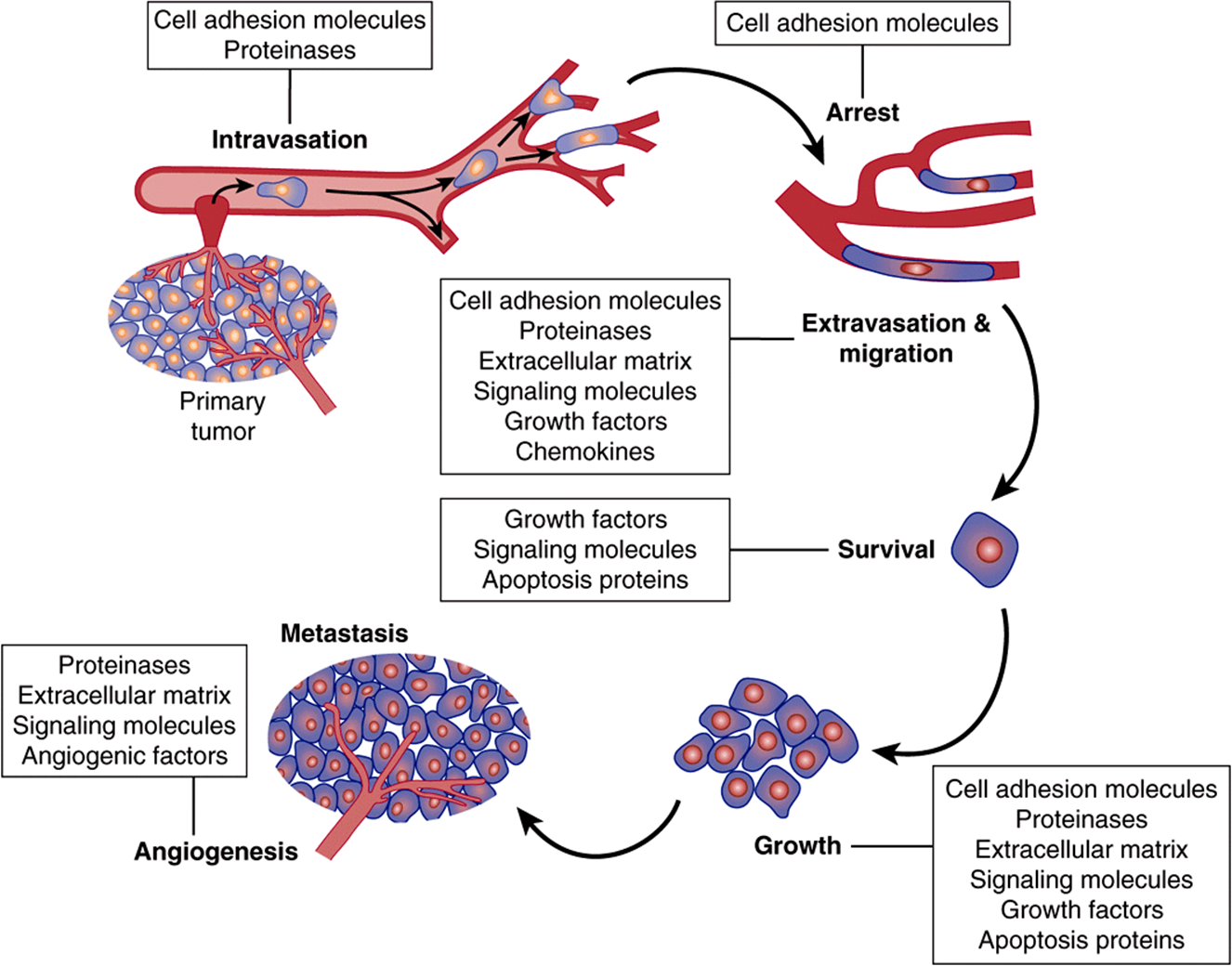
FIGURE 55–14 Simplified scheme of metastasis. Schematic representation of the sequence of steps in metastasis, indicating some of the factors believed to be involved. From Tannock IF et al: The Basic Science of Oncology. 4th ed. McGraw-Hill, 2005.
Many studies have shown changes from normal of molecules on the surfaces of cancer cells. These changes may permit decreased cell adhesion and allow individual cancer cells to detach from the parent cancer. Molecules on cell surfaces involved in cell adhesion are called cell adhesion molecules (CAMs) (see Table 55-12). Decreases of the amounts of E-cadherin, a molecule of major importance in the adhesion of many normal cells, may help to account for the decreased adhesiveness of many cancer cells. Many studies have shown changes in the oligosaccharide chains of cell surface glycoproteins, due to altered activities of various glycosyltransferases (see Chapter 47). One important change is an increase of the activity of GlcNAc transferase V. This enzyme catalyzes transfer of GlcNAc to a growing oligosaccharide chain, forming a β1->6 linkage and allowing further growth of the chain. It has been proposed that such elongated chains participate in an altered glycan lattice at the cell surface. This may cause structural re-organization of receptors and other molecules, perhaps predisposing to the spread of cancer cells.
TABLE 55–12 Some Important Cell Adhesion Molecules (CAMs)

An important property of many cancer cells is that they can release various proteinases into the ECM. Of the four major classes of proteinases (serine, cysteine, aspartate and metallo-), particular interest in cancer has focused on the metalloproteinases (MPs), which constitute a very large family of metal-dependent (usually zinc) enzymes. A number of studies have shown increased activity in tumors of MPs such as MP-2 and MP-9 (also known as gelatinases). These enzymes are capable of degrading proteins in the basement membrane and in the ECM, such as collagen and others, facilitating the spread of tumor cells. Inhibitors of these enzymes have been developed, but so far these have not had any clinical success.
A factor that allows increased movement of cancer cells is epithelial mesenchymal transition. This is a change of cell morphology and function from epithelial to mesenchymal type, perhaps induced by growth factors. The mesenchymal type has more actin filaments, permitting increased movement, an essential property of cells that metastasize.
The ECM plays an important role in metastasis. There is evidence of communication by signaling mechanisms between cancer cells and cells of the ECM. The types of cells in the ECM can also affect metastasis. As mentioned above, proteinases that degrade proteins in the ECM can facilitate spread of cancer cells. In addition, the ECM contains various growth factors that can influence tumor behavior.
On their travels, tumor cells are exposed to various cells of the immune system (such as T cells, NK cells, and macrophages), and must be able to survive exposure to them. Some of these surveillance cells secrete various chemokines, small proteins that can attract various cells such as leukocytes, sometimes causing an inflammatory response to tumor cells.
It has been estimated that only about 1:10,000 cells may have the genetic capacity to successfully colonize. Certain tumor cells show a predilection to metastasize to specific organs (eg, prostate cells to bone); specific cell surface molecules may be involved in this tropism.
Various studies have shown that certain genes enhance metastasis, whereas others act as metastasis suppressor genes. How exactly these genes work is the subject of ongoing studies. However, their existence raises the possibility that increasing the expression of a tumor suppressor gene by a drug or other method might be of therapeutic benefit.
Table 55-13 summarizes some important points regarding metastasis.
TABLE 55–13 Some Important Points Regarding Metastasis

CANCER CELLS EXHIBIT A HIGH RATE OF AEROBIC GLYCOLYSIS
Many aspects of the metabolism of cancer cells (eg, of carbohydrates, lipids, amino acids, and nucleic acids) have been and are being studied. One current approach is analysis of blood and urine by mass spectrometry looking for alterations in metabolite profile that can help detect cancer at an early stage. Here we shall describe only a few of the studies performed on the metabolism of glucose.
Glucose and the amino acid glutamine are two of the most abundant metabolites in plasma, and together they account for much of the carbon and nitrogen metabolism in human cells. In the 1920s, the German biochemist Otto Warburg and his colleagues made a seminal discovery regarding the biochemistry of cancer cells. They found that cancer cells take up large amounts of glucose and metabolize it to lactic acid, even in the presence of oxygen (the so-called Warburg effect). The reasons for this high rate of aerobic glycolysis are still under investigation. Warburg postulated that it could be due to a defect in the respiratory chain, so that tumor cells compensated for this by producing more ATP via glycolysis. Subsequent work has produced a number of other explanations, including that tumor cells have less mitochondria than normal cells and that they contain a mitochondrial-bound isozyme of hexokinase (HK-2) that is not subject to feedback control, allowing increased uptake of glucose. Recent work has focused on the finding that tumor cells exhibit a different isozyme of pyruvate kinase (PK) (see Chapter 18). Normal cells contain PK-1 and tumor cells contain PK-2; they are produced by alternative splicing of the same gene product. For complex reasons that are still to be fully elucidated, this change of isozyme profile is associated with, or leads to less production of ATP (see Figure 55–15). It also is thought to allow increased use of metabolites supplied by glycolysis for building up the biomass (proteins, lipids, nucleic acids, etc) required for proliferation of cancer cells. This offers an explanation for the presumed selective advantage conferred on tumor cells by having a high rate of glycolysis.
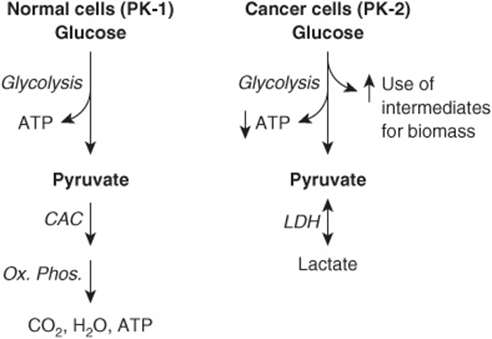
FIGURE 55–15 Pyruvate kinase isozymes and glycolysis in normal and in cancer cells. In normal cells, the major source of ATP is oxidative phosphorylation. Some ATP is obtained from glycolysis. The major pyruvate kinase (PK) isozyme in normal cells is PK-1. In cancer cells, aerobic glycolysis is prominent, lactic acid is produced via the action of lactate dehydrogenase (LDH) and production of ATP from oxidative phosphorylation is diminished (not shown in the Figure). In cancer cells, PK-2 is the major PK isozyme. For complex reasons not as yet fully understood, this change of isozyme profile in cancer cells is associated with decreased net production of ATP from glycolysis, but increased use of metabolites to build up biomass. (CAC, citric acid cycle; OX PHOS, oxidative phosphorylation.)
Despite angiogenesis, many solid tumors have localized areas of poor blood supply, and thus show high rates of anaerobic glycolysis. This leads to excessive production of lactic acid and local acidosis. It has been postulated that local production of acid may allow tumor cells to invade more easily. The low oxygen tension in areas of tumors with poor blood supply stimulates the formation of hypoxia-inducible factor-1 (HIF-1). This transcription factor, whose activity is turned on by low oxygen tension, up-regulates—among other actions—the activities of at least eight genes controlling synthesis of glycolytic enzymes.
The pH and oxygen tension in tumors are important factors affecting the actions of anti-cancer drugs and other treatments. For example, the anti-cancer efficacy of radiation treatment of cancers is significantly lower in hypoxic conditions. Chemicals have been developed to inhibit glycolysis in tumor cells, and perhaps selectively kill them (see Table 55-14). Although found to have variable effectiveness in preclinical studies, so far none of them have attained much clinical use. They include 3-bromopyruvate (an inhibitor of HK-2) and 2-deoxy-D-glucose (an inhibitor of HK-1). Another compound, dichloroacetate (DCA), inhibits the activity of pyruvate dehydrogenase kinase, and thus stimulates the activity of pyruvate dehydrogenase (see Chapter 18), diverting substrate from glycolysis into the citric acid cycle. So far, none of these has attained much clinical use.
TABLE 55–14 Some Compounds that Inhibit Glycolysis and Have Been Found to Display Variable Anticancer Activity

MITOCHONDRIA SHOW ALTERATIONS IN CANCER CELLS
Mitochondria are involved in various aspects of cancer. The possibility that a diminished number of mitochondria are involved in the Warburg effect in cancer cells was mentioned above. Mitochondria generate reactive oxygen species (ROS), which can damage DNA, and they are also involved in apoptosis (see above). Mutations affecting mitochondrial (mt) DNA and thus various mt proteins have also been detected although their precise roles in the biology of cancer cells have not been established. The possible involvement of mitochondrial-bound hexokinase-2 in the Warburg effect was mentioned above. Another mitochondrial enzyme that has attracted considerable attention recently is isocitrate dehydrogenase (IDH) (see Chapter 18). Mutations of this enzyme have been detected in gliomas and acute myeloid leukemia. These cause an elevation of 2-hydroxyglutarate, which may have effects on the tumor cells. Figure 55–16 summarizes some of these points regarding mitochondria.
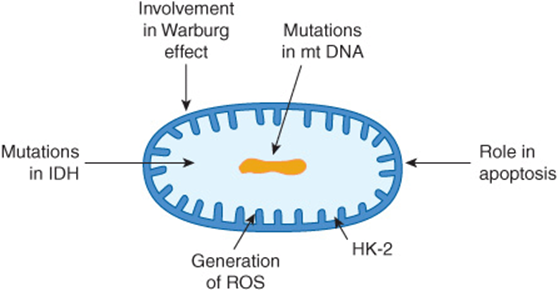
FIGURE 55–16 Some involvements of mitochondria in cancer. Mitochondria are involved in a number of ways in cancer, not all of which are shown here. Possible explanations for the Warburg effect are discussed in the text. Mutations in mtDNA are found in cancer cells, although their significance is not fully understood. Mitochondria play an important role in apoptosis, with release of cytochrome c into the cytoplasm being an important event. Many cancer cells have an isozyme of hexokinase (HK-2), that plays a role in the increased uptake of glucose by tumor cells. Reactive oxygen species (ROS) are generated in mitochondria, and may be important in causing mutations. Recent studies have shown that certain tumor cells have an abnormal isozyme of isocitrate dehydrogenase (IDH), the significance of which is under study.
TUMOR BIOMARKERS CAN BE MEASURED IN BLOOD SAMPLES
Biochemical tests (see Chapter 56) are often helpful in the management of patients with cancer (eg, some patients with advanced cancers may have elevated levels of plasma calcium, which can cause serious problems if not attended to). Many cancers are associated with the abnormal production of enzymes, proteins, and hormones that can be measured in plasma or serum. These molecules are known as tumor biomarkers. Some of them are listed in Table 55-15.
TABLE 55–15 Some Useful Tumor Biomarkers Measurable in Blood
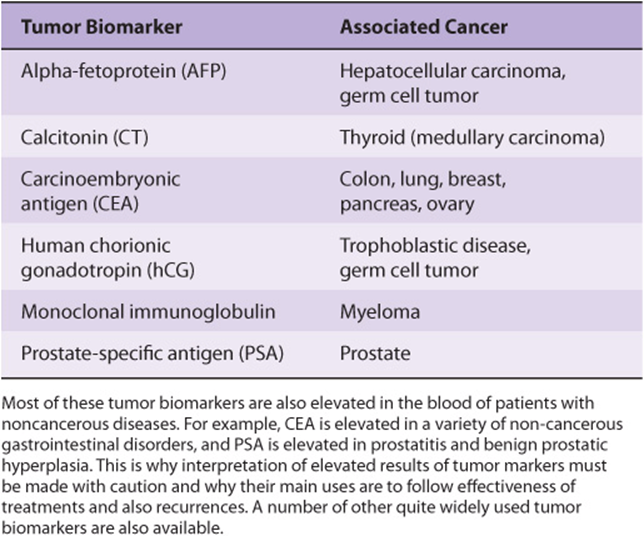
However, significant elevations of some of the biomarkers listed in Table 55-15 also occur in a variety of non-cancerous conditions. For example, elevations of the level of prostate-specific antigen (PSA), a glycoprotein synthesized by prostate cells, occur not only in patients with cancer of the prostate, but also in patients with prostatitis and benign prostatic hyperplasia (BPH). Similarly, elevations of carcinoembryonic antigen (CEA) are found not only in patients with various types of cancer, but also in heavy smokers and people with ulcerative colitis and cirrhosis. The fact that elevations of tumor biomarkers are usually not specific for cancer has meant that measurements of most of them are not used primarily for diagnosis of cancer. Their main uses have been in following the effectiveness of treatments and in detecting early recurrence. The use of CEA in the management of a patient with colorectal cancer is discussed briefly in Chapter 57, Case 4. As with other laboratory tests (Chapter 56), the entire clinical picture must be considered when interpreting the results of measurements of tumor biomarkers.
It is hoped that ongoing research on proteomics will provide new tumor biomarkers of increased sensitivity and specificity, and those capable of alerting one to the presence of cancers at an early stage of their development.
Use of microarray analysis of cancer cells has revealed certain useful biomarkers. It has also been of use in subclassifying tumors (eg, breast) and has provided useful information regarding prognosis and prediction of response to therapy. This is a rapidly expanding area of laboratory analysis.
KNOWLEDGE OF MECHANISMS INVOLVED IN CARCINOGENESIS HAS LED TO THE DEVELOPMENT OF NEW THERAPIES
One of the great hopes of cancer research is that revealing the fundamental mechanisms involved in cancer will lead to new and better therapy. This has already occurred to a certain extent, and it is hoped that ongoing developments will accelerate this process.
Classical chemotherapeutic drugs include alkylating agents, platinum complexes, antimetabolites, spindle poisons, and others. These will not be discussed here.
Among the classes of drugs developed more recently are inhibitors of signal transduction (including tyrosine kinase inhibitors), monoclonal antibodies directed to various target molecules, inhibitors of hormone receptors, drugs that affect differentiation, anti-angiogenesis agents, and biologic response modifiers. Examples of each of these are listed in Table 55-16. A few comments will now be made concerning some of these drugs.
TABLE 55–16 Some Anticancer Agents That are Based on Recent Advances in Knowledge of Cancer Biology

The finding of widespread defects in signaling mechanisms in cancer cells, and in particular the detection of mutations in tyrosine kinases, has led to the development of inhibitors of these enzymes. The most dramatic success has probably been the introduction of imatinib for the treatment of chronic myelocytic leukemia (CML). Imatinib is an orally administered drug that inhibits the tyrosine kinase formed due to the ABL-BCR chromosomal translocation involved in the genesis of CML. This drug has produced complete remissions in many patients. It can be combined with other drugs. Other tyrosine kinase inhibitors have also been developed. Two of these are Erlotinib and Gefinib, which inhibit the epidermal growth factor receptor (EGFR). This molecule is over-expressed in certain lung (eg, non-small cell cancers) and breast cancers, resulting in aberrant signaling. It is important to appreciate that the design of these anti-cancer drugs and of other drugs requires detailed structural knowledge (as provided by X-ray crystallography, NMR studies, model building, etc) of the molecules being targeted. Another class of drugs that have proven useful are monoclonal antibodies to various molecules on the surfaces of neoplastic cells. A few of these are listed in Table 55-16.
Other approaches to treating cancer that are being used or being developed, but are not listed in Table 55-16, include various types of gene therapy (including siRNAs, Chapter 34), immunotherapy (see below), oncolytic viruses (viruses that preferentially invade tumor cells and kill them), inhibitors of the progesterone receptor, aromatase inhibitors (see Chapter 41) (for some breast and ovarian cancers), telomerase inhibitors, applications of nanotechnology (eg, nanoshells and other nanoparticles), phototherapy (see Chapter 31), and drugs that will selectively target cancer stem cells.
It is important to appreciate that all anticancer drugs have side effects, sometimes severe, and that resistance to many of them can develop after variable time periods. The biochemistry of how cancer cells develop resistance to drugs is an important area of research that will not be described here. The overall thrust of drug development for cancer therapy is to use new information emerging from basic studies of cancer biology to develop safer and more effective agents. Understanding genetic differences in handling drugs (see Chapter 53) may also help to personalize anti-cancer drug treatments.
Figure 55–17 summarizes some of the targets for drug therapy and some emerging therapies that have developed from relatively recent studies of basic aspects of cancer.
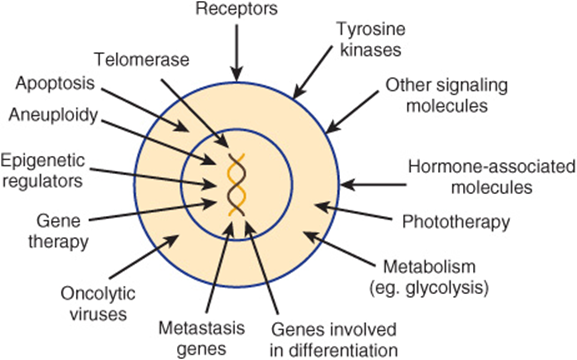
FIGURE 55–17 Some targets for anti-cancer drugs and some emerging therapies, both of which have developed from relatively recent research. Not shown in the Figure are anti-angiogenic agents, applications of nanotechnology, therapies directed against cancer stem cells and immunologic approaches. Most of the targets and therapies indicated are discussed briefly in the text.
THERE ARE MANY IMMUNOLOGIC ASPECTS OF CANCER
Tumor immunology is a large subject. Only a few comments will be made here concerning it. It seems probable that the decline in immune responsiveness that accompanies aging plays a role in the increased incidence of cancer in older people. One long-standing hope has been that immunologic approaches, because of their specificity, might be able to kill off cancer cells selectively. There are many ongoing trials investigating this possibility. These involve the use of various antibodies, vaccines, and the use of various types of T cells that generally have been manipulated in one way or another to increase their ability to kill neoplastic cells.
Chronic inflammation involves aspects of immune function. There is evidence that it can predispose to cancer (eg, the incidence of colorectal cancer is much higher than normal in individuals who have had long-standing ulcerative colitis). Some inflammatory cells produce relatively large amounts of reactive oxygen species, which can cause damage to DNA, and perhaps contribute to oncogenesis. It has also been reported that low doses of aspirinmay lower the risk of development of colorectal cancer, perhaps via its antiinflammatory action.
SUMMARY
![]() Cancer is due to mutations in genes controlling cell multiplication, cell death (apoptosis) and cell-cell interactions (eg, cell adhesion). Other important aspects of cancer are widespread defects in cell signaling pathways, stimulation of angiogenesis, and aneuploidy.
Cancer is due to mutations in genes controlling cell multiplication, cell death (apoptosis) and cell-cell interactions (eg, cell adhesion). Other important aspects of cancer are widespread defects in cell signaling pathways, stimulation of angiogenesis, and aneuploidy.
![]() The great majority of cancers are due to mutations affecting somatic cells. However, many hereditary cancer conditions have been identified.
The great majority of cancers are due to mutations affecting somatic cells. However, many hereditary cancer conditions have been identified.
![]() Major classes of genes involved in cancer are oncogenes and tumor suppressor genes. Mutations affecting genes directing the synthesis and expression of microRNAs are also important.
Major classes of genes involved in cancer are oncogenes and tumor suppressor genes. Mutations affecting genes directing the synthesis and expression of microRNAs are also important.
![]() Epigenetic changes are increasingly being recognized in cancer (and in other diseases); one reason for interest in them is that they may be reversible changes.
Epigenetic changes are increasingly being recognized in cancer (and in other diseases); one reason for interest in them is that they may be reversible changes.
![]() The biology of metastasis is being explored intensively; the discovery of metastasis enhancer and suppressor genes, among other findings, may lead to new therapies.
The biology of metastasis is being explored intensively; the discovery of metastasis enhancer and suppressor genes, among other findings, may lead to new therapies.
![]() The process of apoptosis was described briefly. Cancer cells acquire mutations that permit them to evade apoptosis, thus prolonging their existence.
The process of apoptosis was described briefly. Cancer cells acquire mutations that permit them to evade apoptosis, thus prolonging their existence.
![]() Whole genome sequencing of cancers is helping to reveal the important mutations present in many types of cancer and is yielding new information on the evolution of cancer cells.
Whole genome sequencing of cancers is helping to reveal the important mutations present in many types of cancer and is yielding new information on the evolution of cancer cells.
![]() Cancer cells show various alterations of metabolism. One major finding that has attracted much attention is the high rate of aerobic glycolysis exhibited by many cells. Possible explanations for this phenomenon are described. Mitochondria show some alterations in many cancer cells.
Cancer cells show various alterations of metabolism. One major finding that has attracted much attention is the high rate of aerobic glycolysis exhibited by many cells. Possible explanations for this phenomenon are described. Mitochondria show some alterations in many cancer cells.
![]() Overall, the development of cancer is a multi-step process involving genetic changes that confer selective advantages on clones of cells, some of which eventually acquire the ability to metastasize successfully. Because of the diversity of mutations, it is possible that no two tumors have identical genomes.
Overall, the development of cancer is a multi-step process involving genetic changes that confer selective advantages on clones of cells, some of which eventually acquire the ability to metastasize successfully. Because of the diversity of mutations, it is possible that no two tumors have identical genomes.
![]() Tumor markers may help in the early diagnosis of cancer. They are of particular use in following the response of cancer to treatment and in detecting recurrences.
Tumor markers may help in the early diagnosis of cancer. They are of particular use in following the response of cancer to treatment and in detecting recurrences.
![]() Advances in understanding the molecular biology of cancer cells have led to the introduction of a number of new therapies, with others in the pipeline.
Advances in understanding the molecular biology of cancer cells have led to the introduction of a number of new therapies, with others in the pipeline.
REFERENCES
Berger M, Lawrence, MS, Demechelis F, et al: The genomic complexity of primary human prostate cancer. Nature 2011; 470(7333):214.
Bielas JH, Loeb KR, Rubin BP, et al: Human cancers express a mutator phenotype. Proc Natl Acad Sci USA 2006;103(48):18238.
Couzin-Frankel J: Immune therapy steps up the attack. Science 2010;330:440.
Croce CM: Oncogenes and cancer. N Engl J Med 2008;358(5):502. (One of a series of 12 articles on the molecular origins of cancer published in this journal between Jan 31, 2008 and Dec 17, 2009).
Dick JE: Stem cell concepts renew cancer research. Blood 2008;112:4793.
Fauci AS, Braunwald E, Kasper DL, et al (editors): Harrison’s Principles of Internal Medicine. 17th ed. McGraw-Hill, 2008. (A number of Chapters cover various aspects of cancer, but Chapters 77-81 and 103-106 are particularly relevant to this Chapter).
Green DR: Means to an End: Apoptosis and Other Cell Death Mechanisms. Cold Spring Harbor Press, 2010.
Kaiser J: Epigenetic drugs take on cancer. Science 2010;330:576. (Also see other articles on Epigenetics in the same issue of Science),
Lodish H, Berk A, Kaiser CA, et al: Molecular Cell Biology. 6th ed. WH Freeman & Co, 2008. (Contains a comprehensive chapter on cancer).
Mukerjee S: The Emperor of All Maladies: A Biography of Cancer. Scribner, 2010.
Nussbaum RL, McInnes, RR, Willard HF: Thompson and Thompson Genetics in Medicine. 7th ed. Saunders Elsevier, 2007.
Tannock IF, Hill RP, Bristow RG, Harrington L (editors): The Basic Science of Oncology. 4th ed. McGraw-Hill, 2005.
Thompson SL, Bakhoum SF, Compton DA: Mechanisms of chromosomal instability. Curr Biol 2010; 20(6): R285.
Weinberg R: The Biology of Cancer, Garland Science. 2006.
Yachida S, Jones S, Bozic I, et al: Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010 (Oct 28);467(7319):1114.
Journals dedicated to cancer research include the British Journal of Cancer, Cancer Research, Journal of the National Cancer Institute and Nature Reviews Cancer.
Useful Web Sites:
American Cancer Society. http://www.cancer.org
National Cancer Institute, U.S. National Institute of Health. http://www.cancer.gov
The Cancer Genome Atlas. http://cancergenome.nih.gov
GLOSSARY
Adenomatous polyp: A benign tumor of epithelial origin which has the potential to become a carcinoma. Adenomas are often polypoid. A polyp is a growth that protrudes from a mucous membrane; most are benign, but some polyps can become malignant.
Ames assay: An assay system devised by Dr. Bruce Ames that uses specially designed Salmonella typhimurium to detect mutagens. Most carcinogens are mutagens, but if mutagenicity of a chemical is detected, ideally it should be tested for carcinogenicity by animal testing.
Aneuploidy: Refers to any condition in which the chromosome number of a cell is not an exact multiple of the basic haploid number. Aneuploidy is found in many tumor cells and may play a fundamental role in the development of cancer.
Angiogenesis: The formation of new blood vessels. Angiogenesis is often active around tumor cells, ensuring that they obtain an adequate blood supply. A number of growth factors are secreted by tumor and surrounding cells (eg, vascular endothelial growth factor, VEGF) and are involved in this process.
Apoptosis: Cell death due to activation of a genetic program that causes fragmentation of cellular DNA and other changes. Caspases play a central role in the process. Many positive and negative regulators affect it. The protein p53 induces apoptosis as a response to cell DNA damage. Most cancer cells exhibit abnormalities of apoptosis, due to various mutations that help to ensure their prolonged survival.
Biologic response modifiers: Molecules produced by the body or in the laboratory that when administered to patients alter the body’s response to infection, inflammation and other processes. Examples include monoclonal antibodies, cytokines, interleukins, interferons, and growth factors.
Bloom syndrome: One of the CI syndromes. Due to mutations in a DNA helicase. Subjects are sensitive to DNA damage and may develop various tumors.
Burkitt’s lymphoma: This is a B cell lymphoma, endemic in parts of Africa, where it mainly affects the jaw and facial bones. It is also found elsewhere. A reciprocal translocation involving the C-MYC gene on chromosome 8 and the immunoglobulin heavy chain gene on chromosome 14 is characteristic.
Cancer: A malignant growth of cells.
Cancer stem cell: A cell within a tumor that has the capacity to self-renew and to give rise to the heterogeneous lineages of cancer cells found in the tumor.
Carcinogen: Any agent (eg, a chemical or radiation) capable of causing cells to become cancerous.
Carcinoma: A malignant growth of epithelial origin. A cancer of glandular origin or showing glandular features is usually designated as an adenocarcinoma.
Caspases: Proteolytic enzymes that play a central role in apoptosis, but are also involved in other processes. Some 15 are present in humans. Caspases hydrolyse peptide bonds just C-terminal to aspartate residues.
Cell cycle: The various events pertaining to cell division that occur as a cell goes from one mitosis to another.
Centriole: An array of microtubules that is paired and found in the center of a centrosome. (Also see Centrosome.)
Centromere: The constricted region of a mitotic chromosome where chromatids are joined together. It is in close proximity to the kinetochore. Abnormalities of centromeres may contribute to CI. (Also see Kinetochore.)
Centrosome: A centrally located organelle that is the primary microtubule-organizing center of a cell. It acts as the spindle pole during cell division.
Chromatid: A single chromosome.
Chromatin remodeling: This involves conformational changes of nucleosomes brought about by the actions of multiprotein complexes (such as the SW1/SNF complex). These changes alter gene transcription (turning it on or off, depending on specific conditions). The complexes contain domains homologous to ATP-dependent helicase; these are involved in the changes of conformation. Mutations affecting proteins of the complexes, such as may be found in cancer cells, can affect gene expression. (See also Epigenetics.)
Chromosomal instability (CI): The rate of gain or loss of whole chromosomes or segments of them caused by abnormalities of chromosome segregation during mitosis. (See also Genome instability and Microsatellite instability.) There are a number of disorders that are named CI syndromes because they are associated with chromosomal abnormalities. One such is Bloom syndrome, in which a high frequency of sister chromatid exchanges is observed. An increased incidence of various cancers is found in these conditions.
Chromosomal passenger complex: A complex of proteins that plays a key role in regulating mitosis. At the centromere, it directs alignment of the chromosome and participates in spindle assembly. Its proteins include aurora B kinase and survivin. Mutations affecting its proteins may contribute to CI and aneuploidy.
Chromosomal translocation: When part of one chromosome becomes fused to another, often causing activation of a gene at the site. The Philadelphia chromosome (see below) is one of many examples of a chromosomal translocation involved in the causation of cancer.
Clone: All the cells of a clone are derived from one parent cell.
Copy number variations (CNVs): Variations (because of duplications or deletions) among individuals as to the number of copies they have of particular genes. CNVs are being increasingly recognized for various genes, and some may be associated with various diseases, including certain types of cancer.
Driver mutation: A mutation in a gene that either helps cause cancer or accelerates it. Mutations found in tumors that do not cause cancer or its progression are called passenger mutations.
Epigenetic: Refers to changes of gene expression without change of the sequence of bases in DNA. Factors causing epigenetic changes include methylation of bases in DNA, posttranslational modifications of histones and chromatin remodeling.
FAS receptor: A receptor that initiates apoptosis when it binds its ligand, FAS. FAS is a protein present on the surfaces of activated natural killer cells, cytotoxic T lymphocytes and other sources.
Genome instability: This refers to a number of alterations of the genome, of which the two principal ones are CI and microsatellite instability. In general, it reflects the fact that the genomes of cancer cells are more susceptible to mutations than are normal cells, in part due to impairment of DNA repair systems.
Growth factors: A variety of polypeptides secreted by many normal and tumor cells. These molecules act via autocrine (affects the cells that produce the growth factor), paracrine (affects neighboring cells) or endocrine (travels in the blood to affect distant cells) modes. They stimulate proliferation of target cells via interactions with specific receptors. They also have many other biologic properties.
Hypoxia-inducible factors: A family of transcription factors (at least three) important in directing cellular responses to varying levels of oxygen. Each factor is made up of a different oxygen-regulated α-subunit and a common constitutive β-subunit. At physiological levels of oxygen, the α-subunit undergoes rapid degradation, initiated by prolyl hydroxylases. HIFs have various functions; eg HIF-1 up-regulates various genes encoding enzymes of glycolysis, and also the expression of vascular endothelial growth factor (VEGF).
Kinetochore: A structure that forms on each mitotic chromosome adjacent to the centromere. Mutations affecting the structures of its component proteins could contribute to causing CI. (See also Centromere.)
Leukemias: A variety of malignant diseases in which various white cells (eg, myeloblasts, lymphoblasts, etc) proliferate in an unrestrained manner. Leukemias may be acute or chronic.
Loss of heterozygosity (LOH): This occurs when there is loss of the normal allele (often encoding a tumor suppressor gene) from a pair of heterozygous chromosomes, allowing the results of the defective allele to be manifest clinically.
Lymphoma: A group of neoplasms arising in the reticuloendothelial and lymphatic systems. Major members of the group are Hodgkin and non-Hodgkin lymphomas.
Malignant cells: They are cancer cells—cells with the ability to grow in an unrestrained manner, to invade, and to spread (metastasize) to other parts of the body.
Metastasis: The ability of cancer cells to spread to distant parts of the body and grow there.
Microarray analysis: Analysis using a wafer, often made of silicon, on which thousands of different nucleic acids have been spotted. Samples of cDNA or genomic DNA are applied and allowed to hybridize (binding between complementary sequences). The interactions can be quantitated to assess gene expression or other parameters. Various commercial microarrays (“gene chips”) are available. This technique is being increasingly applied to patients with cancers to help in tumor classification, and in predictions of prognosis and response to drug therapy.
Microsatellite instability: Expansion or contraction of short tandem repeats (microsatellites) due to replication slippage, abnormalities of mismatch repair or of homologous recombination. For Microsatellites, see Chapter 35.
Nanotechnology: The development and application of devices which are only a few nanometers in size. (10-9 m = 1 nm). Some are being applied to cancer therapy. For example, nanoshells (very small spherical particles with a silica core and a gold covering) tuned to near-infrared light have been administered to mice with tumors, in which the nanoshells accumulate. The tumors were subsequently subjected to near infrared laser light. This heated the tumors selectively, killed them, and there was no sign of recurrence on follow-up. (Morton JG et al: Nanoshells for photothermal cancer therapy. Methods Mol Biol 2010;624:101. June 25, 2010).
Necrosis: Cell death induced by chemicals or tissue injury. Various hydrolytic enzymes are released and digest cellular molecules. It is not a genetically programmed process, as is apoptosis. Affected cells usually burst and release their contents, causing local inflammation.
Neoplasm: Any new growth of tissue, benign or malignant
Oncogene: A mutated proto-oncogene whose protein product is involved in the transformation of a normal cell to a cancer cell.
Oncology: The area of medical science that concerns itself with all aspects of cancer (causes, diagnosis, treatment, etc).
Philadelphia chromosome: A chromosome formed by a reciprocal translocation between chromosomes 9 and 22. It is the cause of chronic myeloid leukemia (CML). To form the abnormal chromosome, part of the BCR(breakpoint cluster region) gene of chromosome 22 fuses with part of the ABL gene (encodes a tyrosine kinase) of chromosome 9, directing the synthesis of a chimeric protein that has unregulated tyrosine kinase activity and drives cell proliferation. The activity of this kinase is inhibited by the drug Imatinib (Gleevec), which has been successfully used to treat CML. (See also Chromosomal translocation.)
Procarcinogen: A chemical that becomes a carcinogen when altered by metabolism.
Proto-oncogene: A normal cellular gene, which when mutated can give rise to a product that stimulates the growth of cells, contributing to the development of cancer.
Replication slippage: A process in which, because of misalignment of DNA strands where repeat sequences occur, DNA polymerase pauses and dissociates, resulting in deletions or insertions of repeat sequences.
Retinoblastoma: A rare tumor of the retina. Mutation of the RB tumor suppressor gene plays a key role in its development. Patients with hereditary retinoblastomas have inherited one mutated copy of the RB gene, and need only one further mutation to develop the tumor. Patients with sporadic retinoblastomas are born with two normal copies, and require two mutations to inactivate the gene.
Rous sarcoma virus (RSV): An RNA tumor virus that causes sarcomas in chickens. It was discovered in 1911 by Peyton Rous. It is a retrovirus, using reverse transcriptase in its replication; the DNA copy of its genome subsequently integrates into the host cell genome. It has been widely used in studies of cancer, and its use has led to many important findings.
Sarcoma: A malignant tumor of mesenchymal origin (eg, from cells of the extracellular matrix or other sources).
Telomeres: Structures at the ends of chromosome that contain multiple repeats of specific hexanucleotide DNA sequences. The telomeres of normal cells shorten on repeated cell division, which may result in cell death. The enzyme telomerase replicates telomeres and is often expressed in cancer cells, helping them to evade cell death. Telomerase is usually not detected in normal somatic cells.
Translocation: The displacement of one part of a chromosome to a different chromosome or to a different part of the same chromosome. Classic examples are the translocation found in Burkitt’s lymphoma (see above) and the translocation between chromosomes 9 and 22, which causes the appearance of the Philadelphia chromosome found in chronic myelogenous leukemia. Translocations have been found in many cancer cells.
Transformation: The process by which normal cells in tissue culture become changed to abnormal cells (eg, by oncogenic viruses or chemicals), some of which may be malignant.
Tumor: Any new growth of tissue, but usually refers to a benign or malignant neoplasm.
Tumor suppressor gene: A gene whose protein product normally restrains cell growth, but when its activity is lost or reduced by mutation contributes to the development of a cancer cell.