CHEMICAL BIOLOGY
Natural Products as Anticancer Agents
David G. I. Kingston, Department of Chemistry, Virginia Polytechnic Institute and State University, Blacksburg, Virginia
David J. Newman, Natural Products Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Frederick, Maryland
doi: 10.1002/9780470048672.wecb369
Natural products have provided some of the most effective drugs for the treatment of cancer, including such well-known drugs as paclitaxel (Taxol™; Bristol-Myers Squibb) adriamycin, vinblastine, and vincristine. Natural products have also provided many compounds that have led to the discovery of new biochemical mechanisms. This review summarizes the major natural products in clinical use today and introduces several new ones on the cusp of entering clinical practice. The review is organized by mechanism of action, with compounds that interact with proteins discussed first, followed by compounds that interact with RNA or DNA.
Natural products were the original source of almost all the drugs used by mankind before 1900, and they continue to be a major source of new drugs and drug leads (1, 2). The reasons for the continued importance of natural products are not hard to discover. In the first place, a high correlation exists between the properties of drugs and those of natural products (3, 4). In addition, natural products usually have built-in chirality, and they are thus uniquely suited to bind to complex proteins and other biologic receptors. Finally, natural products have been enormously successful as drugs and drug leads, not only in the anticancer area but also in many other pharmaceutical areas (5). It is thus unsurprising that several authors have gone on record as advocating an increase in the drug discovery effort assigned to natural products (4, 6, 7).
This review summarizes the contributions of natural products to the discovery and development of anticancer agents. It includes information on many natural products and natural product analogs that are in clinical use as anticancer drugs, and it describes some natural product drugs in late-stage clinical trials. Because of space limitations, it is does not provide a comprehensive listing of all natural product and natural product-derived anticancer agents; readers interested in such a listing should consult a recent review (8).
Overview of Natural Products as Anticancer Agents
As of the time of writing, 178 drugs are approved world-wide for the treatment of cancer in all of its manifestations, and 175 of these are listed together with their classifications as to source in a recent review (8). Of the 178 approved antitumor agents, 25 (14%) are natural products, 48 (27%) are modified natural products, and 20 (11%) are synthetic compounds derived from a natural product pharmacophore. Natural products have thus led to 52% of the approved drugs against the collection of diseases that go by the collective name of cancer. Another 20 drugs (11%) are biologics, with the remainder being synthetic compounds. The highly significant contribution of natural products to anticancer drug discovery is clear from these figures.
The term “natural product” describes a broad class of anticancer agents, which range from complex compounds like paclitaxel and vinblastine to relatively simple compounds such as combretastatin-A4. Associated with these different structures are several different mechanisms of action, some of which were only discovered when the corresponding natural product was investigated. The following sections are divided on the basis of the mechanism of action of the drugs rather than their source, and consequently, any given section may include plant, microbial, or marine-derived agents. The broadest division is between those compounds that act by targeting proteins in some way and those compounds that act by direct interactions with DNA or RNA. Thus, these two broad areas provide the two major sections of this review.
Compounds that Target Proteins
The mammalian cell cycle is a complex and carefully regulated biologic process that leads to cell division, and faulty regulation of this cycle is one feature of most cancers. The cell cycle thus offers several targets for therapeutic intervention, and several of the proteins involved either directly or indirectly in controlling this cycle are the targets of some important anticancer agents. The most important targets, in terms of the number of drugs that target them, are the proteins tubulin, topoisomerase I, and topoisomerase II, but other proteins such as the checkpoint kinases chk1 and ckh2 and the heat shock protein Hsp90 are also the targets of some drugs.
The protein tubulin is an interesting and important target. It exists in both α and β forms, and during a normal cell cycle, these two monomeric proteins polymerize into microtubules by noncovalent interaction; these microtubules are then involved in the reorganization of the chromosomes into the nuclei of the mother and daughter cells. After mitosis, the microtubules dissociate to α and β tubulin monomers. Therapeutic intervention, which controls inhibition of mitosis and eventual apoptotic cell death, can occur either by inhibition of the assembly of tubulin monomers into microtubules or by inhibition of the dissociation of microtubules to α and β tubulin monomers. This process is dynamic, and even very small perturbations in assembly and/or disassembly of the monomers/dimers may lead to cell death, frequently via the apoptotic cascade(s), but at agent concentrations that are sometime orders of magnitude lower than those quoted in the literature to give formal inhibition of tubulin in vitro.
The topoisomerases are enzymes that change the topology of DNA. Normal supercoiled DNA needs to unwind to undergo replication, and this unwinding requires that the DNA be cleaved and religated. This cleavage can be brought about by a single-strand break, which is mediated by topoisomerase I, or a double-strand break, which is brought about by topoisomerase II.
Other mechanisms of action involve inhibitors of the heat shock protein Hsp-90, of checkpoint kinases, and inhibitors of protein degradation by interaction with the proteasome.
Compounds that inhibit tubulin assembly
The vinca alkaloids
The antitumor alkaloids vinblastine (1) and vincristine (2) were the first natural products to be used on a large scale as anticancer agents, and they thus blazed the trail for others that came afterward. Vinblastine (as vincaleukoblastine) was isolated from Catharanthus roseus (L.) G. Don, which was formerly known as Vinca rosea L., by two independent teams during the 1950s (9, 10), and vincristine (as leurocristine) was isolated and structurally characterized by Svoboda (11) and Neuss et al. (10) in 1961 and early 1962, respectively. These alkaloids inhibit the polymerization of tubulin to microtubules. Vinblastine is used in combination with other agents for treatment of Hodgkin’s disease and bladder and breast cancers, whereas vincristine is used for treatment of acute lymphocytic leukemias and lymphomas. The semisynthetic analogs vindesine (3) and vinorelbine (4) have been developed more recently. Vindesine, which was first developed in the 1970s, is in clinical use; it seems to be more active than vincristine against non-small-cell lung cancer, but it also has a higher hematological toxicity than vincristine, so its utility is still being evaluated (12). Vinorelbine has been approved for treatment of non-small-cell lung cancer, and the fluorinated analog vinflunine (structure not shown) has entered clinical trials (13). For a recent general review of the vinca alkaloids and their analogs, see Reference 14.
Combretastatin
The first member of this class of compounds, (-)-combretastatin (5), was isolated from Combretum caffrum in 1982 (15). Subsequent studies led to the isolation of many additional combretastatins, including combretastatins A1 (6) and A2 (7), which are two of the most active members of this class, with potent activity as inhibitors of tubulin assembly. Additional development by the Pettit group led to the design of combretastatin A4 phosphate (CA4-P) (8) as a promising drug candidate. This compound is actually a prodrug, which only functions as an inhibitor of tubulin assembly after hydrolysis of the phosphate was subsequently found to be an important member of a new class of compounds known as tumor-specific vascular targeting agents. CA4-P operates by binding to endothelial cell tubulin and causing changes in the morphology of the endothelial cells lining the microvessels feeding the tumors. This process causes disruption of the blood flow, which makes the microvessels unable to deliver oxygen to the tumor and leads ultimately to tumor necrosis. It received orphan drug approval by the Food and Drug Administration (FDA) in 2003 for thyroid cancer, and the FDA has approved it for a “fast track” Phase II clinical trial against anaplastic thyroid cancer (16).
Eribulin (E7389)
The natural product analog E7389 is not yet in clinical use, but it is in Phase III clinical trials and hopefully will enter clinical use within the next few years. The natural product on which it is based, halichondrin B (9), is a member of a relatively large family of congeners with a polycyclic macrolide structure, which is reminiscent of ionophores. Many years of synthetic work by Kishi’s group at Harvard and a group at the Eisai Research Institute in the United States led to the selection of the two lead compounds E7389 (10) and E7390 (11) (17). These compounds were compared in both in vitro and in vivo assays at the National Cancer Institute (NCI) with the natural product obtained by deep-water dredging of a producing sponge. In an example of the skill of synthetic chemists when given what initially seemed to be an almost impossible task, E7389 was prepared in large quantity and entered clinical trials in 2001 as an inhibitor of tubulin assembly. It has a mechanism of action different from that of other tubulin interactive agents; it inhibits microtubule growth, but not shortening, and sequesters tubulin into aggregates (18). It showed good activity against refractory breast carcinoma in Phase II studies and entered Phase III studies for the same indication in late August 2006. Very recently, Hamel’s group at NCI have reported additional investigations on the mechanism of binding of both halichondrin B and E7389 to tubulin, which indicates that these agents may well form small, highly unstable aberrant tubulin polymers rather than the conventional massive stable structures found with vinca alkaloids and the antimitotic peptides (19).
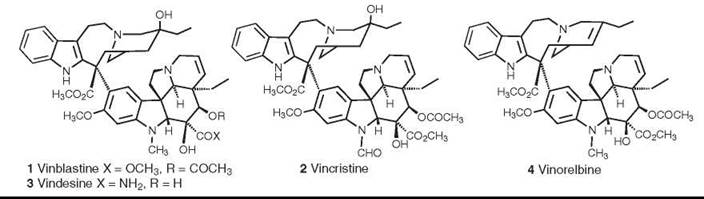
Figure 1. Structures of the vinca alkaloids vinblastine, vincristine, videsine, and vinorelbine.
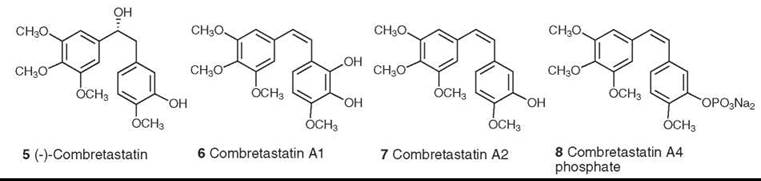
Figure 2. Structures of combretastatin and it analogs.
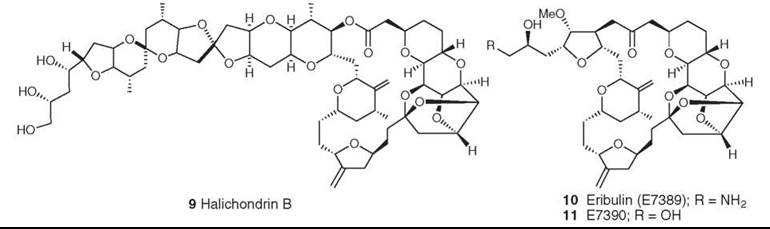
Figure 3. Structures of halichondrin B and its synthetic analogs E7389 and E7390.
Dolastatin
The dolastatins are a class of bioactive peptides isolated by the Pettit group from the Indian Ocean nudibranch, Dolabella auricularia. A total of 18 compounds were isolated over a 20-year period, with structures varying from relatively simple linear peptides to cyclic peptidolactones with nonpeptide components. Dolastatin 10 (12) was the most potent compound, with cytotoxicity in the subnanomolar range. It was shown to be a potent antimitotic agent, binding strongly to the β-subunit of tubulin (20), but it could not be isolated in sufficient quantity because of the scarcity of the source and of the low levels of secondary metabolites in the nudibranch. Pettit and colleagues thus devised many synthetic schemes, which led to the production of enough dolastatin 10 to go into human clinical trials as a tubulin interactive agent. Although it progressed to Phase II, it did not continue further because of a lack of activity and toxicity. However, the base structure led to the synthesis and biologic evaluation by various groups of a large number of related compounds, and synthadotin (also known as tasidotin, 13) emerged as a lead compound (21, 22). Synthadotin is an orally available synthetic derivative of dolastatin 15, and it is in Phase II trials as a tubulin interactive agent. Its discovery provides another example of the skill of synthetic chemists and the potential of novel natural products to be developed into drugs.
Compounds that promote tubulin assembly
Paclitaxel (Taxol)
Paclitaxel (Taxol™; Bristol Myers Squibb 14) and its semisynthetic analog docetaxel (15) are two of the most important anticancer agents of the last 25 years. Paclitaxel was isolated originally by Wall and Wani from Taxus brevifolia (23) and named taxol; this name was later trademarked by Bristol-Myers Squibb (New York, NY), and the name paclitaxel was substituted. The semisynthetic paclitaxel analog docetaxel was prepared by Potier and his collaborators (24). Paclitaxel was selected as a development candidate in 1977 based on its good activity against human tumor xenografts in nude mice. Its drug development was challenging because of problems with solubility and supply, but the supply problem was overcome by a variety of methods (25, 26), and a formulation in ethanol and Cremophor EL enabled clinical work to proceed (27). The discovery of its mechanism of action as a promoter of tubulin assembly (28) was important in maintaining interest in its development, which was a particularly challenging one. As noted, the normal function of a cell requires that microtubules be in dynamic equilibrium with monomeric tubulins. Paclitaxel was the first compound found to disrupt this equilibrium by promoting microtubule assembly.
Paclitaxel and docetaxel (15) are now used, either as single agents or in combination with other drugs such as cisplatin, for the treatment of ovarian cancer (29), breast cancer (30), and non-small-cell lung cancer (31). Paclitaxel has also found an important application as a coating in stents to prevent restenosis (32). It and docetaxel are major drugs, with combined sales of taxane anticancer drugs being over $3 billion in 2004.
Numerous analogs and prodrugs of paclitaxel have been developed, and several of these are in clinical trials. Full coverage of these compounds is beyond the scope of this article, but additional details are provided in a recent review (33). As of early 2007, the only new form of paclitaxel in clinical use is the albumin-bound paclitaxel known as Abraxane (Abraxis BioScience, Schaumberg, IL) (34).
The binding of paclitaxel to microtubules has been studied extensively. The polymeric and noncrystalline nature of the tubulin-taxol complex prevents a direct approach by X-ray crystallography, but Lowe et al. (35) could determine the structure of tubulin at 3.5 A resolution by electron diffraction. Using this structure, various possible binding orientations of paclitaxel on tubulin have been proposed, but recent REDOR NMR studies have established T-Taxol as the most probable conformation (36). The synthesis of the highly active bridged analog britaxel-5 (16), which is constrained to a T-Taxol conformation, confirmed this hypothesis (37).
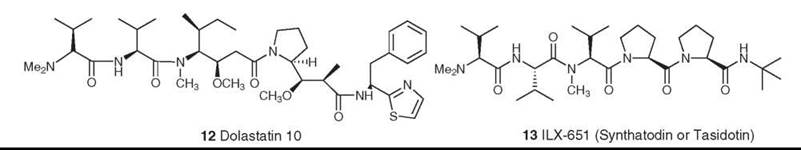
Figure 4. Structures of dolastatin 10 and ILX-651 (synthadotin or tasidotin).
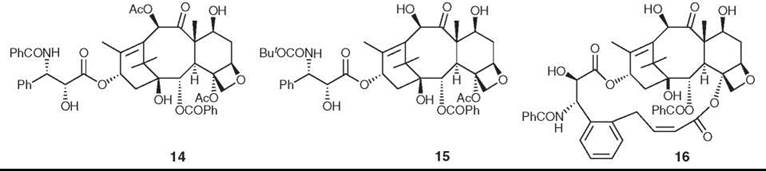
Figure 5. Structures of paclitaxel (Taxol), docetaxel, and britaxel-5.
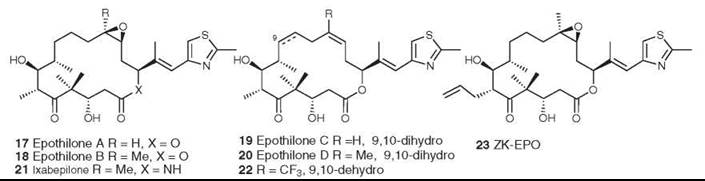
Figure 6. Structures of the epothilones.
Epothilones
The epothilones A-D (17-20) were isolated from the myxobacterium Sorangium cellulosum as antifungal agents (38), but they were found subsequently to have the same mechanism of action as paclitaxel, which promotes the assembly of tubulin into microtubules (39). The epothilones are thus of great interest as potential antitumor agents because of their mechanism of action and because they are also active against some paclitaxel-resistant cell lines. At first glance, they would seem to have a very different shape from paclitaxel, but molecular modeling has shown that some significant common structural features exist in the two base molecules (40). Originally, the epothilones were difficult to obtain in large quantity, and a significant amount of work was performed in academia and industry to synthesize both epothilones A and B and their more active precursors, epothilones C (19) and D (20). However, by using genetic manipulation, Frykman et al. (41) cloned and expressed the polyketide gene cluster that produces epothilones A and B. Subsequent removal of the terminal gene for the P450 enzyme and transfer to a different host enabled them to produce crystalline epothilone D from a large-scale fermentation.
The aza-analog of epothilone B (ixabepilone, 21), which was synthesized by Bristol-Myers Squibb, and epothilone B (patupilone) are in Phase III trials. Epothilone D (KOS-862, 20) and ZK-EPO (23) (42) are in Phase II clinical trials, and the synthetic analog fludelone (22) looks very promising in animal trials (43). However, an evaluation of the epothilones as a class is less optimistic, concluding that “[disappointingly, however, clinical activity has been limited to taxane-sensitive tumor types (prostate cancer and breast cancer) and does not seem to be distinctly different to the activity of taxanes.... Epothilones should certainly not be considered as alternative taxanes, but whether epothilones are here to stay or will fade away has yet to be determined.” (44).
Compounds that inhibit Topoisomerase I
Camptothecin analogs
Camptothecin (24) was isolated from Camptotheca acuminata in 1966 by Wall et al. (45). It had potent anticancer activity in preliminary in vitro and animal assays, but its development was hampered by its extreme insolubility in water. It eventually entered clinical trials in the 1970s as the sodium salt of the carboxylic acid formed by opening the lactone ring, but this proved to have no efficacy and it was dropped from development. Interest in camptothecin was rekindled by the discovery that its primary cellular target was inhibition of topoisomerase I (46). Extensive medicinal chemical studies then led to the development of the two water-soluble derivatives topotecan (Hycamtin; GlaxoSmithKline, Brentford, Middlesex, United Kingdom, 25) and irinotecan (Camptosar; Pfizer, New York, NY, 26). The camptothecins are unique pharmacologically in having topoisomerase I as their only target, and in being able to penetrate mammalian cells readily, and several analogs are in clinical trials (47). Hycamptin and Camptosar are in clinical use for second-line treatment of metastatic ovarian cancer and small-cell lung cancer (topotecan) and for treatment of metastatic colorectal cancer in combination with 5FU/leucovorin (irinotecan) (47, 48).
Rebeccamycin
Rebeccamycin is an indolocarbazole; a comprehensive review of this class has appeared recently (49). Compounds related to rebeccamycin (27) are extremely interesting from a mechanistic standpoint, because relatively simple modifications of the indolocarbazole skeleton generate molecules with enhanced topoisomerase I activity. Thus, active agents can be made by modification of the rebeccamycin skeleton using fluorine substitution, which gives rise to BMS-250749 (28); this compound is headed for Phase I trials as a topoisomerase I inhibitor (47, 50). Second, modification of the base skeleton to include other heterocyclic and carbocyclic rings extends the compounds into previously unexplored chemical space. An example is demonstrated by asymmetric phenyl substitution, which produces compounds such as 29 with significant cytotoxic activity in cell lines, blocking at G2/M or S phase in the cell cycle (51).
Compounds that inhibit Topoisomerase II
Podophyllotoxins
Podophyllotoxin is a major constituent of the rhizome of the American May apple, Podophyllum peltatum. It was shown as early as 1947 to inhibit formation of the mitotic spindle, and its structure (30) was elucidated in 1951 (52).
Podophyllotoxin is too toxic for use as an anticancer agent, but medicinal chemical studies led to the development of etoposide (31) and teniposide (32) as podophyllotoxin analogs. They differ chemically from podophyllotoxin in their stereochemistry and glycosylation at C4 as well as being demethylated at C4’, but their most significant difference is in their mechanism of action. Unlike the parent compound, etoposide and teniposide act as inhibitors of topoisomerase II rather than as inhibitors of tubulin polymerization. Clinically etoposide (31) is used in combination with cisplatin against small-cell cancer, and it is also effective for the treatment of testicular cancer and non-small-cell lung cancer. Teniposide (32) is used in combination with cisplatin against neuroblastoma, with ara-C against acute lymphoblastic leukemia, and with carboplatin against small-cell lung cancer. One problem with these compounds is their lack of water solubility, and the soluble compound Etopophos (Bristol Myers Squibb 33) was developed to circumvent this problem. It can be administered intravenously and is then rapidly converted to etoposide by plasma phosphatase. For a recent general review of the podophyllotoxins, see Reference 53.
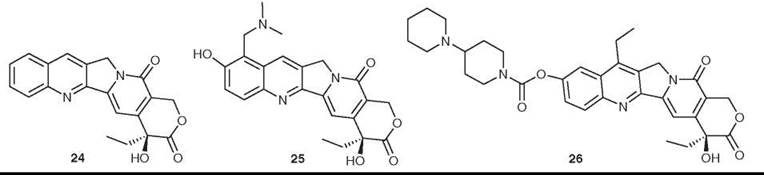
Figure 7. Structures of camptothecin, topotecan, and irinotecan.
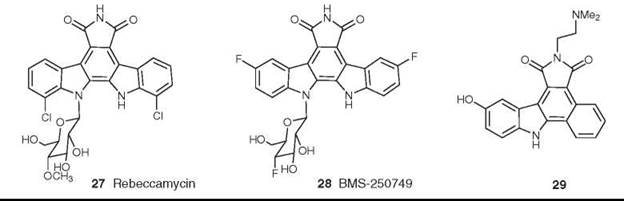
Figure 8. Structures of rebeccamycin and BMS-250749.
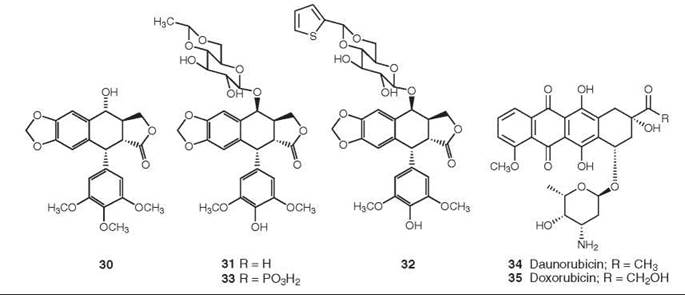
Figure 9. Structures of podophyllotoxin, etoposide, teniposide, etopophos, daunorubicin, and doxorubicin.
Anthracyclines
From the perspective of the number of patients treated, one of the most important classes of topoisomerase II inhibitors is that of the anthracyclines, with daunorubicin (34) and its derivative doxorubicin (adriamycin) (35) being the best known of these agents currently in clinical use. Adriamycin is still a major component of the treatment regimen for breast cancer, despite its known cardiotoxicity (54). The mechanism of action of these molecules is now known to be inhibition of topoisomerase II (55), although they are also effective DNA binders (56). Both drugs are used for the treatment of acute non-lymphocytic leukemia, Hodgkin and non-Hodgkin lymphomas, and sarcomas, in addition to breast cancer. Derivatives of doxorubicin, such as epirubicin, idarubicin, pirirubicin, and valrubicin, have also been approved for clinical use, and the expansion of the efficacy of doxorubicin is being explored through targeted delivery techniques, including both liposomally encapsulated and monoclonal-linked derivatives.
Compounds that interact with other proteins
Geldanamycin
The first signal transduction modulator to enter clinical trials, other than a formal cyclin-dependent kinase or protein kinase C inhibitor, was the microbial product 17-allylamino-geldanamycin (17-AAG, 36). This modulator entered Phase I trials in 2001 and is currently in Phase II trials in a variety of cancers. Geldanamycin and its derivatives bind at the major ATP-binding site of the protein chaperone Hsp-90. The protein chaperones are emerging as attractive targets for cancer chemotherapy, and the reader is referred to three recent reviews for additional information (57-59).
Staurosporine
The indolocarboxazoles first came into prominence with the identification of staurosporine (37) and its simple derivative UCN-01 (38) as inhibitors of components of the eukaryotic cell cycle and of protein kinase C. Although these compounds are related structurally to rebeccamycin, they have very different mechanisms of action, in that they are highly potent but entirely nonselective inhibitors of protein kinases. UCN-01 has entered Phase I/II clinical trials against a variety of cancers, including leukemias, lymphomas, various solid tumors, melanoma, and small-cell lung cancer. Its clinical development has been hampered by its high binding to human plasma proteins (60, 61).
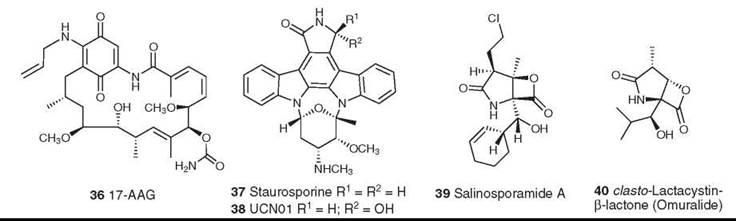
Figure 10. Structures of 17-AAG, staurosporine, UCN01, salinosporamide, and omuralide.
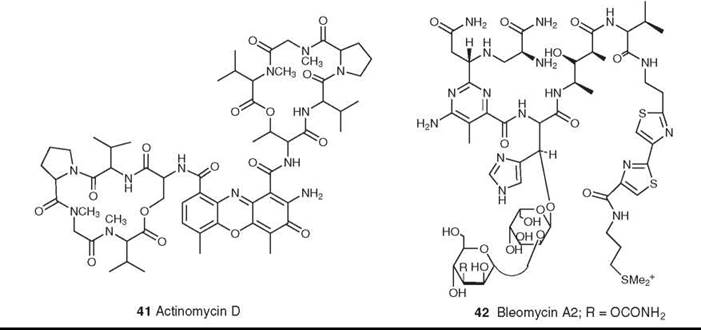
Figure 11. Structures of actinomycin D and bleomycin A2.
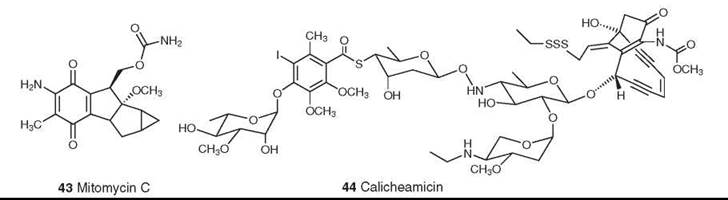
Figure 12. Structures of mitomycin C and calicheamicin.
Salinosporamide
The marine bacterial metabolite salinosporamide A (39) was isolated from the totally new genus Salinispora that mapped to the Micromonosporaceae, which are found in marine sediments across the tropics. It demonstrated activity as a cytotoxic proteasome inhibitor (62) similar to that observed for the structurally related compound omuralide (40) (63, 64), which resulted from a spontaneous rearrangement of the microbial metabolite lactacystin in neutral aqueous media. Salinosporamide has been synthesized (65, 66) and has been fermented in saline media on a large scale under cGMP conditions. It entered Phase I clinical trials in May 2006.
Compounds that target DNA or RNA
The second major class of anticancer agents consists of compounds that act directly on DNA or RNA, either by intercalation, by alkylation, or by cleavage
Actinomycin D
The first microbial-derived agent in clinical use for cancer was actinomycin D (41) (which was systematically named as D-actinomycin C1 and generically named dactinomycin) that was introduced in the early 1960s. Despite extensive research into the preparation of analogs, no derivatives have progressed beyond clinical trials (67). Its mechanism of action is inhibition of DNA-dependent RNA synthesis, which in turn depends on the strong intercalation of actinomycin into double-helical DNA (68, 69). It is used clinically in the treatment of trophoblastic tumors in females, in metastatic carcinoma of the testis, and in Wilms’s tumor in children (67). In recent years there have been reports that actinomycin D may also act on the signal transduction cascade(s) at the level of transcription factor(s), and it will be interesting to see whether these activities rejuvenate interest in this class of molecules (70).
Bleomycins
Another important class is the family of glycopeptolide antibiotics known as bleomycins (e.g., bleomycin A2 and Blenoxane; Nippon Kayaku Co., Ltd., Tokyo, Japan) (42); the bleomycins are structurally related to the phleomycins (71, 72). Bleomycin was originally thought to act through DNA cleavage, because it cleaves both DNA and RNA in an oxidative, sequence-selective, metal-dependent fashion in the presence of oxygen. Recent studies, however, suggest that an alternative mechanism of action may be inhibition of t-RNA from experiments reported recently by the Hecht group (73). Bleomycins are used clinically in combination therapy for the treatment of squamous cell carcinomas and malignant lymphomas.
Mitomycins
The mitomycins (mitosanes) were discovered in the late 1950s, and mitomycin C (43) was approved for clinical use in Japan in the 1960s and in the United States in 1974. Its serious bone marrow toxicity has led to extensive synthetic studies aimed at developing a less toxic analog but without significant success; it remains the only clinically used member of this class. It alkylates DNA only after undergoing a one-electron reduction. The current model postulates that mitomycin C alkylates and cross-links DNA by three competing pathways (74). Clinically it is used primarily in combination with other drugs for the treatment of gastric and pancreatic carcinomas (75).
Calicheamicin
Calicheamicin (44) is a member of a large group of antitumor enediyne antibiotics. It was isolated from Micromonospora echinospora ssp. calichensis by workers at Lederle Laboratories (Pearl River, NY, now Wyeth) (76, 77); the structures of the related esperamicins were published simultaneously (78, 79). Calicheamicin is one of the most potent biologically active natural products ever discovered. It causes single-stranded and double-stranded DNA cleavage through a unique mechanism that involves reductive cleavage of the trisulfide “trigger” followed by Bergman cyclization to a diradical. It proved to be too potent and too toxic for direct clinical use, but it has been used as the “warhead” in the antibody-targeted chemotherapeutic agent Mylotarg (Wyeth Laboratories, Collegeville, PA), which was approved by the FDA in 2000 for clinical use for the treatment of acute myelogenous leukemia (80). Mylotarg is the first such antibody-targeted agent to be approved for use on humans.

Figure 13. Structures of ecteinascidin and cyanosafracin B.
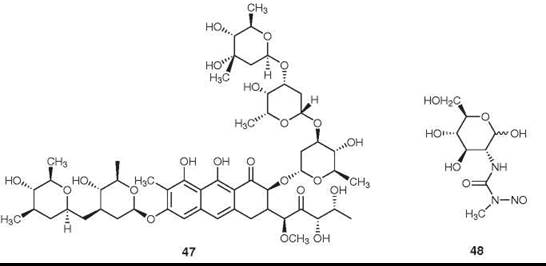
Figure 14. Structures of mithramycin and streptozotocin.
Ecteinascidin
Currently, no approved antitumor drugs directly are derived from marine sources, but ecteinascidin (Yondelis; PharmaMar, Madrid, Spain) was submitted to the EMEA in early August 2006 for approval as an antisarcoma agent and was recommended for approval by the EMEA advisory committee in July 2007 (entered in proof) Approved in September 2007 for Sarcoma. This compound (45) was isolated originally from the Caribbean tunicate, Ecteinascidia turbinata (81, 82). The original supplies for preclinical studies came from a combination of wild harvesting and aquaculture both in sea and on land. The supply problem was finally overcome by the Spanish company, PharmaMar, which developed a 21-step semisynthetic route from the bacterial product cyanosafracin B (46) that could be carried out under cGMP conditions (83). This route provided an adequate source for advanced clinical trials and an assured supply if the drug is approved for clinical use.
Ecteinascidin 743 has a novel mechanism of action, binding to the DNA minor groove and alkylating the N2 position of guanine. This process strongly inhibits the transcription of specific genes. Ultimately it causes a p53-independent cell-cycle block, which leads to apoptosis. It has shown a clinical benefit rate close to 40% in Phase II studies on sarcomas (84).
Other agents
Mithramycin (47) is an antitumor antibiotic isolated from Streptomyces plicatus. It currently is used to a limited extent for the treatment of embryonal cell carcinoma of the testes and of cancer-related hypercalcemia (85). It is reported to be a specific inhibitor of the Sp1 transcription factor in hematopoietic cells (86).
Streptozotocin (48) is an N-nitroso urea isolated from Streptomyces achromogenes. It acts as a DNA-alkylating agent (87), and it is recommended for use in combination with doxorubicin (35) as the drug of choice for the chemotherapy for patients with malignant neuroendocrine pancreatic tumors (88).
Conclusions
As noted in the Introduction, natural products have served historically as the major source of drugs and lead compounds for the treatment of cancer, and the examples provided in this short review indicate that important discoveries in this area are still being made. Despite this impressive track record, many pharmaceutical companies have deemphasized natural product-based drug discovery efforts in favor of approaches such as combinatorial chemistry. Sadly de novo combinatorial chemistry, which was expected to be a panacea for the discovery of small-molecule drug leads over the last 15 or so years, has so far yielded only one drug for antitumor therapy. This drug is the orally active multikinase inhibitor, Sorafenib from Bayer AG (Leverkusen, Germany), which was approved in 2005 (89). The comments of Ortholand and Ganesan (4) are appropriate here
The early years of combinatorial chemistry suffered from an excess of hype, and a major victim was natural-product screening. Many organizations went through an irreversible shift in policy, and prematurely discontinued their efforts in this area. We are now seeing the backlash from this knee-jerk reaction. The early combinatorial strategies were flawed and unproven, and have yet to deliver any blockbuster drugs. Meanwhile, we have lost the uniqueness of screening natural-product space as a complement to synthetic compounds. If past indicators are any guide, there are undoubtedly many more unique and potent biologically active natural products waiting to be discovered.
The data in this review support this statement and show clearly that natural products continue to provide both tools to probe biologic mechanisms and skeletons upon which to “improve” on the properties of the natural product. Scientists are still discovering or rediscovering the truth that natural products are the best lead structures from which to begin a search for novel mechanisms and novel treatments for a multitude of diseases, not just cancer.
References
1. Kingston DGI, Newman DJ. The search for novel drug leads for predominately antitumor therapies by utilizing mother nature’s pharmacophoric libraries. Curr. Opin. Drug Disc. Develop. 2005; 8:207-227.
2. Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981-2002. J. Nat. Prod. 2003; 66:1022-1037.
3. Feher M, Schmidt JM. Property distributions: differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci. 2003; 43:218-227.
4. Ortholand J-Y, Ganesan A. Natural products and combinatorial chemistry: back to the future. Curr. Opin. Chem. Biol. 2004; 8:271-280.
5. Butler MS. The role of natural product chemistry in drug discovery. J. Nat. Prod. 2004; 67:2141-2153.
6. Clardy J, Walsh C. Lessons from natural molecules. Nature 2004; 432:829-837.
7. Nielsen J. Combinatorial synthesis of natural products. Curr. Opin. Cell. Biol. 2002; 6:297-305.
8. Newman DJ, Cragg GM. Natural products as sources of drugs over the last 25 years. J. Nat. Prod. 2007; 70:461-477.
9. Noble RL, Beer CT, Cutts JH. Role of chance observation in chemotherapy: Vinca rosea. Ann. N.Y. Acad. Sci. 1958; 76:882- 894.
10. Neuss N, Cone NJ, Gorman M, Boaz HE. Vinca alkaloids. II. Structures of leurocristine (LCR) and vincaleukoblastine (VLB). J. Am. Chem. Soc. 1962; 84:1509-1510.
11. Svoboda GH. Alkaloids of Vinca rosea (Catharanthus rosea).9. Extraction and characterization of leurosidine and leurocristine. Lloydia 1961; 24:173-178.
12. Joel S. The comparative clinical pharmacology of vincristine and vindesine: Does vindesine offer any advantage in clinical use? Cancer Treat. Rev. 1995; 21:513-525.
13. Duflos A, Kruczynski A, Barret J-M. Novel aspects of natural and modified vinca alkaloids. Curr. Med. Chem.: Anti-Cancer Agents 2002; 2:55-70.
14. Gueritte F, Fahy J. The vinca alkaloids. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, eds. 2005. Taylor and Francis, Boca Raton, FL. pp. 123-135.
15. Pettit GR, Cragg GM, Herald DL, Schmidt JM, Lohavanijaya P. Isolation and structure of combretastatin. Can. J. Chem. 1982; 60:1374-1376.
16. Pinney KG, Jelinek C, Edvardsen K, Chaplin DJ, Pettit GR. The discovery and development of the combretastatins. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, eds. 2005. Taylor and Francis, Boca Raton, FL. pp. 23-46.
17. Zheng W, Seletsky BM, Palme MH, Lydon PJ, Singer LA, Chase CE, Lemelin CA, Shen Y, Davis H, Tremblay L, et al. Macrocyclic ketone analogues of halichondrin B. Bioorg. Med. Chem. Lett. 2004; 14:5551-5554.
18. Jordan MA, Kamath K, Manna T, Okouneva T, Miller HP, Davis C, Littlefield BA, Wilson L. The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol. Cancer Ther. 2005; 4:1086-1095.
19. Dabydeen DA, Burnett JC, Bai R, Verdier-Pinard P, Hickford SJ, Pettit GR, Blunt JW, Munro MHG, Gussio R, Hamel E. Comparison of the activities of the truncated halichondrin B analog NSC 707389 (E7389) with those of the parent compound and a proposed binding site on tubulin. Mol. Pharmacol. 2006; 70:1866-1875.
20. Bai R, Pettit GR, Hamel E. Binding of dolastatin-10 to tubulin at a distinct site for peptide antimitotic agents near the exchangeable nucleotide and vinca alkaloid sites. J. Biol. Chem. 1990; 265:17141-17149.
21. Cunningham C, Appleman LJ, Kirvan-Visovatti M, Ryan DP, Regan E, Vukelja S, Bonate PL, Ruvuna F, Fram RJ, Jekunen A, et al. Phase I and pharmacokinetic study of the dolastatin-15 analogue tasidotin (ILX651) administered intravenously on days 1, 3, and 5 every 3 weeks in patients with advanced solid tumors. Clin. Cancer Res. 2005; 11:7825-7833.
22. Ebbinghaus S, Rubin E, Hersh E, Cranmer LD, Bonate PL, Fram RJ, Jekunen A, Weitman S, Hammond LA. A phase I study of the dolastatin-15 analogue tasidotin (ILX651) administered intravenously daily for 5 consecutive days every 3 weeks in patients with advanced solid tumors. Clin. Cancer Res. 2005; 11:7807-7816.
23. Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971; 93:2325-2327.
24. Gueritte-Voegelein F, Senilh V, David B, Guenard D, Potier P. Chemical studies of 10-deacetyl baccatin III. Hemisynthesis of taxol derivatives. Tetrahedron 1986; 42:4451-4460.
25. Denis J-N, Greene AE, Guenard D, Gueritte-Voegelein F, Mangatal L, Potier P. A highly efficient, practical approach to natural taxol. J. Am. Chem. Soc. 1988; 110:5917-5919.
26. Holton RA, Biediger RJ, Boatman PD. Semisynthesis of taxol and taxotere. In: Taxol: Science and Applications. Suffness M, ed. 1995. CRC Press, Inc., Boca Raton, FL. pp. 97-121.
27. Suffness M, Wall ME. Discovery and development of taxol. In: Taxol: Science and Applications. Suffness M, eds. 1995. CRC Press, Inc., Boca Raton, FL. pp. 3-25.
28. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by taxol. Nature 1979; 277:665-667.
29. Piccart MJ, Cardoso F. Progress in systemic therapy for breast cancer: an overview and perspectives. Eur. J. Can. 2003; 1(supp):56-69.
30. Ozols RF. Progress in ovarian cancer: an overview and perspective. Eur. J. Can. 2003; 1(supp):43-55.
31. Davies AM, Lara PN, Mack PC, Gandara DR. Docetaxel in non-small cell lung cancer: a review. Exp. Opin. Pharmacother. 2003; 4:553-565.
32. Kamath KR, Barry JJ, Miller KM. The taxus drug-eluting stent: a new paradigm in controlled drug delivery. Adv. Drug Deliv. Rev. 2006; 58:412-436.
33. Kingston DGI, Newman DJ. Taxoids: cancer-fighting compounds from nature. Curr. Opin. Drug Disc. Dev. 2007; 10:130-144.
34. Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Exp. Opin. Pharmacother. 2006; 7:1041-1053.
35. Lowe J, Li H, Downing KH, Nogales E. Refined structure of a β-tubulin at 3.5 A resolution. J. Mol. Biol. 2001; 313:1045-1057.
36. Paik Y, Yang C, Metaferia B, Tang S, Bane S, Ravindra R, Shanker N, Alcaraz AA, Johnson SA, Schaefer J, et al. REDOR NMR distance measurements for the tubulin-bound paclitaxel conformation. J. Am. Chem. Soc. 2007; 129:361-370.
37. Ganesh T, Guza RC, Bane S, Ravindra R, Shanker N, Lakdawala AS, Snyder JP, Kingston DGI. The bioactive taxol conformation of ^-tubulin: Experimental evidence from highly active constrained analogs. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:10006-10011.
38. Hofle G, Bedorf N, Steinmetz H, Schomburg D, Gerth K, Reichenbach H. Epothilone A and B - novel 16-membered macrolides with cytotoxic activity: Isolation, crystal structure, and conformation in solution. Angew Chem Int. Ed. 1996; 35:1567-1569.
39. Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995; 55:2325-2333.
40. Giannakakou P, Gussio R, Nogales E, Downing KH, Zaharevitz D, Bollbuck B, Poy G, Sackett D, Nicolaou KC, Fojo T. A common pharmacophore for epothilone and taxanes: Molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:2904-2909.
41. Frykman S, Tsuruta H, Lau J, Regentin R, Ou S, Reeves C, Carney J, Santi D, Licari P. Modulation of epothilone analog production through media design. J. Ind. Microbiol. Biotech. 2002; 28:17-20.
42. Klar U, Buchmann B, Schwede W, Skuballa W, Hoffmann J, Lichtner RB. Total synthesis and antitumor activity of ZK-EPO: the first fully synthetic epothilone in clinical development. Angew Chem Int. Ed. 2006; 45:7942-7948.
43. Wu K-D, Cho YS, Katz J, Ponomarev V, Chen-Kiang S, Danishefsky SJ, Moore MAS. Investigation of antitumor effects of synthetic epothilone analogs in human myeloma models in vitro and invivo. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:10640-10645.
44. de Jonge M, Verweij J. The epothilone dilemma. J. Clin. Oncol. 2005; 23:9048-9050.
45. Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. Plant antitumor agents. 1. The isolation and structure of camp- tothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J. Am. Chem. Soc. 1966; 88:3888-3890.
46. Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985; 260:14873-14878.
47. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 2006; 6:789-802.
48. Rahier NJ, Thomas CJ, Hecht SM. Camptothecin and its analogs. Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, eds. 2005. Taylor and Francis, Boca Raton, FL. pp. 5-21.
49. Sanchez C, Mendez C, Salas JA. Indolocarbazole natural products: occurrence, biosynthesis, and biological activity. Nat. Prod. Rep. 2006; 23:1007-1045.
50. Saulnier MG, Balasubramanian BN, Long BH, Frennesson DB, Ruediger E, Zimmerman K, Eummer JT, St. Laurent DR, Stoffan KM, Naidu BN, et al. Discovery of a fluoroindolo[2,3-a]carbazole clinical candidate with broad spectrum antitumor activity in preclinical tumor models superior to the marketed oncology drug, CPT-11. J. Med. Chem. 2005; 48:2258-2261.
51. Routier S, Merour J-Y, Dias N, Lansiaux A, Bailly, C, Lozach, O, Meijer, L. Synthesis and biological evaluation of novel phenylcarbazoles as potential anticancer agents. J. Med. Chem. 2006; 49:789-799.
52. Hartwell JL, Schrecker AW. Components of podophyllin. V. The constitution of podophyllotoxin. J. Am. Chem. Soc. 1951; 73:2909-2916.
53. Lee K-H, Xiao Z. Podophyllotoxin and analogs. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, eds. 2005. Taylor and Francis, Boca Raton, FL.pp.71-87.
54. Arcamone F. Anthracyclines. In: Anticancer agents from natural products. Cragg GM, Kingston DGI, Newman DJ, eds. 2005. Taylor and Francis, Boca Raton, FL. pp. 299-320.
55. Zunino F, Capranico G. DNA topoisomerase II as the primary target of anti-cancer anthracyclines. Anticancer Drug Des. 1990; 5:307-317.
56. Frederick CA, Williams LD, Ughetto G, Van der Marel GA, Van Boom JH, Rich A, Wang AHJ. Structural comparison of anticancer drug-DNA complexes: adriamycin and daunomycin. Biochemistry 1990; 29:2538-2549.
57. Isaacs JS. Heat-shock protein 90 inhibitors in antineoplastic therapy: is it all wrapped up? Exp. Opin. Investig. Drugs 2005; 14:569-589.
58. Janin YL. Heat shock protein 90 inhibitors. A text book example of medicinal chemistry J. Med. Chem. 2005; 48:7503-7512.
59. Chiosis G. Targeting chaperones in transformed systems - a focus on hsp90 and cancer. Exp. Opin. Ther. Targ. 2006; 10:37-50.
60. Prudhomme M. Staurosporines and structurally related indolocarbazoles as antitumor agents. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, eds. 2005. Taylor and Francis, Boca Raton, FL. pp. 499-517.
61. Sanchez C, Mendez C, Salas JA. Indolocarbazole natural products: Occurrence, biosynthesis, and biological activity. Nat. Prod. Rep. 2006; 23:1007-1045.
62. Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew Chem. Int. Ed. 2003; 42:355-357.
63. Corey EJ, Li WD. Total synthesis and biological activity of lactacystin, omuralide and analogs. Chem. Pharm. Bull. 1999; 47:1-10.
64. Corey EJ, Li WD, Nagamitsu T, Fenteany G. The structural requirements for inhibition of proteasome function by the lactacystin-derived β-lactone and synthetic analogs. Tetrahedron 1999; 55:3305-3316.
65. Reddy LR, Saravanan P, Corey EJ. A simple stereocontrolled synthesis of salinosporamide A. J. Am. Chem. Soc. 2004; 126:6230-6231.
66. Reddy LR, Fournier J-F, Reddy BVS, Corey EJ. New synthetic route for the enantioselective total synthesis of salinosporamide A and biologically active analogues. Org. Lett. 2005; 7:2699-2701.
67. Mauger AM, Lackner H. The actinomycins. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, eds. 2005. Taylor and Francis, Boca Raton, FL. pp. 281-297.
68. Kawamata J, Imanishi M. Interactions of actinomycin with deoxyribonucleic acid. Nature 1960; 187:1112-1113.
69. Kersten W, Kersten H, Rauen HM. Action of nucleic acids on the inhibition of growth by actinomycin of Neurospora crassa. Nature 1960; 187:60-61.
70. Gniazdowski M, Denny WA, Nelson SM, Czyz M. Transcription factors as targets for DNA-interacting drugs. Curr. Med. Chem.: Anti-Cancer Agents 2003; 10:909-924.
71. Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Antitumor antibiotics: bleomycin, enediynes, and mitomycin. Chem. Rev. 2005; 105:739-758.
72. Hecht SM. Bleomycin group antitumor agents. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, eds. 2005. Taylor and Francis, Boca Raton, FL. pp. 357-381.
73. Tao Z-F, Konishi K, Keith G, Hecht SM. An efficient mammalian transfer RNA target for bleomycin. J. Am. Chem. Soc. 2006; 128:14806-14807.
74. Kumar SG, Lipman R, Cummings J, Tomasz M. Mitomycin c-DNA adducts generated by dt-diaphorase. Revised mechanism of the enzymatic reductive activation of mitomycin C. Biochemistry 1997; 36:14128-14136.
75. Remers WA. The mitomycins. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, eds. 2005. Taylor and Francis, Boca Raton, FL. pp. 475-497.
76. Lee MD, Dunne TS, Siegel MM, Chang CC, Morton GO, Borders DB. Calichemicins, a novel family of antitumor antibiotics. 1. Chemistry and partial structure of calichemicin y J. Am. Chem. Soc. 1987; 109:3464-3466.
77. Lee MD, Dunne TS, Chang CC, Ellestad GA, Siegel MM, Morton GO, McGahren WJ, Borders DB. Calichemicins, a novel family of antitumor antibiotics. 2. Chemistry and structure of calichemicin Y J. Am. Chem. Soc. 1987; 109:3466-3468.
78. Golik J, Clardy J, Dubay G, Groenewold G, Kawaguchi H, Konishi M, Krishnan B, Ohkuma H, Saitoh K, Doyle TW. Esperamicins, a novel class of potent antitumor antibiotics. 2. Structure of esperamicin X. J. Am. Chem. Soc. 1987; 109:3461-3462.
79. Golik J, Dubay G, Groenewold G, Kawaguchi H, Konishi M, Krishnan B, Ohkuma H, Saitoh K, Doyle TW. Esperamicins, a novel class of potent antitumor antibiotics. 3. Structures of esperamicins A1, A2, and A1b. J. Am. Chem. Soc. 1987; 109:3462-3464.
80. Hamann PR, Upeslacis J, Borders DB. Enediynes. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, eds. 2005. Taylor and Francis, Boca Raton, FL. pp. 451-474.
81. Rinehart KL, Holt TG, Fregeau NL, Stroh JG, Keifer PA, Sun F, Li LH, Martin DG. Ecteinascidins 729, 743, 745, 759a, 759b, and 770: potent antitumor agents from the caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990; 55:4512-4515.
82. Wright AE, Forleo DA, Gunawardana GP, Gunasekera SP, Koehn FE, McConnell OJ. Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinata. J. Org. Chem. 1990; 55:4508-4512.
83. Cuevas C, Perez M, Martin M, Chicharro JL, Fernandez-Rivas C, Flores M, Francesch A, Gallefo P, Zarzuelo M, de la Calle F, et al. Synthesis of ecteinascidin ET-743 and phthalascidin PT-650 from cyanosafracin B. Org. Lett. 2000; 2:2545-2548.
84. Fayette J, Coquard IR, Alberti L, Ranchere D, Boyle H, Blay J-Y. ET-743: a novel agent with activity in soft tissue sarcomas. Oncologist 2005; 10:827-832.
85. Lynch GR, Lane M. Other antitumor antibiotics. In: Cancer Management in Man. Woolley PV, ed. 1989. Kluwer Academic Publishers, Dordrecht. pp. 134-146.
86. Koschmieder S, Agrawal S, Radomska HS, Huettner CS, Tenen DG, Ottmann OG, Berdel WE, Serve HL, Muller-Tidow C. Decitabine and vitamin D3 differentially affect hematopoietic transcription factors to induce monocytic differentiation. Int. Oncol. 2007; 30:349-355.
87. Dolan ME. Inhibition of DNA repair as a means of increasing the antitumor activity of DNA reactive agents. Adv. Drug Deliv. Rev. 1997; 26:105-118.
88. Arnold R, Rinke A, Schmidt C, Hofbauer L. Chemotherapy. Best Pract. Res. Clin. Gastroent. 2005; 19:649-656.
89. Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nature Rev. Drug Discov. 2006; 5:835-844.
Further Reading
Cragg GM, Kingston DGI, Newman DJ, eds. Anticancer Agents from Natural Products. 2005. CRC Press; Boca Raton, FL.
See Also
Cancer, Chemical Biology of
Chemistry of Protein Reactive Natural Products
Natural Products in Marine Organisms, Chemical Diversity of
Natural Products in Microbes, Chemical Diversity of
Natural Products in Plants, Chemical Diversity of
Natural Products, Common Biological Targets for
Natural Products, Inhibition of Protein Biosynthesis by