CHEMICAL BIOLOGY
Recent Progress in Drug Discovery Based on Plant-Derived Natural Products Research
Kuo-Hsiung Lee, Hideji Itokawa, Toshiyuki Akiyama and Susan L. Morris-Natschke, Natural Products Research Laboratories, School of Pharmacy, University of North Carolina, Chapel Hill, North Carolina
doi: 10.1002/9780470048672.wecb373
Numerous important bioactive compounds have been, and continue to be, isolated worldwide from natural sources. These compounds include both primary and secondary metabolites isolated mainly from plants, as well as from the animal and mineral kingdoms. The recent development of new bioassay methods has facilitated progress in the BDFI (bioactivity-directed fractionation and isolation) of many useful bioactive compounds from natural sources (1). These active principles could be developed or additionally modified to enhance the biologic profiles as clinical trials candidates. Many natural pure compounds have become medicines, dietary supplements, and other useful commercial products. This article summarizes research on many different useful compounds isolated or developed from plants with an emphasis on those discovered recently by the laboratories of the authors as antitumor and anti-HIV clinical trial candidates.
From ancient times, many crude herbs have been used as remedies for various diseases. In Asia, these therapies include traditional Chinese medicine (TCM), Japanese-Chinese medicine (Kampo), Korean-Chinese medicine, Jamu (Indonesia), and Ayurvetic medicine (India). In Europe, phytotherapy and homeopathy have found medicinal use. These herbal therapies generally are classified as “alternative medicines” in America. Alternative medicine, which comes mainly from the aforementioned traditional and folk medicines used worldwide, also is being combined with conventional medicine (Western medicine), which results in integrative medicine. In TCM, crude herbal drugs formerly were divided into three categories: upper-, middle-, and lower-class medicines. Upper-class medicines usually are not toxic, have moderate physiologic effects, and often are used to maintain good health. Thus, they sometimes are called supplementary drugs. Both upper- and middle-class medicines are used as therapeutic drugs, but the former medicines are not as toxic as the latter. Lower-class medicines can contain very toxic substances, which can be used judiciously as medicines. TCM prescriptions usually mix herbs that belong to these three categories, according to a unique principle (2). The experience gained by using TCM for centuries provides a rich source of information for modern research in drug discovery.
Anticancer and Antitumor Compounds
Suffness and Douros (3) suggested the following definitions to avoid confusion of terminology. Cytotoxicity is used when compounds or extracts show in vitro (in cells) activity against tumor cell lines. Antitumor or antineoplastic indicates that the materials are effective in vivo (in animals) in experimental systems. Anticancer refers to compounds that are active clinically against human cancer.
The development of novel, clinically useful anticancer agents is highly dependent on the bioassay screening systems, as well as the sample sources. Two bioassay types have been used: cell-based and mechanism of action-(MOA)-based. Initially, cell-based assays used primarily L1210, P388, and nasopharyngeal (KB) cells in preliminary screening for antitumor activity. Screening against a panel of human cancer cell lines was implemented to discover agents active against different types of cancer. For active agents, the in vitro studies are followed by in vivo xenograft studies to ensure efficacy. New MOA-based bioassay systems aimed at particular molecular targets also have revolutionized the discovery of potential drug candidates. Important anticancer drug targets include tubulin, DNA topoisomerases I and II (topo I and topo II), cyclin-dependent kinases (CDKs), growth and transcription factors, and so forth.
Higher plants have yielded many effective, clinically useful anticancer drugs, including those derived from Catharanthus alkaloids, Taxus diterpenes, Camptotheca alkaloids, and Podophyllum lignans. Research in this area has been reviewed extensively (3-13).
The seminal discoveries of taxol (tubulin-interactive) and camptothecin (topo I-interactive) by Wall and Wani (14-16) represent how natural products have influenced the additional development of natural product-derived and synthetic entities. The following discussion of the discovery and development of current important antineoplastic compounds will be organized by plant species.
Catharanthus species
Catharanthus alkaloids, particularly vinblastine (A1) and vincristine (A2), are well known anticancer drugs, which are used clinically to treat Hodgkin’s lymphoma and acute childhood lymphoblastic leukemia, respectively. These alkaloids interact with tubulin, a protein necessary for cell division, and are inhibitors of mitosis (the process of cell division).
Originally, these compounds were isolated from Catharanthus roseus (L.) G. Don [formerly known as Vinca rosea (Apocynaceae family)], which is used as folk medicine in Madagascar to inhibit milk secretion and as a hypotensive agent, astringent, and emetic. Moreover, native people in England and the West Indies have used this species to lower blood sugar.
Numerous synthetic analogs have been designed to have activity against other tumor types or to have fewer side effects. Among them, navelbine (vinorelbine) (A3) was developed by Burroughs Wellcome (17) and is used against non-small cell lung and advanced breast cancers. This synthetic analog of A1 has an eight-membered, rather than a nine-membered, C ring, and a dehydrated D ring (18). Eldisine (vindesine) (A4) is another structurally modified analog, which is used against acute lymphoblastic leukemia, breast cancer, and malignant melanoma.
EC145 (A5), a folic acid conjugate of desacetyl vinblastine monohydrazide, represents a new generation of receptor-specific targeted chemotherapy and is undergoing Phase I anticancer clinical trials (19). Phase II trials for bladder and kidney cancers are underway with vinflunine (A6), a bifluorinated vinorelbine derivative (20, 21).

Figure 1. Structures of natural and synthetic Catharanthus alkaloids.
Taxus species
Taxol (paclitaxel) (B1), a taxane diterpene isolated from the bark of the Pacific yew tree Taxus brevifolia Nutt. (Taxaceae family), is used extensively in patients with advanced and metastatic ovarian and breast tumors, particularly tumors that are refractory to standard chemotherapy. Initially, supply problems severely limited the full exploration of its antineoplastic potential. However, the semisynthesis of B1 from 10-deacetylbaccatin III (B2), which is isolated from needles of the European yew tree, Taxus baccata L., provided an alternative renewable resource to resolve the supply problems.
Wall and Wani (22) are the pioneers in taxol discovery. To date, around 400 taxoids have been isolated from the Taxus species. Taxus alkaloids were reviewed recently in the book Taxus, genus Taxus edited by Itokawa and Lee (23). Biologic activity and the chemistry of taxoids from the Japanese yew also have been reviewed (24).
B1 interacts with cellular tubulin via promotion of microtubule assembly and inhibits mitosis. It is active against breast, brain, tongue, endometrial, and ovarian, as well as other, cancers (25, 26). The clinically used analog docetaxel (taxotere) (B3) is synthesized from the more readily available B2. Taxotere is used particularly against non-small cell lung cancers.
Extensive structure-activity relationship (SAR) studies have led to many related antineoplastic taxane analogs, including ortataxel (B4), an orally administered taxoid in Phase II clinical trials (27). SAR studies of ring C-secotaxoids were published recently (28).
In addition, conjugates between taxol and various other compounds, such as 3,17β-estradiol (29), various fatty acids (30), or a biodegradable polymer (poly-L-glutamic acid, paclitaxel polyglumex) (31, 32), seek to improve drug targeting or tissue distribution. The laboratories of the authors have conjugated taxoids with other anticancer agents, including epipodophyllotoxins (B5) (33) and camptothecin (B6) (34). A recent review provides an overview of novel formulations of taxanes, including many in clinical trials, developed to overcome the solubility issues with B1 and B3 (35).

Figure 2. Structures of natural and synthetic taxoids.
Camptotheca species
Camptothecin (CPT, C1) is a potent antitumor pentacyclic alkaloid isolated from Camptotheca acuminata Decne. (Nyssaceae family) and originating in China (36, 37). Interest in CPT was sparked by the discovery that its primary cellular target is DNA topo I (38). 10-Hydroxycamptothecin (C2), which also occurs naturally, has a better therapeutic index and is used in China for treating many cancers.
Poor water solubility of the natural products led to the development of the semisynthetic, more water-soluble analogs topotecan (Hycamptin, C3) and irinotecan (Camptosar, C4), which are used primarily for the treatment of advanced ovarian and metastatic colorectal cancers, respectively (39). Some CPT analogs, such as C5, synthesized in the laboratories of the authors showed greater topo I inhibition than CPT (40). Additional CPT analogs also are of interest in combination regimens as radiation sensitizers (41). Two epipodophyllotoxin-campothecin conjugates, C6 and C7, from the laboratories of the authors exhibit dual mechanisms of action, being both topoisomerase I/II-inhibitory (42). They have improved in vitro anticancer profiles (42) and are active against etoposide- and camptothecin-resistant KB cells (KB7D and KB/CPT100, respectively).
Clinical application and perspectives on the CPTs have been discussed in several excellent reviews (43-45). New CPT analogs in anticancer clinical trials include DB-67 (a silatecan or 7-silylcamptothecan, C8) (46) and rubitecan (9-nitrocamptothecin, C9) (47).

Figure 3. Structures of natural and synthetic camptothecins.
Podophyllum species
The genus Podophyllum (Berberidaceae family), including the American P. peltatum L. (American mayapple) and Indian or Tibetan P. emodi Wall (syn. P. hexandrum Royle), has been used for centuries for its medicinal properties. Podophyllin, a resin obtained from an alcoholic extract of Podophyllum rhizome, has been used for a long time as a remedy for warts and was listed in the U.S. Pharmacopoeia from 1820 to 1942, when it was removed because of undesirable toxicity (48).
Podophyllotoxin (D1) is an aryltetralinlactone cyclolignan with a flat, rigid five-ring skeleton; it was isolated in 1880 from rhizomes of P. peltatum. It was found to show antineoplastic activity, but it is highly toxic and failed the U.S. National Cancer Institute (NCI) Phase I clinical trials as an antitumor drug in the 1970s. Chemical modification of D1 led to successful development of the clinically useful anticancer drugs etoposide (D2) and teniposide (D3). These compounds inhibit cellular DNA topo II and are used to treat small cell lung and testicular cancers and lymphomas/leukemias. Etopophos (etoposide phosphate, D4) is a clinically used, water-soluable phosphate ester of etoposide. It lessens the chance of precipitation of the drug during intravenous administration.
Limitations of D2, including myelosuppression, drug resistance development, and poor water solubility, prompted extensive SAR studies. Using drug improvement principles, several series of 4-alkylamino and 4-arylamino epipodophyllotoxin analogs were synthesized and showed increased inhibition of DNA topo II activity and increased cytotoxicity in D2-resistant cell lines (49-51). From the preclinical development in the laboratories of the authors, GL-331 (D5) (52), which contains a p-nitroanilino moiety at the 4β position of D2, emerged as a clinical trials candidate. Compared with D2, GL-331 has advantages of better water solubility, easier manufacturability, and fewer side effects. It also shows cytotoxic activity against D2-resistant cancer cell lines. GL-331 progressed to Phase IIa clinical trials as an anticancer drug (53).
The rational design of improved D2-analogs has made use of several new computational strategies (52, 54-56). In 2004, Gordaliza et al. (57) reviewed the distribution, sources, application, and new cytotoxic derivatives of D1. More recently, Lee and Xiao (58) also have reviewed podophyllotoxins and related analogs, including GL-331, to demonstrate how plant natural products can lead to successful preclinical drug candidates.

Figure 4. Structures of natural and synthetic podophyllotoxins.
Cephalotaxus species
The genus Cephalotaxus (Cephalotaxaceae family) contains coniferous evergreen trees and shrubs that are indigenous to Asia. Historically, the bark has long been used in TCM for various indications.
Powell et al. (59-61) originally isolated the antitumor alkaloids homoharringtonine (E1) and harringtonine (E2). Subsequently, Chinese investigators discovered that alkaloids from C. fortunei Hook. F possess antitumor activity (62). Consequently, E1 was obtained from the Chinese evergreen tree C. harringtonia K. Koch var. harringtonia (63), and other active alkaloids were isolated from various Cephalotaxus species (64, 65). Interestingly, the parent compound, cephalotaxine (E3), is devoid of antitumor activity.
E1 has been investigated in anticancer clinical trials at the U.S. National Cancer Institute (NCI), particularly for the treatment of myeloid leukemia (66, 67). However, its side effects still remain an issue. Thus, the authors have continued to study new natural constituents of Cephalotaxus species and to develop new analogs on the basis of SAR studies, as presented in a review by Itokawa et al. (68).
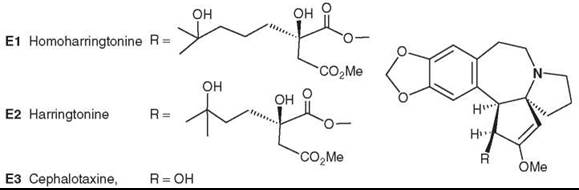
Figure 5. Structures of Cephalotaxus alkaloids.
Colchicum species
The alkaloid colchicine (F1) isolated from the medicinal plant Colchicum autumnale L. (Liliaceae family) still is used to treat gout and familial Mediterranean fever. F1 and thiocolchicine (F2) (SCH3rather than OCH3 at C-10), which is more stable and more potent but slightly more toxic, are mitotic inhibitors that inhibit polymerization of tubulin (69). Although they show antileukemic activity, they are too toxic to use as anticancer agents, which prompts the synthesis of new, less toxic analogs.
Replacing the C-7 acetoamide group on the B ring with various oxygen-containing groups [ketone (F3, thiocolchicone), hydroxyl (F4), and ester (F5, F6)] (70) led to compounds that were equally or more active in vitro than F2. The C-ring contracted colchinol-7-one thiomethyl ether or allo-ketone (F7) was equipotent with the seven-membered ring natural product F1. Three-related, ring-contracted colchicinoids (F8-F10) showed excellent activity in drug-sensitive and drug-resistant KB cell lines (71).
The above synthetic colchinoids have three methylated phenolic groups in the A ring, which are needed for full potency as tubulin/mitotic inhibitors. Removing one or two of the methyl groups reduces potency, and tridemethylated colchicines and thiocolchicines (F11-F14) no longer interact with tubulin but rather are a new class of DNA topo II inhibitory agents (72). They show in vitro activity against bone and breast cancers (73).

Figure 6. Structures of colchicine and related compounds.
Salvia species
Salvia officinalis L. (Labiatae family) is native to Europe and America, but the roots and rhizome of S. miltiorrhiza Bunge (called Tanshen) have been used widely in China for treating various cardiac (heart) and vascular (blood vessel) disorders, such as atherosclerosis or blood-clotting abnormalities. This plant exhibits hypotensive effects, causes coronary artery vasodilation, and inhibits platelet aggregation. Accordingly, it should not be used in combination with warfarin. Tanshen also has been applied for hemorrhage, dysmenorrhea, miscarriage, swelling, inflammation, chronic hepatitis, and insomnia (74, 75). Clinically available preparations of a S. miltiorrhiza/ Dalbergia odorifera mixture may have potential as an antianginal drug (76).
Tanshinone diterpenoids, including tanshinone I (G1) and tanshinone IIA (G2), are bioactive compounds from S. miltiorrhiza (77). Sodium tanshinone sulfate (G3) is a water-soluble derivative of G2, is used clinically to treat angina pectoris and myocardial infarction, and exhibits strong membrane-stabilizing effects on red blood corpuscles. In addition, novel secoabietane rearranged diterpenoids were isolated recently from this species (78).
Other studies have shown that S. miltiorrhiza exerts clear cytotoxic effects and strongly inhibits the proliferation of liver cancer cells (79). Various tanshinones were tested in SAR studies against several human tumor cells, namely nasopharyngeal (KB), cervical (Hela), colon (Colon 205), and laryngeal (Hep-2) cell systems (74, 75).
Neo-tanshinlactone (G4), also originally isolated from Tanshen, has a unique and different structure compared with other compounds from S. miltiorrhiza Bunge. Preliminary studies of this compound showed unique specific activity against the MCF-7 breast cancer cell line but insignificant activity against other cell lines in the tested panel. Extended bioassay studies showed that G4 is quite active against estrogen receptor positive (ER+) human breast cancer cell lines (MCF-7 and ZR-75-1) but inactive against ER negative (ER-) human breast cancer lines (MDA MB-231 and HS 587-T) (80). This finding is significant because more than 60% of breast cancer cases in postmenopausal women are ER+. Compared with the breast cancer drug tamoxifen citrate, G4 was 10-fold more potent and 20-fold more selective against two ER+ cell lines (80). It also was potent against an ER- cell line that overexpresses the protein HER2+, which plays a key role in regulating cell growth and affects 25-30% of breast cancer patients (80). These results indicate that G4 is an excellent candidate for additional development toward anti-breast cancer clinical trials.
To that end, synthetic analog studies have identified a compound (G5) with comparable or better anticancer activity and certain structural features that are critical to the anticancer activity of this compound type (81).
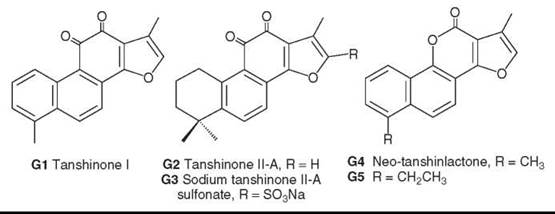
Figure 7. Structures of tanshinlactones and neo-tanshinlactone.
Cocculus species
Cocculus trilobus DC. (Menispermaceae family), found in the mountains of east Asia, has folkloric uses as a diuretic, an analgesic, and an anti-inflammatory crude drug.
Sinococuline (H1) was isolated as an antitumor principle from the stems and rhizomes (82). It also has in vivo activity against P388 leukemia. H1 likely is a general cytotoxic rather than a cell-specific agent (83).

Figure 8. Structure of sinococuline.
Maytenus species
Maytenus illicifolia Mart. ex Reiss. (Celastraceae family), more commonly known as “Cangorosa,” is found in South America where it is used for its analgesic, antipyretic, antiseptic, and anticancer properties and for birth control, particularly in Paraguay.
Bioactivity-directed fractionation and isolation by various research groups has led to the isolation of various active principles. Kupchan et al. (84, 85) first identified antileukemic maytansinoids, for example, maytansine (I1), from the African plant M. ovatus [later renamed M. serrata (Hochst. ex A. Rich.) R. Wilczek]. Although it advanced to Phase II clinical trials, testing then was suspended because of neurotoxicity. The related maytanprine (I2), isolated from M. diversifolia (Maxim.) Ding Hou, has been investigated for growth-inhibiting and apoptosis-inducing activities in K562 leukemia cells (86). Cytotoxic monotriterpenes include pristimerin (I3) and isotingenone III (I4) from M. illicifolia (87), as well as the triterpene dimers dihydroisocangorosin A (I5) and cangorosin B (I6) (88, 89). The authors also have identified cytotoxic sesquiterpene pyridine alkaloids, including emarginatines B (I7) and F (I8), from M. emarginata (Willd.) Ding Hou (90, 91).
Recently, new triterpenes were isolated from M. chuchuhuasca R. Hamet and Colas (92), along with sesquiterpenes from the same plant (93). New compounds also were reported from M. ilicifolia (94). In addition, antitumor promoting sesquiterpenes were isolated from M. cuzooina Loes. (95).

Figure 9. Structures of Maytenus natural products.
Curcuma species (turmeric)
The turmeric rhizome is a main ingredient of curry powder. It gives color and flavor to food, and it has aromatic, stimulant, and carminative properties. This herb is used traditionally in India to treat biliary disorders, anorexia, cough, diabetic wounds, liver disorders, rheumatism, and sinusitis and in China for abdominal pains and jaundice. Turmeric has a protective effect on the liver, stimulates bile secretion in animals, and is recommended for use in liver disorders.
The major pigment in Curcuma species (Zingiberaceae family) is the yellow phenolic diarylheptanoid curcumin (J1). Curcumin and its analogs have potent antioxidant and antiinflammatory effects, cytotoxicity against tumor cells, and antitumor-promoting activity (96). The biologic effects and targets of curcumin, as well as its possible roles in cancer prevention and therapy, have been reviewed recently (97, 98).
Several synthetic curcumin analogs, including J2, showed potent antiandrogenic activities against two human prostate cancer cell lines, PC-3 and DU-145 (99). In expanded in vitro testing, these synthetic curcumin derivatives showed antiprostate cancer activity superior to that of hydroxyflutamide, the currently available and preferred anti-androgen for treating prostate cancer (100). In continuing work in the laboratories of the authors, dimethoxy-4-ethoxycarbonylethylenyl-curcumin (J3) showed potent anti-androgenic activity and therefore is a promising prostate cancer drug candidate (101, 102). A recent review from the laboratories of the authors discusses the design and development of curcumin analogs as promising candidates for chemotherapy of prostate cancer (103). In addition, the sesquiterpene curcumol (J4), obtained from Curcuma aromatica Salisb., was effective against cervical cancer (104).

Figure 10. Structures of curcuminoids and curcumol.
Euphorbia species
Euphorbia kansui Liou (Euphorbiaceae family) is distributed widely in northwest China. The dried roots of the plant are known as “kansui” and classified as a lower-class medicine. It is used as an herbal remedy for ascites (abdominal fluid accumulation) and cancer in China.
Ingenol diterpenoids are among the bioactive chemical constituents. Kansuiphorins A-D (K1-K4) were isolated as cytotoxic principles of E. kansui by the laboratories of the authors (105, 106). In particular, K1 and K2 demonstrated potent antileukemic activity against P-388 leukemia in mice (105). A related ingenol-type diterpene (DBDI, K5) showed unique suppression of mast cell activation, a process that occurs during inflammation, and thus, might have the potential to treat allergic diseases (107). In other pharmacological studies, two isolated ingenols from an immuno-enhancing E. kansui extract increased immune activity in a dose-dependent manner (108). Three new cytotoxic diterpenoids, yuexiandajisus D (K6), E (K7), and F (K8), were isolated from the species E. ebrateolata Hayata. (109). In additional studies, K6 showed moderate cytotoxicity against additional (HCT-8 and Bel-7402) cell lines (110).

Figure 11. Structures of Kansui natural products.
Anti-HIV Compounds
Acquired immunodeficiency syndrome (AIDS), a degenerative disease of the immune system, is caused by the human immunodeficiency virus (HIV) and results in life-threatening opportunistic infections and malignancies. Antiviral and immunomodulating natural products have been investigated as treatments for AIDS (111).
Lomatium suksdorfii (coumarin derivatives)
Suksdorfin (L1), a dihydroseselin-type angular pyranocoumarin isolated from Lomatium suksdorfii Coult. Rose (Apiaceae family) was identified as a lead anti-HIV natural product through BDFI (112). Substitution of two camphanoyl esters for the acetate and isovaleroyl groups in the natural product lead to the extremely potent lead compound 3'R,4'R-di-O-(-)-camphanoyl-(+)-cis-khellactone (DCK) (L2) (113).
After additional synthetic modification to improve potency, 4-methyl DCK (L3) and then 3-hydroxymethyl-4-methyl DCK (L4) were found. The latter compound was selected as a clinical trial candidate (114).
Furthermore, a positional isomer of DCK, 3'R,4'R-di-O-(-)-camphanoyl-2',2'dimethyldihydropyrano(2,3-f)chromone (DCP) (L5) is even more promising because most DCP analogs are active against drug-resistant HIV strains, although DCK analogs are not. Adding an ethyl group at the 2-position of DCP decreased toxicity to cells compared with DCP, so, to date, the most likely clinical trials candidate in the DCP series is 2-ethyl DCP (L6) (114, 115).
The DCK and DCP compounds exert their antiviral activity by blocking the HIV reverse transcriptase (RT), however, at a later step than that affected by the clinically approved RT inhibitors, such as AZT. Thus, these compounds have a novel mechanism of action compared with current drugs. DCK and DCP compounds could be useful in the treatment of AIDS, although additional investigation is merited and needed (116).

Figure 12. Structures of anti-HIV coumarins.
Syzigium claviflorum (triterpene betulinic acid derivatives)
Triterpenes represent a structurally varied class of natural products existing in many plant species. Thousands of triterpenes have been reported with hundreds of new derivatives described each year. Some naturally occurring triterpenes exhibit moderate anti-HIV-1 activity and, therefore, provide good leads for additional drug development because of their unique mode of action and chemical structures. Research has identified anti-HIV triterpenes that block HIV entry, including absorption (glycyrrhizin) and membrane fusion (RPR103611); inhibit viral reverse transcriptase (RT) (mimuscopic acid) and protease (ganoderiol, geumonoid); and act during viral maturation (DSB, see more description below) (117).
Two lupane triterpenes, betulinic acid (M1) and platanic acid (M2), from Syzigium claviflorum Wall. (Myrtaceae family) were reported first to reduce HIV IIIB reproduction by 50% in H9 lymphocytes (118). Afterwards, many derivatives were synthesized and studied for anti-HIV activity. Dimethyl succinyl betulinic acid (DSB, M3) was found to be the most useful candidate as an anti-HIV agent (119, 120).
DSB is the first in a new class of drugs to treat HIV infection. Its novel viral target—maturation—is unlike that of any currently approved anti-AIDS drug. DSB disrupts the late-stage viral maturation processes of HIV and causes the viral core structure to be defective and noninfectious (121).
DSB was discovered originally by the Natural Products Research Laboratories (NPRL), University of North Carolina, directed by the author (K.H.L.) (119), and then licensed to Pana- cos Pharmaceuticals, Inc. (Watertown, MA) for development. Panacos has named the compound Bevirimat and lists it as the lead antiviral product of the company.
During 2004, two Phase I studies and a Phase I/II study of M3 were completed. In the Phase I studies, the drug was well tolerated and showed good anti-HIV levels in the body. In the Phase I/II study, M3 showed activity in HIV-infected patients and significantly reduced viral blood levels (known as viral load) (122, 123). Another 2004 milestone was that the U.S. Food and Drug Administration (FDA) granted Fast Track Status for M3.
The Phase IIa study demonstrated the antiviral potency of M3, following once-daily oral dosing for 10 days in HIV-infected subjects not on other antiretroviral therapy. Viral load was reduced significantly compared with placebo. On day 11, following complete dosing, the median reduction at the 200-mg dose was a 91% decrease. In the Phase IIa trial, M3 was well tolerated, all adverse experiences were mild or moderate, and no dose-limiting toxicity was identified (122, 123).
Subsequently, studies have shown that M3 can be administered successfully in a tablet form rather than by an oral solution. Also, two drug interaction clinical trials of M3, in combination with the approved HIV drugs ritonavir and atazanavir, have been completed and showed little likelihood of significant adverse drug-drug interactions when used in combination therapy (124).
In summary, M3 shows potent viral load reduction, a strong safety profile (with no evidence of organ toxicity or clinical intolerance), no evidence of clinically significant drug interactions, and, quite importantly, no evidence of rapid resistance development, which is a primary cause for antiretroviral treatment failure (125-127).
Phase IIb clinical trials began in 2006 and still are ongoing. One of the trial goals will be to determine an optimal dose of M3. These trials will involve HIV-infected patients who are failing current therapy and will be randomized, blinded, and placebo-controlled (124).
In Phase III clinical trials targeted for 2007/2008, combination therapy studies will be performed in a total of 300 to 500 patients at a commercial dose. The target for New Drug Application (NDA) is 2008/2009 (124). The efficient clinical trials progress of M3 continues to mark it as a leading new treatment for AIDS.

Figure 13. Structures of anti-HIV triterpenes.
Active Compounds Isolated from Well-Known Folkloric Medicine
In addition to anticancer and anti-HIV agents, various types of active compounds that are active against other diseases and disorders (e.g., malaria, inflammation, and so forth.) also have been isolated from natural sources, especially well-known folkloric medicine. These compounds and their plant sources are described below.
Artemisia annua (qinghao, artemisinin derivatives)
Qinghao (Sweet Wormwood) is the dried aerial parts of the herb Artemisia annua L. (Asteraceae family), which has been used in China for centuries to treat fever and malaria. Artemisinin (N1) (Qing Hao Su) (128), the active principle, directly kills Plasmodium falciparum (malaria parasites) with little toxicity to animals and humans. Thus, it is a clinically effective, safe, and rapid antimalarial agent (129, 130). The novel endo-peroxide link is essential for the antimalarial activity.
Artemether (N2) and arteether (N3) are the most well-studied analogs among many synthetic derivatives and are used in malaria-prone regions, particularly India (131). Artemether and sodium artesunate (a hemisuccinate derivative of dihydroartemisinin) (N4) have been added by the World Health Organization to its Model List of Essential Medicines (132).
The laboratory of the authors has synthesized analogs related to artemisinin (133). Recently, an antimalarial synthetic trioxolane drug development candidate called OZ-277 (N5, also known as RBx11160) (134) has sparked great interest and has progressed to Phase II clinical trials in India, Thailand, and Africa. Modification and pharmacological studies are ongoing (135-137). A recent review discusses artemisinin and related antimalarials (138).

Figure 14. Structures of artemisinin and related compounds.

Figure 15. Structures of Panax ginsenosides.
Ginsengs: Asian, American, Sanchi, and Siberian
Ginseng is the root of Panax ginseng C.A. Meyer (Asian ginseng) (Araliaceae family). In Oriental medicine, it has enjoyed a strong reputation since ancient times for being tonic, regenerating, and rejuvenating. The genus name Panax is formed from the Greek pan (all) and akos (remedy). This panacea (panakeia) was believed to be the universal remedy. Wild ginseng is scarce and has been replaced by cultivated ginseng or “true” ginseng. Species include American ginseng (P. quinquefolium L.), cultivated in North America; Japanese ginseng (P. japonicus (Nees.) C.A. Mey., widely distributed in Japan; San-chi ginseng (P. notoginseng (Burk.) F.H. Chen), reputed as a tonic and hemostatic in China; and Siberian ginseng (Eleutherococcus senticosus Maxim.).
Asian ginseng (Panax ginseng C.A. Meyer)
M any compounds have been isolated from the root of Asian ginseng: polysaccharides, glycopeptides (panaxanes), vitamins, sterols, amino acids and peptides, essential oil, and polyalkynes (139-141). About 30 saponins (called ginsenosides) isolated from the root are dammarane triterpenoids, which generally contain three or four hydroxyl (OH) groups [a 3β,12β,20(S) trihydroxylated-type (protopanaxadiol-type) and a 3β,6α,12β, 20(S) tetrahydroxylated-type (protopanaxatriol-type)], which are attached to various sugars. The individual saponins (e.g., ginsenosides Rb1-2, Rc-f, Rg1-3, and Rh1-2, O1-O11) differ in the mono-, di-, or tri-saccharide nature of the two sugars attached at the C-3 or C-6 and C-20 hydroxy groups. In some cases, the C-20 hydroxy group is absent (e.g., in ginsenoside Rg5, O12) or the C-12 hydroxy group also is attached to a sugar.
Traditionally, ginseng is used to restore normal pulse and remedy collapse, to benefit the spleen and liver, to promote production of body fluid, to calm nerves, and to treat diabetes and cancer. Regarding the last two effects, ginsenoside Rh2 (O11) has been found to lower plasma blood glucose in streptozotocin-induced diabetic rats (142), and recently, ginseng and its constituents have been studied for cancer prevention and anticarcinogenic effects against chemical carcinogens. Ginsenoside Rg3 (O9) and Rg5 (O12) were found to reduce significantly lung tumor incidence, and Rg3 (O9), Rg5 (O12), and Rh2 (O11) showed active anticarcinogenic activity (143). In addition, effects of ginseng on quality of life have been discussed (144).

Figure 16. Structures of two different ginsenosides in P. ginseng and P. quinquefolium.
American ginseng (Panax quinquefolium L.)
American ginseng contains almost the same components as Panax ginseng (139). Thus, it could be used for the same medical conditions as Asian ginseng. However, in Chinese theory, some differences exist: American ginseng is “cool” and is used mainly to reduce the internal heat and promote the secretion of body fluids, whereas Asian ginseng is “warm.” Correspondingly, differences in biologic activity also exist (140): American ginseng stimulates the production of human lymphocytes, whereas Asian ginseng does not have a significant effect, and Siberian ginseng enhances production.
The main chemical difference between Asian and American ginseng is in the presence or absence of ginsenoside Rf (O6) and 24(R)-pseudoginsenoside F11 (O13). Ginsenoside Rf is found in Asian ginseng but not in American ginseng, and although 24(R)-pseudoginsenoside F11 is abundant in American ginseng, only trace amounts are present in Asian ginseng (141, 145-147).
Sanchi ginseng [Panax notoginseng (Burk.) F.H. Chen]
This ginseng exerts a major effect on the cardiovascular system. It dilates the coronary vessels and reduces vascular resistance, which results in increased coronary flow and decreased blood pressure. Chinese traditional medicine used this ginseng to arrest bleeding, remove blood stasis, and relieve pain. Recent studies have shown that, in the treatment of angina pectoris, this herb can produce a 95.5% improvement in symptoms. The herb usually can stop bleeding in cases of respiratory bleeding or vomiting of blood.
P. notoginseng contains saponins similar to those found in P. ginseng, both ginsenosides and notoginsenosides. Certain saponins of both types, including notoginsenosides K, Ri, and U (O14-O16), showed immunological adjuvant activity (stimulated immune response against antigens) (148, 149). Another important bioactive constituent of P. notoginseng is the non-protein amino acid dencichine (O17), which can increase platelets and stop bleeding (150).
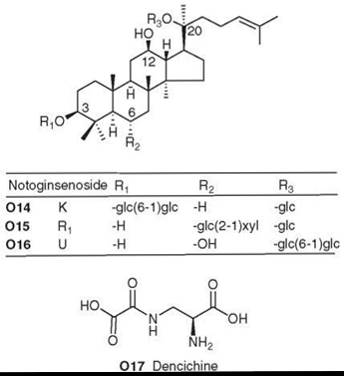
Figure 17. Structures of P. notoginseng natural products.
Siberian ginseng (Eleutherococcus senticosus maxim.)
Siberian ginseng is harvested from its natural habitat in Russia and northeast China and has been used in China for over 2000 years. It is not a true ginseng like Panax ginseng or P. quin- quefolia, but it does belong to the same Araliaceous family. Siberian ginseng has its own bioactive ingredients with unique and proven medicinal values. It possesses significant adapto- genic action (antistress and antifatigue) and is recommended as a general tonic. Because of its nonspecific mechanisms of action, Siberian ginseng has a broad range of clinical applications.
The root contains polysaccharides, phenolics (coumarins, lignans, and phenylpropionic acids), and glycosides. Some members (e.g., eleutheroside K, O18) of the latter group are specifically triterpenoid in nature, whereas others, including isofraxoside (eleutheroside B1, O19), glycosides of syringaresinol (e.g., eleutheroside E, O20), and the ethyl ether of galactose (eleutheroside C, O21), belong in a miscellaneous series. Eleutherosides E (O20) and B (O22, also called syringin) are two major glycosides and typically are used as marker compounds associated with bioactivity, particularly antifatigue action (151). A new lignan glycoside eleutheroside E2 (O23) was isolated recently from E. senticosus (152).

Figure 18. Structures of compounds in Siberian ginseng.
Ganoderma lucidum (fungus)
Chinese people consider Ganoderma lucidum (Polyporaceae family) as the “Miraculous King of Herbs.” It is highly regarded for its medicinal properties, which include promoting the healing ability of the human body, strengthening the immune system, and increasing longevity. Accordingly, Ganoderma works in the treatment of cancer because it helps cleanse the body from toxins and strengthen the immune system. It also enhances liver detoxification, thus improving liver function and stimulating the regeneration of liver cells.
The chemical composition includes polysaccharides and lanosteroid triterpenes. In the former class, many polysaccharides have been linked to immune-stimulating effects (153-155). The latter class contains over 130 different triterpenoids with diverse pharmacological activities (156). Examples of the polyoxygenated triterpene structures are ganoderic acids B (P1), C2 (P2), and D (P3), as well as ganoderiol F (P4), ganodermanondiol (P5), and ganodermanontriol (P6).
Because of its widespread use as a health food as well as for medicinal purposes, the quality control of G. lucidum is quite important, and modern analytical methods are being established for the quantitative determination of major triterpenoids, including P1-P3, as marker compounds (157).
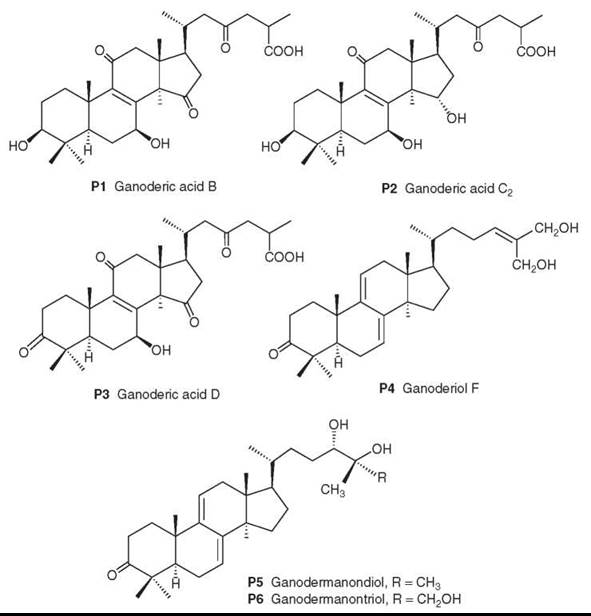
Figure 19. Structures of Ganoderma triterpenes.
Cordyceps sinensis (Tung Chung Hsia Tsao)
Vegetable caterpillars are called “Tung Chung Hsia Tsao” in Chinese, which translates as “winter worm and summer grass.” They result from the infection of large underground caterpillars by a fungus. An entomopathogenic fungus grows in the larva of the sphinx moth in autumn. Although the larva hibernates underground through the winter, the fungus kills the infected host and grows throughout the cadaver. In summer, a rod-like stroma of the fungus grows out from the mummified shell of the dead host, looking like a sprouting, dark blue to black grass. The mycelia of Cordyceps sinensis (Berk.) Saccar. (Clavicipitaceae family) that colonize the larvae of Hepialus armoricans (Hepialidae family) are representative vegetable caterpillars and are highly valued in the Chinese traditional medicinal system.
Initial records of using vegetable caterpillars as medicine date back to the Ming dynasty in China and appear in “Pen-Tsao-Kang-Mu” (Compilation of MateriaMedica) in 1596. This fungus is regarded as a popular and effective medicine for treating numerous illnesses, promoting longevity, relieving exhaustion, and increasing athletic power. More recently, their physiologic activities, including immunostimulating and antitumor effects, have encouraged their medicinal use.
In traditional medicinal practices, wild-harvested mycelia are considered to have higher therapeutic benefits and, therefore, command higher prices than cultivated fungus. Native occurrence of the fungus is confined to the highlands of the Himalayan region, in addition to some provinces of China with cold, arid environments. Several mycelial formation products grown in artificial media are available commercially as health food supplements in the United States and Canada.
A comprehensive review (158) recently discussed the chemical constituents and pharmacological actions of the Cordyceps species. Chemical constituents include cordycepin (Q1, 3'-deoxyadenosine) and other adenosine analogs, ergosterol derivatives (Q2-Q4), and peptides, including cyclic peptides such as cordycepeptide A (Q5) (159). These compounds likely contribute to the antitumor, antibacterial, antifungal, and antiinflammatory activities. Regarding the latter activity, Q1 has been found to inhibit platelet aggregation (160). C. sinensis also contains polysaccharides, which account for anti-inflammatory, antioxidant, antitumor, antimetastatic, immunomodulatory, hyperglycemic, steroidogenic, and hypolipidemic effects (158, 159). New diphenyl ethers (cordyols A-C, Q6-Q8) were discovered recently in Cordyceps (161). Q6 was associated with significant anti-HSV (herpes simplex virus) activity (161).

Figure 20. Structures of Cordyceps natural products.
Conclusion
Based on the above successful examples, new drugs derived from natural product leads will be discovered continuously, and modern medicinal chemistry-based molecular modification will play an important role in developing the new leads into useful drug candidates. Highly efficient bioactivity-directed fractionation and isolation, characterization, analog synthesis, and mechanistic studies are prerequisites for the development of new plant-derived compounds as clinical candidates for world-class new medicines. Drug discovery also will benefit from the discovery of new biologic targets and the continual improvement of bioassay technology (162-169). The development of new, effective, and safe world-class drugs from medicinal plants, which have been appreciated for centuries for treating illness, will be long lasting, as they are the best and most effective source for generating new medicines by use of modern scientific technology.
Acknowledgment
This investigation was supported by grants from the National Cancer Institute, the National Institutes of Health (NIH) (CA-17625), and the National Institute of Allergy and Infectious Diseases, NIH (AI-33066) that were awarded to K.H. Lee. This article is No. 257 in the series “Antitumor Agents” and No. 71 in the series “Anti-AIDS Agents.”
References
1. Balunas MJ, Kinghorn AD. Drug discovery from medicinal plants. Life Sci. 2005; 78:431-441.
2. Lee KH, Itokawa H, Kozuka M. Asian herbal products: the basis for development of high-quality dietary supplements and new medicines. In: Asian Functional Foods. Shi J, Ho CT, Shahidi F, eds. 2005.Marcel Dekker/CRC Press, Boca Raton, FL. pp. 21-72.
3. Suffness M, Douros J. Current status of the NCI plant and animal product program. J. Nat. Prod. 1982; 45:1-14.
4. Itokawa H. Research on antineoplastic drugs from natural sources especially from higher plants. Yakugaku Zasshi 1988; 108:824-841.
5. Lee KH. Antineoplastic agents and their analogues from Chinese traditional medicine. In: Human Medicinal Agents from Plants. Kinghorn AD, Balandrin M, eds. 1993. Amer. Chem. Soc. Symposium Series 534. pp. 170-190.
6. Itokawa H, Takeya K, Hitotsuyanagi Y, Morita H. Antitumor compounds isolated from higher plants. Yakugaku Zasshi 1999; 119:529-583.
7. Itokawa H, Takeya K, Hitotsuyanagi Y, Morita H. Antitumor compounds isolated from higher plants. In: Studies in Natural Products Chemistry. Ur-Rahman A, ed. 2000. Elsevier, Amsterdam, pp. 269-350.
8. Itokawa H, Takeya K, Lee KH. Anticancer compounds from higher plants. In: Biomaterials from Aquatic and Terrestrial Organisms. Fingerman M, Nagabhushanam R, eds. 2006. Science Publishers, Enfield, NH, pp. 255-283.
9. Tang W, Hemm I, Bertram B. Recent development of antitumor agents from Chinese herbal medicines; Part I. Low molecular compounds. Planta Med. 2003; 69:97-108.
10. Tang W, Hemm I, Bertram B. Recent development of antitumor agents from Chinese herbals medicines; Part II. Low molecular compounds. Planta Med. 2003; 69:193-201.
11. Lee KH. Current developments in the discovery and design of new drug candidates from plant natural product leads. J. Nat. Prod. 2004; 67:273-283.
12. Mukherjee AK, Basu S, Sarkar N, Ghosh AC. Advances in cancer therapy with plant based natural products. Curr. Med. Chem. 2001; 8:1467-1486.
13. Cragg GM, Newman DJ. Discovery and development of antineoplastic agents from natural sources. Cancer Invest. 1999; 17:153-163.
14. Cragg GM, Newman DJ. A tale of two tumor targets: topoisomerase I and tubulin. The Wall and Wani contribution to cancer chemotherapy. J. Nat. Prod. 2004; 67:233-244.
15. Wall ME, Wani MC. Camptothecin and taxol: from discovery to clinic. J. Ethnopharmacol. 1996; 51:239-253.
16. Oberlies NH, Kroll DJ. Camptothecin and taxol: historic achievements in natural products research. J. Nat. Prod. 2004; 67:129-135.
17. Potier P. The synthesis of navelbine: prototype of a new series of vinblastine derivatives. Sem. Oncol. 1989; 16:2-4.
18. Jenks S, Smigel K. Updates: cancer drug approved; new leukemia treatment. J. Nat. Cancer Inst. 1995; 87:167-170.
19. Vlahov IR, Santhapuram HKR, Kleindl PJ, Howard SJ, Stanford KM, Leamon CP. Design and regioselective synthesis of a new generation of targeted chemotherapeutics. Part I: ED145, a folic acid conjugate of desacetylvinblastine monohydrazide. Bioorg. Med. Chem. Lett. 2006; 16:5093-5096.
20. Okouneva T, Hill BT, Wilson L, Jordan MA. The effects of vinflunine, vinorelbine, and vinblastine on centromere dynamics. Mol. Cancer Ther. 2003; 2:427-436.
21. Kruczynski A, Barret JM, Erievant C, Colpaert F, Fahy J, Hill BT. Antimitotic and tubulin-interacting properties of vinflunine, a novel fluorinated Vinca alkaloid. Biochem. Pharmacol. 1998; 55:635-648.
22. Wani MC, Tayler HI, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemia and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971; 93:2325-2327.
23. Itokawa H, Lee KH, eds. Taxus: Genus Taxus. 2003. Taylor & Francis, London.
24. Shigemori H, Kobayashi J. Biological activity and chemistry of taxoids from the Japanese yew, Taxus cuspidate. J. Nat. Prod. 2004; 67:245-256.
25. Cragg GM, Suffness M. Metabolism of plant-derived anticancer agents. Pharmcol. Ther. 1988; 37:425-461.
26. Kingston DG. The chemistry of taxol. Pharmcol. Ther. 1991; 52:1-34.
27. Geney R, Chen J, Ojima I. Recent advances in the new generation taxane anticancer agents. Med. Chem. 2005; 1:125-139.
28. Appendino G, Bettoni P, Noncovich A, Sterner O, Fontana G, Bombardelli E, Pera P, Bernack RJ. Structure-activity relationship of ring C-secotaxoids. 1. Acylative modifications. J. Nat. Prod. 2004; 67:184-188.
29. Liu C, Strobl JS, Bane S, Schilling JK, McCracken M, Chatterjee SK, Rahim-Bata R, Kingston DGI. Design, synthesis, and bioactivities of steroid-linked taxol analogues as potential targeted drugs for prostate and breast cancer. J. Nat. Prod. 2004; 67:152-159.
30. Kuznetsova L, Chen J, Sun L, Wu X, Pepe A, Veith JM, Pera P, Bernacki RJ, Ojima I. Syntheses and evaluation of novel fatty acid-second generation taxoid conjugates as promising anticancer agents. Bioorg. Med. Chem. Lett. 2006; 16:974-977.
31. Raez LE, Lilenbaum R. New developments in chemotherapy for advanced non-small cell lung cancer. Curr. Opin. Oncol. 2006; 18:156-161.
32. Singer JW, Shaffer S, Baker B, Gbernareggi A, Stromatt S, Nienstedt D, Besman M. Paclitaxel poliglumex (XYOTAX; CT-2103): an intracellularly targeted taxane. Anticancer Drugs 2005; 16:243-254.
33. Shi Q, Wang HK, Bastow KF, Tachibana Y, Chen K, Lee FY, Lee KH. Antitumor agents 210. Synthesis and evaluation of taxoid-epipodophyllotoxin conjugates as novel cytotoxic agents. Bioorg. Med. Chem. 2001; 9:2999-3004.
34. Ohtsu H, Nakanishi Y, Bastow KF, Lee FY, Lee KH. Antitumor agents 216. Synthesis and evaluation of paclitaxel-camptothecin conjugates as novel cytotoxic agents. Bioorg. Med. Chem. 2003; 11:1851-1857.
35. Hennenfent KL, Govindan R. Novel formulations of taxanes: a review. Old wine in a new bottle? Ann. Oncol. 2006; 17:735-749.
36. Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. Plant antitumor agents. 1. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminate. J. Am. Chem. Soc. 1966; 88:3888-3890.
37. Wall ME. In: Chronicles of Drug Discovery, Volume 3. Lednicer D, ed. 1993. American Chemical Society, Washington D.C., pp. 327-348.
38. Covey JM, Jaxel C, Kohn KW, Pommier Y. Protein-linked DNA strand breaks induced in mammalian cells by camptothecin, an inhibitor of topoisomerase I. Cancer Res. 1989; 49:5016-5022.
39. Vanhoefer U, Harstrick A, Achterrath W, Cao S, Seeber S, Rustum YM. Irinotecan in the treatment of colorectal cancer: clinical overview. J. Clin. Oncol. 2001; 19:1501-1518.
40. Wang HK, Liu SY, Hwang KM, Taylor G, Lee KH. Synthesis of novel water-soluble 7-(aminoacylhydrazone)-formyl camptothecins with potent inhibition of DNA topoisomerase I. Bioorg. Med. Chem. 1994; 2:1397-1402.
41. Dallavalle S, Merlini L, Morini G, Muso L, Penco S, Beretta GL, Tinelli S, Zunino F. Synthesis and cytotoxic activity of substituted 7-aryliminomethyl derivatives of camptothecin. Eur. J. Med. Chem. 2004; 39:507-513.
42. Bastow KF, Wang HK, Cheng YC, Lee KH. Antitumor agents 173. Synthesis and evaluation of camptothecin-4β-amino-4’-O-demethyl epipodophyllotoxin conjugates as inhibitors of mammalian DNA topoisomerases and as cytotoxic agents. Bioorg. Med. Chem. 1997; 5:1481-1488.
43. O’Leary J, Muggia FM. Camptothecins: a review of their development and schedules of administration. Eur. J. Cancer 1998; 34:1500-1508.
44. Garcia-Carbonero R, Supko JG. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin. Cancer Res. 2002; 8:641-661.
45. Thomas CJ, Rahier NJ, Hecht SM. Camptothecin: current perspectives. Bioorg. Med. Chem. 2004; 12:1585-1604.
46. Du W, Kaskar B, Blumbergs P, Subramanian PK, Curran DP. Semisynthesis of DB-67 and other silatecans from camptothecin by thiol-promoted addition of silyl radicals. Bioorg. Med. Chem. 2003; 11:451-458.
47. Clark JW. Rubitecan. Expert Opin. Investig. Drugs 2006; 15:71-79.
48. Imbert TF. Discovery of podophyllotoxins. Biochimie 1998; 80:207-222.
49. Lee KH, Imakura Y, Haruna M, Beers SA, Thurston LS, Dai HJ, Chen CH, Liu SY, Chan YC. New cytotoxic 4-alkylamino analogues of 4’-demethyl-epipodophyllotoxin as inhibitors of human DNA topoisomerase II. J. Nat. Prod. 1989; 52:606-613.
50. Chang JY, Han FS, Liu SY, Wang HK, Lee KH, Cheng YC. Effect of 4-β-arylamino derivatives of 4’-O-demethylpodophyllotoxin on human DNA topoisomerase II, tubulin polymerization. KB cells, and their resistant variants. Cancer Res. 1991; 51:1755-1759.
51. Wang ZW, Kuo YH, Schnur D, Bowen JP, Liu SY, Han FS, Chan JY, Chen YC, Lee KH. New 4β-arylamino derivatives of 4’-O-demethylepipodophyllotoxin and related compounds as potent inhibitors of human DNA topoisomerase II. J. Med. Chem. 1990; 33:2660-2666.
52. Cho SJ, Tropsha A, Suffness M, Cheng YC, Lee KH. Three dimensional quantitative structure-activity relationship study of 4’-O-demethylepipodophyllotoxin analogs using the modified CoMFA/q2-GRS approach. J. Med. Chem. 1996; 39:1383-1385.
53. Communication between Genelabs Technologies, Inc. and Fossella FV. MD. University of Texas MD Anderson Cancer Center, July 18, 1994.
54. Zheng J,Wang HK, Bastow KF, Zhu XK, Cho SJ, Cheng YC, Lee KH. Antitumor agents. 177. Design, syntheses, and biological evaluation of novel etoposide analogs bearing pyrrolecarboxamidino group as DNA topoisomerase II inhibitors. Bioorg. Med. Chem. Lett. 1997; 7:607-612.
55. Zhu XK, Guan J, Tachibana Y, Bastow KF, Cho SJ, Cheng HH, Cheng YC, Gurwith M, Lee KH. Antitumor agents. 194. Synthesis and biological evaluations of 4β-mono-, -di-, and -trisubstituted aniline-4’-O-demethylpodophyllotoxin and related compounds with improved pharmacological profiles. J. Med. Chem. 1999; 42:2441-2446.
56. Xiao Z, Xiao YD, Feng J, Golbraikh A, Tropsha A, Lee KH. Modeling of epipodophyllotoxin derivatives using variable selection k nearest neighbor QSAR method. J. Med. Chem. 2002; 45:2294-2309.
57. Gordaliza M, Garcia PA, del Corral JMM, Castro MA, Gomez-Zurita MA. Podophyllotoxin: distribution, sources, application, and new cytotoxic derivatives. Toxicon 2004; 44:441-459.
58. Lee KH, Xiao Z. The podophyllotoxins and analogs. In: Antitumor Agents from Natural Sources. Kingston DGI, Cragg GM, Newman DJ, eds. 2005. CRC Press, Boca Raton, FL.
59. Powell RG, Madrigal RV, Smith CR, Mikolajczak KL. Alkaloids of Cephalotaxus harringtonia var. drupacea. 11-Hydroxycephalotaxine and drupacine. J. Org. Chem. 1974; 39:676-680.
60. Powell RG, Weisleder D, Smith CR. Antitumor alkaloids for Cephalotaxus harringtonia: structure and activity. J. Pharm. Sci. 1972; 61:1227-1230.
61. Powell RG, Weisleder D, Smith CR, Rohwedder WK. Structures of harringtonine, isoharringtonine, and homoharringtonine. Tetrahedron Lett. 1970; 11:815-818.
62. Huang CC, Han CS, Yue XF, Shen CM, Wang SW, Wu FG, Xu B. Cytotoxity and sister chromatid exchanges induced in vitro by six anticancer drugs developed in the People’s Republic of China. J. Natl. Cancer Inst. 1983; 71:841-847.
63. Spencer GF, Plattner RD, Powell RG. Quantitative gas chromatography and gas chromatography-mass spectrometry of Cephalotaxus alkaloids. J. Chromatogr. 1976; 120:335-341.
64. Grem JL, Cheson BD, King SA, Leyland-Jones B, Suffness M. Cephalotaxine esters: antileukemic advance or therapeutic failure? J. Natl. Cancer Inst. 1988; 80:1095-1103.
65. PaudlerWW, Kerley GI, McKay J. The alkaloids of Cephalotaxus drupacea and Cephalotaxus fortunei. J. Org. Chem. 1963; 28:2194-2197.
66. Luo CY, Tang JY, Wang YP. Homoharringtone: a new treatment option for myeloid leukemia. Hematology 2004; 9:259-270.
67. Kantarjian HM, Cortes J. New strategies in chronic myeloid leukemia. Intl. J. Hematol. 2006; 83:289-293.
68. Itokawa H,Wang X, Lee KH. Homoharringtonine and related compounds. In: Antitumor Agents from Natural Sources. Kingston DGI, Cragg GM, Newman DJ, eds. 2005. CRC Press, Boca Raton, FL.
69. Caprard HG,Brossi A. Tropolonic colchicum alkaloids. In: The Alkaloids. Brossi A, ed. 1984. Academic Press, New York. pp. 48-54.
70. Shi Q, Verdier-Pinard P, Brossi A, Hamel E, McPhail AT, Lee KH. Antitumor agents 172. Synthesis and biological evaluation of novel deacetamidothiocolchicin-7-ols and ester analogs as antitubulin agents. J. Med. Chem. 1997; 40:961-966.
71. Shi Q, Chen K, Brossi A, Verdier-Pinard P, Hamel E, McPhail AT, Lee KH. Antitumor agents 184. Syntheses and antitubulin activity of compounds derived from reaction of thiocolchicone with amines: lactams, alcohols, and esters analogs of allothiocolchicinoids. Helv. Chim. Acta 1998; 81:1023-1037.
72. Guan J, Zhu XK, Tachibana Y, Bastow KF, Brossi A, Hamel E, Lee KH. Antitumor agents. 185. Synthesis and biological evaluation of tridemethylthiocolchicine analogues as novel topoi- somerase II inhibitors. J. Med. Chem. 1998; 41:1956-1961.
73. Bastow KF, Tatematsu H, Bori ID, Fukushima Y, Sun L, Goz G, Lee KH. Induction of reversible protein-linked DNA breaks in human osteogenic sarcoma cells by novel cytocidal colchicine derivatives which inhibit DNA topoisomerase II in vitro: absence of cross-resistance in a colchicine-resistant sub-clone. Bioorg. Med. Chem. Lett. 1993; 3:1045-1050.
74. Wu WL, Chang WL, Chen CF. Cytotoxicity activities of tanshinones against human crcinoma cell lines. Am. J. Chinese Med. 1991; 14:207-216.
75. Ryu SY, Lee CO, Choi SU. In vitro cytotoxicity of tanshinones from Salvia miltiorrhiza. Planta Med. 1997; 63:339-342.
76. Sugiyama A, Zhu BM, Takahara A, Satoh Y, Hashimoto K. Cardiac effects of Salvia miltoirrhiza/Dalbergia odorifera mixture, an intravenously applicable Chinese medicine widely used for patients with ischemic heart disease in China. Circulat. J. 2002; 66:182-184.
77. Li HB, Chen F. Preparative isolation and purification of six diterpenoids from the Chinese medicinal plant Salvia miltiorrhiza by high-speed counter-current chromatography. J. Chromatogr. A 2001; 925:109-114.
78. Chang J, Xu J, Li M, Zhao M, Ding J, Zhang JS. Novel cytotoxic seco-abietane rearranged diterpenoids from Salvia prionitis. Planta Med. 2005; 71:361-366.
79. Liu J, Shen HM, Ong CN. Salvia militiorrhiza inhibits cell growth and induces apoptosis in human hepatoma HepG(2) cells. Cancer Lett. 2000; 153:85-93.
80. Wang X, Bastow KF, Sun CM, Lin YL, Yu HJ, Don MJ, Wu TS, Nakamura S, Lee KH. Antitumor Agents. 239. Isolation, structure elucidation, total synthesis, and anti-breast cancer activity of neo-tanshinlactone from Salvia miltiorrhiza. J. Med. Chem. 2004; 47:5816-5819.
81. Wang X, Nakagawa-Goto K, Bastow KF, Don MJ, Lin YL, Wu TS, Lee KH. Antitumor agents. 254. Synthesis and biological evaluation of novel neo-tanshinlactone analogues as potent anti-breast cancer agents. J. Med. Chem. 2006; 49:5631-5634.
82. Itokawa H, Tsuruoka S, Takeya K, Mori N, Sonobe T, Kosemura S, Hamanaka T. An antitumor morphinane alkaloid, sinococuline, fromCocculus trilobus. Chem. Pharm. Bull. 1987; 35:1660-1662.
83. Liu WK, Wang XK, Che CT. Cytotoxic effects of sinococuline. Cancer Lett. 1996; 99:217-224.
84. Kupchan SM, Komoda Y, Court WA, Thomas GJ, Smith RM, Karim A, Gilmore CJ, Haltiwanger RC, Bryan RF. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J. Am. Chem. Soc. 1972; 94:1354-1356.
85. Kupchan SM, Sneden AT, Branfman AR, Howie GA, Rebhun LI, McIvor WE, Wang RW, Schanitman TC. Structural requirements for antileukemic activity among the naturally occurring and semisynthetic maytansinoids. J. Med. Chem. 1978; 21:31-37.
86. Nakao H, Senokuchi K, Umebayashi C, Kanemaru K, Masuda T, Oyama Y, Tonemori S. Cytotoxic activity of maytanprine isolated from M. diversifolia in human leukemia K562 cells. Biol. Pharm. Bull. 2004; 27:1236-1240.
87. Itokawa H, Shirota O, Ikuta H, Morita H, Takeya K, Iitaka Y. Triterpenes from rhizomes of Maytenus ilicifolia. Phytochemistry 1991; 30:3713-3716.
88. Itokawa H, Shirota O, Morita H, Takeya K, Tomioka N, Iitaka Y. Triterpene dimers from Maytenus ilicifolia. Tetrahedron Lett. 1990; 31:6881-6882.
89. Shirota O, Morita H, Takeya K, Itokawa H. Revised structures of cangorosins, triterpene dimers from Maytenus ilicifolia. J. Nat. Prod. 1997; 60:111-115.
90. Kuo YH,Chen CH, Kuo LM, King ML, Wu TS, Haruna M, Lee HK. Antitumor agents 112. Emarginatine B, a novel potent cytotoxic sesquiterpene pyridine alkaloid from Maytenus emarginata. J. Nat. Prod. 1990; 53:422-428.
91. Kuo YH, King ML, Chen CF, Chen HY, Chen CH, Chen K, Lee HK. Two new macrolide sesquiterpene pyridine alkaloids from Maytenus emarginata: emarginatine G and the cytotoxic emarginatine F. J. Nat. Prod. 1994; 57:262-269.
92. Shirota O,Sekita S, Satake M, Morita H, Takeya K, Itokawa H. Nine regioisomeric and stereoisomeric triterpene dimers from Maytenus chuchuhuasca. Chem. Pharm. Bull. 2004; 52:739-746.
93. Shirota O, Sekita S, Satake M, Morita H, Takeya K, Itokawa H. Two new sesquiterpene pyridine alkaloids from Maytenus chuchuhuasca. Heterocycle 2004; 63:1891-1896.
94. Ohsaki A, Imai Y, Naruse M, Ayabe S, Komiyama K, Takashima J. Four new triterpenoids from Maytenus ilicifolia. J. Nat. Prod. 2004; 67:469-471.
95. Gonzalez AG, Tincusi BM, Bazzocchi IL, Tokuda H, Nishino H, Konoshima Y, Jimenez IA, Ravelo AG. Anti-tumor promoting effects of sesquiterpenes from Maytenus cuzcoina (Celastraceae). Bioorg. Med. Chem. 2000; 8:1773-1778.
96. Aggarwal BB, Kumar A, Bharti AC. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res. 2003; 23:363-398.
97. Shishodia S, Sethi G, Aggarwal BB. Curcumin: getting back to the roots. Ann. NY Acad. Sci. 2005; 1056:206-217.
98. Singh S, Khar A. Biological effects of curcumin and its role in cancer chemoprevention and therapy. Anticancer Agents Med. Chem. 2006; 6:259-270.
99. Ohtsu H, Xiao Z, Ishida J, Nagai M, Wang HK, Itokawa H, Su CY, Shih C, Lee Y, Tsai MY, Chang C, Lee KH. Antitumor agents 217. Curcumin analogues as novel androgen receptor antagonists with potential as anti-prostate cancer agents. J. Med. Chem. 2002; 45:5037-5042.
100. Ohtsu H, Itokawa H, Su CY, Shih C, Chiang T, Chang E, Lee YF, Chiu SY, Chang C, Lee KH. Antitumor agents 222. Synthesis and anti-androgen activity of new diarylheptanoids. Bioorg. Med. Chem. 2003; 11:5083-5090.
101. Lin L, Shi Q, Su CY, Shih CCY, Lee KH. Antitumor agents 247. New 4-ethoxycarbonylethylcurcumin analogs as potential antiandrogenic agents. Bioorg. Med. Chem. 2006; 14:2527-2534.
102. Lin L, Shi Q, Nyarko AK, Bastow KF, Wu CC, Su CY, Shih CCY, Lee KH. Antitumor agents 250. Design and synthesis of new curcumin analogues as potential anti-prostate cancer agents. J. Med. Chem. 2006; 49:3963-3972.
103. Lin L, Lee KH. Structure-activity relationships of curcumin and its analogs with different biological activities. In: Studies in Natural Products Chemistry, Volume 33. Ur-Rahman A, ed. Elsevier, New York, pp. 785-812.
104. Lee KH. Antineoplastic agents and their analogues from Chinese traditional medicine. In: Human Medicinal Agents from Plants. Kinghorn AD, Balandrin M, eds. 1993. Amer. Chem. Soc. Symposium Series 534, pp. 170-190.
105. Wu TS, Lin YM, Haruna M, Pan DJ, Shingu T, Chen YP, Hsu HY, Nakano T, Lee KH. Antitumor agents, 119. Kansuiphorins A and B, two novel antileukemic diterpene esters from Euphorbia kansui. J. Nat. Prod. 1991; 54:823-829.
106. Pan DJ, Hu CQ, Chang JJ, Lee TTY, Chen YP, Hsu HY, McPhail DR, McPhail AT, Lee KH. Kansuiphorin-C and -D, cytotoxic diterpenes from Euphorbia kansui. Phytochemistry 1991; 30:1020-1023.
107. Nunomura S, Kitanaka S, Ra C. 3-O-(2,3-Dimethylbutanoyl)-13-O-decanoylingenol from Euphorbia kansui suppresses IGE-mediated mast cell activation. Biol. Pharm. Bull. 2006; 29:286-290.
108. Matsumoto T, Cyong JC, Yamada H. Stimulatory effects of ingenols from Euphorbia kansui on the expression of macrophage Fc receptor. Planta Med. 1992; 58:255-258.
109. Shi HM, Williams ID, Sung HHY, Zhu HX, Ip NY, Min ZD. Cytotoxic diterpenoids from the roots of Euphorbia ebracteolata. Planta Med. 2005; 71:349-354.
110. Fu GM, Qin HL, Yu SS, Yu BY. Yuexiandajisu D, a novel 18-nor-rosane-type dimeric diterpenoid from Euphorbia ebracteolata Hayata. J. Asian Nat. Prod. Res. 2006; 8:29-34.
111. Cos P, Maes L, Berghe DV, Hermans N, Pieters L, Vlietinck A. Plant substances as anti-HIV agents selected according to their putative mechanism of action. J. Nat. Prod. 2004; 67:284-293.
112. Lee TT, Kashiwada Y, Huang L, Sneider J, Cosentino M, Lee KH. Suksdorfin: an anti-HIV principle from Lomatium suks- dorfii, its structure-activity correlation with related coumarins, and synergistic effects with anti-AIDS nucleosides. Bioorg. Med. Chem. 1994; 2:1051-1056.
113. Huang L, Kashiwada Y, Cosentino LM, Fan S, Chen CH, McPhail AT, Fujioka T, Mihashi K, Lee KH. Anti-AIDS agents 15. Synthesis and anti-HIV activity of dihydroseselins and related analogs. J. Med. Chem. 1994; 37:3947-3955.
114. Yu D, Suzuki M, Xie L, Morris-Natschke SL, Lee KH. Recent progress in the development of coumarin derivatives as potent anti-HIV agents. Med. Res. Rev. 2003; 23:322-345.
115. Yu D, Chen CH, Brossi A, Lee KH. Anti-AIDS agents 60. Substituted 3’R,4’R-di-O-(-)-camphanoyl-2’2’-dimethyldihydropyrano [2,3-f.chromone (DCP) analogs as potent anti-HIV agents. J. Med. Chem. 2004; 47:4072-4082.
116. Yu D, Lee KH. Anti-AIDS agents 63. Recent progress and prospects on plant-derived anti-HIV agents and analogs. In: Medicinal Chemistry of Bioactive Natural Products. Liang XT, Fang WS, eds. 2006 Wiley, Inc., Hoboken, NJ, pp. 357-398.
117. Kashiwada Y, Hashimoto F, Cosentino LM, Chen CH, Garrren PE, Lee KH. Beturinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. J. Med. Chem. 1996; 39:1016-1017.
118. Fujioka T, Kashiwada Y, Kilkuskie RE, Cosentino LM, Ballas LM, Jiang JB, Janzen WP, Chen IS, Lee KH. Anti-AIDS agents 11. Betulinic acid and platonic acid as anti-HIV principles from Syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J. Nat. Prod. 1994; 57:243-247.
119. Lee KH, Kashiwada Y, Hashimoto F, Cosentino LM, Manak M. Betulinic acid derivatives and antiviral use. University of North Carolina, at Chapel Hill and Biotech, Research Laboratories, PCT Int. Appl. WO 9639033. December 12, 1996.
120. Sun IC, Kashiwada Y, Morris-Natschke SL, Lee KH. Plant- derived terpenoids and analogues as anti-HIV agents. Curr. Top. Med. Chem. 2002; 3:155-169.
121. Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway FP, Freed EO, Wild CT. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:13555-13560.
122. Results of the Phase IIa study were presented as an oral late breaker presentation at the 45th Interscience Conference on Antimicrobial Agents and Chemotherapy, 2005.
123. Triangle Business Journal, September 9, 2005.
124. Data provided by Panacos, Inc.
125. Yu D, Wild CT, Martin DE, Morris-Natschke SL, Chen CH, Allaway G, Lee KH. Anti-AIDS agents 64. The discovery of maturation inhibitors and their potential in the therapy of HIV. Expert Opin. Investig. Drugs 2005; 14:681-693.
126. Yu D, Lee KH. Anti-AIDS agents 63. Recent progress and prospects on plant-derived anti-HIV agents and analogs. In: Medicinal Chemistry of Bioactive Natural Products. Liang XT, Fang WS, eds. 2006. John Wiley & Sons, Hoboken, NJ, pp. 357-398.
127. Yu D, Morris-Natschke SL, Lee KH. Anti-AIDS agents 67. New developments in natural products-based anti-AIDS research. Med. Res. Rev. 2007; 27:133-148.
128. Li Y, Huang H, Wu YL. Qinghaosu (Artemisinin) - A fantastic antimalarial drug from a traditional Chinese medicine. In: Medicinal Chemistry of Bioactive Natural Products. Liang XT, Fang WS, eds. 2006. John Wiley & Sons, Hoboken, NJ, pp. 183-256.
129. Meshnick SR. In: Antimalarial Chemotherapy. Rosenthal PJ, ed. 2001. Humana Press, Totowa, NJ, pp. 191-201.
130. Avery MA, McLean G, Edwards G, Ager A. Structure-activity relationships of peroxide-based artemisinin antimalarials. In: Biologically Active Natural Products: Pharmaceuticals. Cutler SJ, Cutler HG, eds. 2000. CRC Press, Boca Raton, FL, pp. 121-132.
131. Pareek A, Nandy A, Kochar D, Patel KH, Mishra SK, Mathur PC. Efficacy and safety of β-arteether and α/β-arteether for treatment of acute Plasmodium falciparum malaria. Am. J. Trop. Med. 2006; 75:139-142.
132. http://mednet3.who.int/EMLib/.
133. Imakura Y, Yokoi T, Yamagishi T, Koyama J, Hu H, McPhail DR, McPhail AT, Lee KH. Synthesis of deethanoqinghaosu, a novel analog of the antimalarial qinghaosu. J. Chem. Soc. Commun. 1988; 372.
134. Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FC, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, McIntosh K, Padmanilayam M, Santo Tomas J, Scheurer C, Scorneaux B, Tang Y, Urwyler H, Wittlin S, Charman WN. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 2004; 430:900-904.
135. Dong Y, Chollet J, Matile H, Charman SA, Chiu FC, Charman WN, Scorneaux B, Urwyler H, Santo Tomas J, Scheurer C, Snyder C, Dorn A, Wang X, Karle JM, Tang Y, Wittlin S, Brun R, Vennerstrom JL. Spiro and dispiro-1,2,4-trioxolanes as antimalarial peroxides: charting a workable structure-activity relationship using simple prototypes. J. Med. Chem. 2005; 48:4953-4961.
136. Perry CS, Charman SA, Prankerd RJ, Chiu FC, Dong Y, Vennerstrom JL, Charman WN Chemical kinetics and aqueous degradation pathways of a new class of synthetic ozonide anti- malarials. J. Pharm. Sci. 2006; 95:737-747.
137. Dong Y, Tang Y, Chollet J, Matile H, Wittlin S, Charman SA, Charman WN, Tomas JS, Scheurer C, Snyder C, Scorneaux B, Bajpai S, Alexander SA, Wang X, Padmanilayam M, Cheruku SR, Brun R, Vennerstrom JL. Effect of functional group polarity on the antimalarial activity of spiro and dispiro-1,2,4-trioxolanes. Bioorg. Med. Chem. 2006; 14:6368-6382.
138. Haynes RK. From artemisinin to new artemisinin antimalarials: Biosynthesis, extraction, old and new derivatives, stereochemistry and medicinal chemistry requirements. Curr. Top. Med. Chem. 2006; 6:509-537.
139. Shibata S, Fujita M, Itokawa H, Tanaka O. Studies on the constituents of Japanese and Chinese crude drugs. XI. Panaxadiol, a sapogenin of Ginseng roots. Chem. Pharm. Bull. 1963; 11:759.
140. Shibata S. Chemistry and cancer preventing activities of ginseng saponins and some related triterpenoid compounds. J. Korean Med. Sci. 2001; 16(suppl S):S28-S37.
141. Tanaka O. Ginseng and its congeners - Traditional oriental food drugs. In: Food Phytochemicals for Cancer Prevention II, ACS Symposium Series 1994; 547:335-341.
142. Lai DM, Tu YK, Liu IM, Chen PF, Cheng JT. Mediation of β-endorphin by ginsenoside Rh2 to lower plasma glucose in streptozotocin-induced diabetic rats. Planta Med. 2006; 72:9-13.
143. Yun TK, Choi SY. The relationship between cancer and ginseng intake. Intl. J. Epidemiol. 1990; 19:871-876.
144. Ellis JM, Reddy P. Effects of Panax ginseng on quality of life. Ann. Pharmacother. 2002; 36:375-379.
145. Yun TK, Lee YS, Lee YH, Kim SI, Yun HY. Anticarcinogenic effect of Panax ginseng CA Meyer and identification of active compounds. J. Korean Med. Sci. 2001; 16(suppl S)S6-S18.
146. Attele AS, Wu JA, Yuan CS. Ginseng pharmacology. Biochem. Pharmacol. 1999; 58:1685-1693.
147. Surh YJ, Na HK, Lee JY, Keum YS. Molecular mechanisms underlying anti-tumor promoting activities of heat-processed Panax ginseng CA Meyer. J. Korean Med. Sci. 2001;16(suppl S):S38-S41.
148. Sun H, Ye Y, Pan Y. Immunological-adjuvant saponins from the roots of Panax notoginseng. Chem. Biodivers. 2005; 2:510-515.
149. Sun H, Chen Y, Ye Y. Ginsenoside Re and notoginsenoside R1: immunological adjuvants with low haemolytic effect. Chem. Biodivers. 2006; 3:718-726.
150. Xie GX, Qiu YP, Qiu MF, Gao XF, Liu YM, Jia W. Analysis of dencichine in Panax notoginseng by gas chromatography-mass spectrometry with ethyl chloroformate derivatization. J. Pharm. Biomed. Anal. 2007; 43:920-925.
151. Kimura Y, Sumiyoshi M. Effects of various Eleutherococcus senticosus cortex on swimming time, natural killer activity and corticosterone level in forced swimming stressed mice. J. Ethnopharmacol. 2004; 95:447-453.
152. Li XC, Barnes DL, Khan IA. A new lignan glycoside from Eleutherococcus senticosus. Planta Med. 2001; 67:776-778.
153. Bao XF, Zhen Y, Ruan L, Fang JM. Purification, characterization, and modification of T lymphocyte-stimulating polysaccharides from spores of Ganoderma lucidum. Chem. Pharm. Bull. 2002; 50:623-629.
154. Bao XF, Wang XS, Dong Q, Fang JN, Li XY. Structural features of immunologically active polysaccharides from Ganoderma lucidum. Phytochemistry 2002; 29:175-181.
155. Lin YL, Lee SS, Lou SM, Chiang BL. Polysaccharide purified from Ganoderma lucidum induces gene expression changes in human dendritic cells and promotes T helper 1 immune response in BALB/c mice. Mol. Pharmacol. 2006; 70:637-644.
156. Shiao MS. Natural products of the medicinal fungus Ganoderma lucidum: occurrence, biological activities, and pharmacological functions. Chem. Rec. 2003; 3:172-180.
157. Wang XM, Yang M, Guan SH, Liu RX, Xia JM, Bi KS, Guo DA. Quantitative determination of six major triterpenoids in Ganoderma lucidum and related species by high performance liquid chromatography. J. Pharm. Biomed. Anal. 2006; 41:838-844.
158. Ng TB, Wang HX. Pharmaceutical actions of Cordyceps, a prized folk medicine. J. Pharm. Pharmacol. 2005; 57:1509-1519.
159. Li SP, Yang FQ, Tsim KWK. Quality control of Cordyceps sinensis, a valued traditional Chinese medicine. J. Pharm. Biomed. Anal. 2006; 41:1571-1584.
160. Ho HJ, Cho JY, Rhee MH, Park HJ. Cordycepin (3’-deoxyadenosine) inhibits human platelet aggregation in a cyclic AMP- and cyclic GMP-dependent manner. Eur. J. Pharmacol. 2007; 558: 43-51.
161. Bunyapaiboonsri AT, Yoiprommarat S, Intereya K, Kocharin K. New diphenyl ethers from the insect pathogenic fungus Cordyceps sp. BCC 1861. Chem. Pharm. Bull. 2007; 55:304-307.
162. Kaelin WG Jr. Taking aim at novel molecular targets in cancer therapy. J. Clin. Invest. 1999; 104:1495.
163. Keshet E, Ben-Sasson SA. Anticancer drug targets: approaching angiogenesis. J. Clin. Invest. 1999; 104:1497-1501.
164. Kaelin WG Jr. Choosing anticancer drug targets in the postgenomic era. J. Clin. Invest. 1999; 104:1503-1506.
165. Shapiro GI, Harper JW. Anticancer drug targets: cell cycle and checkpoint control. J. Clin. Invest. 1999; 104:1645-1653.
166. Sellers WR, Fisher DE. Apoptosis and cancer drug targeting. J. Clin. Invest. 1999; 104:1655-1661.
167. Gibbs JB. Anticancer drug targets: growth factors and growth factor signaling. J. Clin. Invest. 2000; 105:9-13.
168. Reddy A, Kaelin WG Jr. Using cancer genetics to guide the selection of anticancer drug targets. Curr. Opin. Pharmacol. 2002; 2:366-373.
169. Cummings J, Ward TH, Ranson M, Dive C. Apoptosis pathway- targeted drugs-from the bench to the clinic. Biochim. Biophys. Acta 2004; 1705:53-66.
See Also
Drug Discovery
Natural Products as Anticancer Agents
Natural Products in Plants, Chemical Diversity of
Natural Products, Common Biological Targets of
Natural Products: An Overview