CHEMICAL BIOLOGY
Nuclear Magnetic Resonance (NMR) Spectroscopy: Overview of Applications in Chemical Biology
Gerd Nielsen*, Max Stadler*, Henry Jonker, Marco Betz and Harald Schwalbe, Institute for Organic Chemistry and Chemical Biology, Center for Biomolecular Magnetic Resonance, Frankfurt, Germany
doi: 10.1002/9780470048672.wecb393
Nuclear Magnetic Resonance (NMR) spectroscopy is a powerful method to determine the structure of biomacromolecules and their complexes in solution. It allows determination of the dynamics of proteins, RNA, DNA, and their complexes at atomic resolution. Therefore, NMR spectroscopy can monitor the often transient weak interactions in the interactome of proteins and the interaction between proteins and small-molecule ligands. In addition, intrinsically unstructured proteins can be investigated, and first reports of structure determination of membrane proteins in the immobilized state (solid state) are developing. This review will introduce the fundamental NMR observables as well as the methods to investigate structure and dynamics, and it will discuss several examples where NMR spectroscopy has provided valuable information in the context of Chemical Biology.
___________________________
* Both authors have contributed equally.
Chemical Biology
Research in Chemical Biology is dedicated to the design and targeted synthesis of novel molecules of small or large molecular weight to investigate how they maintain, modulate, regulate, or interfere with cellular function or even change the function, morphology, and differentiation status of entire cells. The structure-function relationship that leads to the understanding and prediction of cellular function is at the heart of chemical biology. Molecules exert function through their chemical properties, which are determined by the dynamic spatial arrangement of their atoms, their interactions with other molecules, their stabilities against degradation, and their localization within the cell.
Chemical Biology encompasses more aspects than the development of novel high-affinity ligands for targets of pharmaceutical interest alone. However, if the latter is understood as the art of developing novel chemical entities toward new drugs, then Chemical Biology has a tremendous impact on the understanding of key properties of molecules that may induce specific cellular responses or help maintain cellular function.
NMR Spectroscopy
Nuclear magnetic resonance (NMR) spectroscopy is a noninvasive and nondestructive spectroscopic technique that allows determination of the constitution and relative configuration of molecules, the characterization of the dynamic three-dimensional (3D) conformation of molecules, and their interaction with other molecules. NMR spectroscopy detects the characteristics of nuclear spins; the most commonly studied nuclei are the spin-1/2-particles 1H, 13C, 15N, and 31P. NMR observables sensitively depend on their chemical surroundings of individual atoms. Therefore, NMR spectroscopy can derive information about the conformational dynamics and interactions of molecules in solution and at ambient temperature. In addition, thermodynamic and kinetic information about the interaction of molecules can be derived on a per-atom basis.
Biomolecular NMR spectroscopy is applicable to both liquid- and solid-state samples. Liquid-state NMR spectroscopy, in which molecules are dissolved in a variety of different solvents and studied at ambient temperatures, is a powerful tool to derive information on the structure of proteins and nucleic acids, as well as their complexes with each other and small molecules, ions, and solvents. Liquid-state NMR can be applied not only to native folded states of proteins, but also to intrinsically unstructured proteins as well as proteins in their unfolded state and under nonphysiological conditions (i.e., in organic solvents). Figure 1 provides an overview on the number of protein structures determined by liquid-state NMR spectroscopy.
NMR spectroscopy can detect the conformational dynamics, which are also referred to as conformational switching of RNA molecules that coexist in different stable states. Solid-state NMR spectroscopy investigates molecules as powders or crystals and has become a powerful tool for the investigation of membrane proteins and their complexes with small peptide agonists. In addition, protein amyloid fibers and polymers can be investigated.
Furthermore, NMR spectroscopy can study intrinsically unstructured proteins and noncoding RNAs uniquely, which includes their structural transitions induced when encountering molecular targets. The technique can be applied to systems under defined conditions (in vitro), but proteins can also be investigated within cells (in vivo). NMR can monitor the concentration fluxes of complex mixtures, which are extracted from tissue or within body fluids. These fluxes are important for understanding the metabolic state of an organism (metabonomics). In this context, NMR has been applied to characterize the metabonome of entire organisms such as Caenorhabditis elegans.
In this review, we present the basic observables and tools in liquid-state NMR spectroscopy followed by examples of the application of NMR in chemical biology. We will focus on the application of NMR spectroscopy to study proteins in solution.

Figure 1. Number of protein structures solved by NMR spectroscopy from protein database http://www.pdb.org/; structures are sorted by molecular weight of the complex.
Basic NMR Observables
In NMR spectroscopy, signals of NMR-active nuclei with spin 1/2 (e.g., 1H, 13C, 15N, 19F, and 31P) have Lorentzian line shapes with center peak positions referred to as chemical shift δi. Their linewidths at half height Гi1/2 depend on the overall correlation time for rotational tumbling TC of the molecule that is proportional to the size of the molecule, and on possible local conformational or chemical exchange processes. Figure 2 shows the one-dimensional (1D) proton spectrum of a small molecule, which is an inhibitor of the kinase p38, dissolved in dimethyl sulfoxide (DMSO). The integral of each signal corresponds to the number of different protons that cannot be superimposed by symmetry operations or that are equivalent because of dynamic averaging. Magnetically nonequivalent atoms generate higher-order spectra; this phenomenon will not be discussed here.
Expansions of three regions in the spectrum on the right hand side of (Fig. 2) show how protons display different line shapes according to their chemical environments. The protons in exchange with residual water in the DMSO solvent, such as the urea protons (H1 and H2), exhibit exchange broadened signals (Fig. 2a), whereas nonexchangeable protons (e.g., H3) (Fig. 2b) have sharp signals. The aromatic protons are coupled to each other through scalar J-couplings (vide infra), which results in the higher-order spectrum with a complex peak pattern shown in (Fig. 2c).
In the following five paragraphs, the basic NMR observables will be introduced briefly, for a more thorough introduction we recommend “Spin dynamics, basics of nuclear magnetic resonance” by Malcolm H. Levitt (1) or “NMR: the toolkit” by Peter Hore et al (2).

Figure 2. 1H spectrum of a ligand in deuterated DMSO. The size of the integrals represents the corresponding number of protons. Residual water in the solvent shows up at 3.3 ppm.
Chemical shift
The most important NMR parameter is the chemical shift δi, which is derived from the Larmor frequency of a given nucleus i that resonates in a magnetic field. The chemical shift is measured in parts per million (ppm) relative to a reference compound.
The Larmor frequency of a given nucleus depends on the gyromagnetic ratio γ (a physical property of the nucleus) and the magnetic field. The electron density that surrounds the nucleus results in an additional magnetic field that opposes the external field and thereby slightly alters its Larmor frequency. This modulation of the main field is referred to as shielding, and the shift of the NMR frequency leads to characteristic chemical shift values for the different functional groups or certain environments.
The NMR signal of a nucleus will appear at an average chemical shift value. In case of the three protons of a methyl group, such averaging comes about from fast rotation around the CC-bond that connects the methyl group to the rest of the molecule; the rotation leads to identical chemical shifts for the three protons. Slow molecular motions on the NMR timescale (see the section entitled “Dynamic information from line shape analysis”) can lead to one nucleus having two peaks due to two different conformations. On the intermediate timescale, the averaged NMR signal will be broadened and may disappear. Thus, the NMR chemical shift values are very sensitive probes of the local structure and conformation.
Different methods exist that predict protein secondary structure elements from chemical shift values [e.g., Chemical Shift Index (CSI) (3) and Probability-based protein Secondary Structure Identification using combined NMR chemical-shift data (PSSI)] (4). These methods are based on the statistics of database analysis of chemical shift values from a range of peptides and proteins of known secondary structure. Chemical shifts have also been used to predict backbone torsion angles by software such as Torsion Angle Likelihood Obtained from Shifts and Sequence similarity (TALOS) (5).
More recently, software has been developed that predicts 1H, 13C, and 15N chemical shift values of proteins from either 3D structure files, for example, SHIFTS (6), SHIFTX (7), and SPARTA (8), or from the mere amino acid sequence using SHIFTY (9). First results have been reported on the de novo structure determination of proteins using fragment-based chemical shift predictions and molecular modeling (10, 11).
Spin-spin coupling (J-coupling constant)
The nJ-coupling, which is also known as the indirect spin-spincoupling or scalar coupling, develops from interactions between electrons and nuclear spins of atoms n bonds apart, where there is a slight energetic preference for the nuclear spin to have the same direction as the nearest electron spin. Briefly, this phenomenon is known as the Pauli Exclusion Principle (stating that two fermions cannot occupy the same quantum chemical state simultaneously) that leads to tiny but measurable energy differences depending on the spin states of covalently bonded atoms. The J-coupling will split the NMR signal of each nucleus into multiplets depending on its number of NMR active neighbors (i.e., the amount of different spin states available within the coupled spin system). Typically, scalar couplings are observed for nuclei that are connected by up to three bonds. In rigid systems such as aromatic rings, however, they may even be observable across up to five bonds. The size of the coupling between two nuclei is termed the J-coupling constant. The multiplicity of a signal is determined by the number of chemically equivalent nuclei that couple to the nucleus of interest by through-bond interactions, usually one, two, or three bonds apart.
Vicinal 3J-coupling constants provide valuable information for the determination of biomacromolecule conformation. The structural information is derived from Karplus equations (12) that provide empirical relationships between dihedral bond angles, and the 3J-coupling. Karplus equations obey the following general formula:
![]()
where ф defines the torsion angle; and A, B, and C are either empirically calibrated or calculated from quantum chemistry. A given 3J-coupling may correspond to up to four different torsion angles, but this ambiguity can be resolved by measuring complementary 3J couplings that describe the same torsion angle. For example, the protein backbone torsion angles ф and ψ, which are indicative of secondary structure, can be defined and stereochemistry can be determined for diastereotopic groups.
Interestingly, J-couplings can also be conveyed via hydrogen bonds. In this case, the J-couplings are referred to as 2hJ-couplings where “2h” signifies the correlation of two noncovalently bound nuclei. The occurrence of 2hJ-couplings can be used to detect hydrogen bonds directly in proteins (13, 14) as well as in double-stranded DNA and RNA (15).
Dipolar coupling and nuclear overhauser effect
The Nuclear Overhauser Effect (NOE) (16) provides distance information about atoms that are spatially close. The magnetization is transferred through space and develops from dipolar interactions between noncovalently bound nuclei. The efficiency of this transfer is measured by the dipolar coupling DAB, which is also known as the direct dipole-dipole coupling. Magnetic transfer develops from interactions between two nuclei each possessing a magnetic moment that creates a local magnetic field that will be sensed by the neighboring nucleus. The interaction strength of two magnetic dipoles depends on the internuclear distance (1/rAB3) and the orientation (![]() where θAB is the angle between the internuclear vector that connects A and B and the external magnetic field.
where θAB is the angle between the internuclear vector that connects A and B and the external magnetic field.
In isotropic solution, the dipolar coupling is averaged to zero through molecular rotational tumbling with a characteristic correlation time TC. Yet, on shorter timescales, fixed orientations of the internuclear vector in the magnetic field persist and induce dipolar relaxation effects. This interaction between dipoles causes relaxation by transfer of magnetization between different NMR active nuclei and is called the NOE. The NOE decreases with the internuclear distance by rAB—6 and increases with the gyromagnetic ratio (γAγB)2. Therefore, the NOE effect is strongest between protons as they have large gyromagnetic ratios, and NOEs are generally observed between protons that are situated within 5 A of each other.
Residual dipolar coupling
In contrast to NOEs that provide short-range intermolecular distance information, Residual Dipolar Couplings (RDCs) provide long-range information about the relative orientation between bond vectors. Dipolar couplings that are averaged out in isotropic solution can be partially reintroduced by dissolving the (asymmetric) biomolecule of interest in an anisotropic medium. The interaction between the biomolecule and the anisotropic medium will weakly align the biomolecule relative to the external magnetic field. Initially, the anisotropy of the magnetic susceptibility of biomolecules, often with paramagnetic centers, was exploited to align the biomolecule; now, external alignment media include liquid crystals, phages, stretched gels, and lipid bicelle solutions (17). However, the development of paramagnetic tags is also becoming a tool for biomolecular NMR investigations (18-20).
Partial alignment introduces a residual dipolar coupling on a spin pair like the backbone 1H-13C bond. A 1H-13C spin pair in isotropic solution will, in a coupled spectrum, yield a doublet with a splitting the size of the 1J-coupling. In an anisotropic solution, however, the splitting observed is the sum of the scalar coupling (1JCH) and the residual dipolar coupling (1DCH). The size of the residual dipolar coupling depends on the orientation of the bond vector relative to the molecular alignment tensor.
One important application of RDC measurements is the structural refinement of biomolecules that consist of several domains that are connected by more or less flexible linkers. Because of the flexibility of the linker and the distance between domains, J-couplings and NOE restraints will frequently not be sufficient for correct determination of the relative domain orientation. The addition of RDC restraints in structure calculation not only refines the biomolecular structure but also allows the relationship between structure and function to be studied. Interactions with other biomolecules and ligand binding may induce an intramolecular rearrangement of the relative orientation of domains that is detectable through RDC measurements (21, 22).
Relaxation
Relaxation, or the return of nuclear spin magnetization to the equilibrium state, occurs essentially by two different physical processes that allow the nuclear spins to exchange energy with their surroundings: one that occurs parallel to the direction of the external magnetic field (longitudinal) and one that is transverse to the external field. The longitudinal relaxation time T1, or spin-lattice relaxation, develops from interactions with neighboring unexcited nuclei (the lattice) that affects the component of magnetization in the direction of the external magnetic field. The transverse relaxation time T2, or spin-spin relaxation, develops from dispersion of magnetization between excited neighboring spins that have magnetic moments orthogonal to the external field. The longitudinal relaxation time in the rotating frame, T1p, is the relaxation of magnetization that has been spin-locked perpendicular to the external field (23, 24).
T1 reports on fast dynamics on a timescale of ps-ns, whereas T2 relaxation depends on both fast and slower dynamics (ps-ns and ps-ms). The experimentally measured T2 relaxation times include an exchange contribution that can be measured by a Carr-Purcell-Meiboom-Gill (CPMG) pulse train (25, 26) or an effective spin-lock field (27-29). The combination of T2 and T1p measurements allows determination of the contribution of chemical exchange to the relaxation time. Furthermore, relaxation dispersion experiments have been developed to measure slow time-scale ps-ms dynamic processes (30-35).
Heteronuclear NOEs (e.g., {1H}-15N or {1H}-13C hetNOEs) are obtained by measuring HSQC-type spectra (see the section entitled “Two-dimensional heteronuclear correlation experiments”) with and without proton saturation. The hetNOE is extracted from the difference in the signal amplitude of these measurements and reports on the fast dynamics of the heteronuclear bonds (ps to ns timescale). Maximal hetNOE values are observed when the bond vector tumbles at the same frequency as the entire protein, whereas faster motion with respect to overall tumbling leads to smaller hetNOEs.
The conformational dynamics of a biomolecule can be determined by measuring the relaxation properties of the heteronuclear bond vectors (e.g., the protein backbone 1H-15N bond). Regions of a protein that are unstructured or flexible (e.g., loops and tails) will typically show larger T2 and T1p values and smaller T1 values and hetNOEs than the rigid well structured core of the molecule. Distinct changes in the flexibility of a biomolecular structure caused by ligand binding can therefore be probed using a combination of heteronuclear relaxation experiments.
Relaxation is strongly dependent on molecular motions. The overall random molecular tumbling, which is expressed in the rotational correlation time Tc, governs the overall relaxation process. Larger molecules have slower tumbling motions that lead to higher TC values. However, local dynamics and independent domains can modulate the relaxation parameters, which account for differences in their flexibility and mobility.
Typically, relaxation times are interpreted in the framework of a model-free analysis (36-38), and the general order parameter S2 of a given heteronuclear bond can be extracted. S2 defines the spatial restriction on a per-residue basis of a target protein. Unique information can be obtained about the changes that occur to a target molecule on ligand binding by measuring S2 before and after the binding event. The order parameter varies between 0 and 1, which spans from completely unrestricted internal motion of the bond vector (S2 = 0) to complete rigidity (S2 = 1). Dynamics that are equal to or faster than the overall correlation time can be measured by the order parameter (39, 40).
Experimental Techniques
NMR spectroscopy provides important information on molecular structure and dynamics, but certain limitations apply for the study of biomolecules. The sample conditions need to be optimized toward stability at high concentrations without exceeding a salt concentration of roughly 300 mM (leading to increased NMR pulse length). To reduce N-H exchange, the solution should be buffered to a pH between 5 and 7. Distinct chemical shifts allow monitoring of individual atom types, but dispersion is rather low (on the sub-ppm scale) and easily produces spectral overlaps especially for larger molecules. The size of the molecular target (e.g., protein or RNA) also restricts NMR experiments because of relaxation effects. It is, however, possible to reduce these relaxation effects greatly by partial or selective deuteration.
To reduce spectral overlap, 1D NMR can be extended to higher dimensional NMR experiments. Two-dimensional (2D) experiments that correlate protons through J-couplings or dipolar couplings include 1H-1H COSY (41), 1H-1H TOCSY (42), and 1H-1H NOESY (43, 44). Particularly helpful are 2D heteronuclear experiments, which correlate protons with directly bound carbon or nitrogen atoms. Furthermore, a variety of 3D and higher-dimensional experiments has been developed that are essential for assigning large biomolecules (45). In contrast to protons, NMR-active and stable isotopes 13C and 15N both have low natural abundances, and proteins are therefore typically expressed in Escherichia coli bacteria that grow in a minimal medium that contains only 13C, 15N labeled precursors, which allows proteins to be studied in isotope labeled form. Recently, expression media have also been established to express isotope-labeled proteins in sf9 cells (46) or HEK293 cells (47). Typical protein concentrations are between 0.1-1 mM in 0.3-0.5 mL of buffered solutions that correspond to approximately 1015-1017 molecules per NMR sample for detailed structural studies, but it can be as low as 1012 for more analytical purposes.
Sensitivity can furthermore be increased up to fourfold by using cryogenic probe technology, in which the radio frequency (rf) transmitter and preamplifier coils are super-cooled to 20K by helium gas that ensures higher signal-to-noise of the electrical signal. Vacuum insulation around the rf coil allows the NMR sample situated only millimeters away from the coil to be measured at ambient temperature.
The development of higher magnetic field strength has also been vital to improve both resolution of NMR spectra (proportional to B0n, where n is the dimensionality of the NMR experiment) and sensitivity (proportional to B05/2).
Another way of overcoming spectral overlap for proteins is selective isotope labeling of one or several amino acid types, which results in less-crowded 1H-15N spectra with cross-peaks from the labeled residues only. The same is possible for RNA where nucleotides can be selectively isotope enriched. In addition, methods for segmental labeling of parts of the biomolecules have been developed for proteins (48, 49) and for RNA (50).
One-dimensional NMR spectroscopy
The very high resolution of the chemical shifts gives information on a per-residue basis, and it also leads to spectral crowding in simple experiments. Figure 3 shows three very different 1D proton spectra of the 14-kDa protein called α-lactalbumin, in three different states: native folded, molten globule, and unfolded (urea denatured).
The folded state (Fig. 3a) shows sharp lines and a large degree of chemical shift dispersion. The side-chain methyl groups positioned in the interior of the protein can be in close contact with aromatic rings that induce ring current effects. Methyl groups are often shifted toward very low ppm values (between 0.5 and -0.5 ppm). The molten globule state (Fig. 3b) is a partially folded, highly dynamic protein state that contains some stable secondary structure elements, but with a dynamic tertiary structure. The time scale of the interconversion between different states is typically slow, of the order of milliseconds, which leads to significant line broadening. No side-chain methyl groups are as well shielded as in the folded state, and therefore they show chemical shift values larger than 0.5 ppm. The same is observed for the unfolded state (Fig. 3c), but the total lack of secondary structure elements leads to sharper lines than in the molten globule state.
Protons have large gyromagnetic ratios and are therefore highly sensitive NMR nuclei. The high sensitivity in combination with their high abundance in biomolecules has made protons the nuclei of choice in biomolecular NMR. However, other nuclei can also be detected directly for specific purposes. 31P is a moderately sensitive NMR nucleus present in the nucleic acid backbone and in the side chains of phosphorylated proteins. Its chemical shift is highly sensitive, and 31P is therefore a useful probe of structural changes.
19F is almost as sensitive as 1H and therefore is well suited for direct detection. However, for biomolecular NMR, it is of limited interest, as it does not occur naturally in biomolecules; instead, it has to be introduced by chemical modification or labeled precursors for biosynthesis. In small drugs, 19F can be used as a metabolic tracer for interaction studies or as a contrast agent in imaging applications. NMR active isotopes 13C and 15N are not only scarce but also have the disadvantage of low NMR sensitivity. Traditionally, they have therefore only been observed indirectly through neighboring protons. Recently, experiments have been developed to detect either 13C or 15N directly both for proteins (51) and RNA (52, 53).

Figure 3. ID proton spectra of the side-chain region of α-lactalbumin in (a) folded state, (b) partially folded (molten globule), (c) unfolded.
Two-dimensional homonuclear correlation experiments
A 2D NMR experiment consists of a series of 1D experiments, in which the magnetization is transferred from one nucleus to another. This experiment is followed by a delay called the evolution time, where the spins are allowed to precess freely. Incrementing the evolution time in successive experiments results in a 2D spectrum that correlates protons that have exchanged magnetization during the evolution time.
In the 1H-1H COSY (correlation spectroscopy) experiment (41), magnetization is transferred via the J-coupling and shows correlations between protons three bonds apart. The cross peak usually shows a characteristic antiphase-square pattern, but it may be split even more by additional passive couplings that lead to spectral crowding and loss of intensity. For small molecules, the COSY-spectrum may suffice for assignment of the proton resonances. For larger molecules, such as peptides, the COSY-experiment is used in combination with other homonuclear experiments, especially because the spectral region of the aliphatic protons is often too crowded to allow unambiguous assignment.
In the 1H-1H-TOCSY (Total Correlation Spectroscopy) experiment (42), magnetization is also transferred via the J-coupling, but an additional isotropic mixing step leads to the correlation of all spins within a given spin system. In the case of a small peptide, the amide backbone protons show cross peaks to all side-chain protons of the same amino acid residue, which facilitates the identification of amino acid type by their characteristic peak patterns and circumvents the problem of overcrowding in the COSY spectrum. The individual amino acid residues (spin systems) are finally connected by distance information obtained from the 1H-1H-NOESY (Nuclear Overhauser Effect Spectroscopy) experiments (44), in which the correlation is transferred via the dipolar coupling. Cross-peaks may be observed for protons that are up to 5 A apart from each other. For a small peptide (Fig. 4), this information is sufficient for a sequential assignment. A closely related experiment is ROESY (Rotational frame nuclear Overhauser Effect Spectroscopy) (44, 54), which is complementary to the NOESY in respect to its dependency on the correlation time of the molecule studied. With increasing size, the NOESY cross-peaks display a sign change from positive to negative, and the signal intensity is very low close to the transition point. This low NOE signal intensity is often the case for medium-sized peptides in which case the ROESY experiment is preferable. The signal intensity in the ROESY spectrum is also dependent on the size of the molecule, but the sign is always negative.
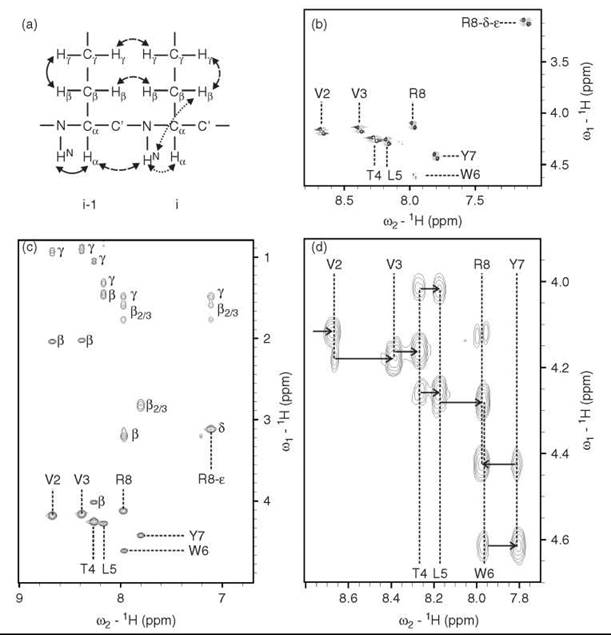
Figure 4. Homonuclear 2D spectra of the 8mer peptide EVVTLYWR in 90 % H2O, 10 % D2O. (a) Representation of coherence transfer pathways for COSY (solid arrows), TOCSY (dotted arrows) and NOESY (dashed arrows) experiments. (b) Section of COSY spectrum displaying the backbone HN-Ha-correlations, additionally the side-chain HN-Ha-correlations of R8 is visible in the upper right-hand corner. The HN-Ha-correlation of E1 cannot be detected because of solvent-exchange broadening of the N-terminal amino group. (c) Section of TOCSY spectrum that displays the correlations of the backbone HN with all protons within the amino acid side chain. (d) Section of ROESY spectrum that displays correlations between backbone HN-Ha intraresidual as well as to the neighboring (i-1) amino acid. The cross peaks to the (i-1) amino acid have higher intensities. Thus, a ''sequential walk'' is possible (arrows) that allows identification of the position of amino acids within the peptide chain. Additionally the Ha of E1 can be assigned (first arrow on the left).
Two-dimensional heteronuclear correlation experiments
Introducing a heteronuclear dimension reduces the signal overlap by using the additional chemical shift dispersion of the heteronuclei and facilitates assignment of biomolecules. The Heteronuclear Single Quantum Correlation (HSQC) experiment yields a spectrum that correlates the chemical shift of a 1H spin with that of a covalently bound 13C or 15N spin (55). In a 1H-15N HSQC spectrum, every peak represents the correlation of an amide 1H-15N bond, which shows correlations for both backbone and side-chain amides of proteins and nucleotide imino protons of RNA. Equivalently to the 1D case (Fig. 3), different folding states of a protein will lead to distinctly different 1H-15N HSQC spectra; these states are shown for α-lactalbumin in folded, molten globule and unfolded states in (Fig. 5(a-c)). Chemical shift dispersion is clearly much larger in the folded state than in the molten globule or unfolded states. This increased chemical shift dispersion facilitates analysis of larger proteins in their native, folded state, as much less signal overlap is observed compared with the unfolded states. The highly flexible molten globule shows significant line broadening of signals; therefore, many peaks are not visible. In the unfolded state, most peaks reappear, but the signal overlap in the proton dimension is significantly worse than in the folded case as all amide protons are equally solvent accessible and therefore experience similar degrees of shielding from the external magnetic field. Contrarily, the nonexchanging 15N nuclei typically retain high chemical shift dispersion in the unfolded and molten globule states.
Similar to the HSQC experiment, multiple quantum coherences can be used to correlate protons with J-coupled heteronuclei. The information content of the Heteronuclear Multiple Quantum Correlation (HMQC) experiment (56) is equivalent to the HSQC, but the sensitivity can be improved in certain cases. Additionally, by proper tuning of delays and phase cycling, it can be transformed into the heteronuclear multiple bond correlation experiment (57-59), which results in correlations between 2J- and 3J-coupled nuclei.
For large biomolecules, fast relaxation of the transverse magnetization becomes a serious problem that leads to poor spectral resolution with broad or lacking signals. In a coupled 1H-15N HSQC spectrum, cross-peaks will appear as multiplets, in which the different components of the multiplet have significantly different line widths because of constructive or destructive interactions of the two main relaxation mechanisms: dipole-dipole relaxation and relaxation caused by the chemical anisotropy. The TROSY (60) experiment (Transverse Relaxation Optimized Spectroscopy) overcomes much of the problem of fast transverse relaxation by selecting the one component of the multiplet, in which these relaxation effects almost cancel each other out, which thereby renders spectra with single sharp peaks even for very large biomolecules. The TROSY experiment relies on the fact that the chemical shift anisotropy scales with the magnetic field whereas dipole-dipole relaxation is field independent, and the maximal TROSY effect is only obtained at high magnetic field strength (above 14,1 Tesla), where it has significantly extended the range of biomolecules amenable to NMR spectroscopy.
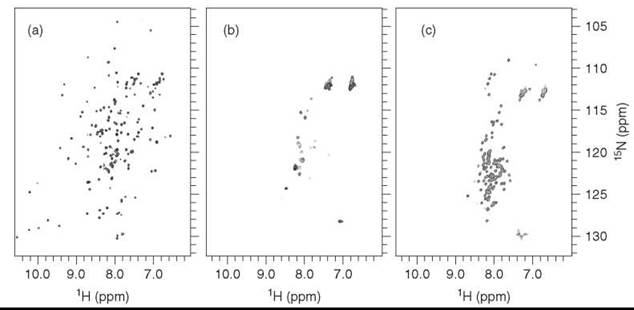
Figure 5. 1H-15N HSQC spectra of α-lactalbumin in (a) folded state, (b) partially folded (molten globule), and (c) unfolded state.
Multidimensional NMR experiments
Two-dimensional experiments may be extended to higherdimensional experiments by including additional incremented evolution times into the pulse sequence separated by mixing sequences. The basic building blocks for these experiments are usually 2D homonuclear techniques like TOCSY (42) and NOESY (44) or the heteronuclear INEPT-transfer experiment (Insensitive Nuclei Enhanced by Polarization Transfer) (61). Combinations of these experiments alleviate the problem of spectral crowding and allow for new assignment strategies for proteins (45) and nucleic acids (62). Experiments such as TOCSY-HSQC and NOESY-HSQC combine homonuclear and heteronuclear techniques, whereas triple-resonance experiments such as HNCO, HNCA, or HNCACB (Fig. 6) correlate three different NMR-active nuclei (e.g., along a protein backbone and are therefore used in suitable combinations to assign proteins) (45).

Figure 6. Backbone Assignment: Five sections (strips) of the 1H-13C-plane of a HNCACB experiment of cdc25A that show the correlations of residues 174 to 1 78 as an example of the general backbone assignment strategy. The strips show correlations from the HN to the Ca (black) and C (gray) intraresidually as well as to the (i-1) amino acid, which allows assignment by a sequential walk. Ambiguous assignments can be resolved by comparison with other triple resonance experiments.
Applications of NMR Spectroscopy to Chemical Biology
In this article, some examples are shown of the application of NMR spectroscopy in chemical biology. We put emphasis on experiments that characterize interactions of proteins and small molecular weight ligands. These interactions can be mapped either by characterizing the target protein (protein-observed experiments) or the ligand (ligand-observed experiments). Finally, the method of structure calculation based on NMR-derived data is briefly introduced.
This introductory review cannot possibly cover the whole range of techniques in a field as multifaceted as NMR spectroscopy. We therefore refer to review articles published during the last decade (63-67).
Protein-observed experiments
Binding of a ligand will cause diverse effects on a protein, many of which can be probed by NMR spectroscopy of isotopically enriched protein. Depending on the used isotopes and the labeling pattern, such studies can be both costly and time consuming. Nevertheless, protein-observed experiments offer more detailed information about the binding event than what can be obtained from ligand-observed methods. Especially, they map out the dynamic and often remote (e.g., allosteric) response of the protein receptor on binding of a small molecule. Changes in the chemical environment of each nucleus are indicated by chemical shift perturbations, either directly induced by ligand binding or via induction of structural changes. This method allows immediate distinction of different binding sites and is a particular advantage of NMR screening experiments that is not possible in most, if not all, alternative-screening techniques. Hydrogen-deuterium exchange experiments provide information about solvent accessibility and participation in hydrogen-bonding networks. Changes in the line shape of NMR signals may elucidate folding intermediates and pathways, whereas relaxation studies probe thermodynamic and kinetic properties.
Chemical shift perturbations (CSPs)
The location of a ligand-binding site can be determined for small to medium-sized proteins that are uniformly 15N labeled and where the amide signals have been assigned by NMR spectroscopy. 1H-15N HSQC spectra are recorded in the absence and presence of a ligand (NMR titration experiments), and the observed changes in the amide chemical shift or signal intensity of the amino acid residues that bind the ligand indicate the location of the binding pocket.
This approach can be extended to larger targets by the TROSY experiment in combination with different labeling strategies (e.g., selective labeling of specific amino acids or deuteration). Deuteration replaces the nonexchangeable 1H atoms by 2H, which thereby reduces T2 relaxation significantly (68-70). To map a ligand-binding site by CSPs, it is not necessary to have a complete assignment of the protein. Per-atom information about binding events can be gained for large, only partially assigned proteins, in which structure calculation based on NMR data is not yet possible.
Arginine side chains are often involved in intermolecular contacts, therefore assignment of the 1He-15Ne side-chain groups allows mapping of ATP binding on kinases to be studied by CSPs as well as binding of RNA or DNA (71).
The relation between ligand binding and CSPs is not only dependent on the proximity of the ligand to the residues that show chemical shift changes. Conformational rearrangements of the protein/biomolecule as well as allosteric effects may cause significant CSPs of residues distant from the binding pocket. Reliable determination of the binding pocket is possible by comparing the CSPs induced by a series of structurally similar ligands (72).
An interesting example of the application of CSP studies is the ribosomal L11 protein (Fig. 7). Complex formation with its natural RNA substrate results in very large CSPs of the amide resonances that correspond to the binding surface on the C-terminal domain of the L11 protein. Addition of the antibiotic thiostrepton induces tighter binding of the N-terminal domain to the RNA, which again results in significant chemical shift changes (73).

Figure 7. Overlay of the 1H-15N TROSY spectra of (a) L11 in its free form with the RNA bound form, (b) L11 in the RNA bound form with the RNA and thiostrepton bound form. The backbone assignments for the major shifting peaks are indicated by arrows. (c) The L11 interaction sites are indicated in red for the RNA ( > 1.0 ppm) and green for thiostrepton ( > 0.3 ppm) on the combined ribbon/surface representation of the L11-RNA complex (PDB: 1 MMS). (d) Diagram of the combined amide 1H and 15N CSPs in L11 caused by addition of RNA (red) and thiostrepton (green). See color insert.
Another interesting example of the effect of ligand binding on protein backbone 1H-15N signals is the p38a kinase. Certain ligands can bind not only to the ATP binding site but also to an allosteric hydrophobic pocket situated in an Asp-Phe-Gly (DFG) loop. This loop is highly conserved among kinases, and ligands that bind this allosteric site are called DFG-out ligands, as they require the kinase to undergo a conformational change. X-ray structures of p38a show that the Phe side chain of the DFG motive is displaced by 10A when bound to DFG-out ligands compared with the apo state (74, 75). This displacement is not observed when the kinase is complexed with ligands that only bind to the ATP binding site (DFG-in ligands) (76).
Investigations of this DFG-in/out phenomenon by NMR spectroscopy reveal dynamics that are not obtainable from static X-ray crystallographic studies (77). The kinase in its free form is in a slow DFG-in/out equilibrium. This equilibrium is not disturbed by a DFG-in ligand, whereas a DFG-out ligand disrupts the motion forcing the kinase into the DFG-out conformation. Cross peaks that develop from residues in the DFG-loop disappear because of conformational interconversion on the intermediate NMR timescale that leads to extensive line broadening. This mechanism is illustrated in (Fig. 8) of two 1H-15N TROSY spectra of p38a selectively labeled with 15N phenylalanine. The first spectrum of the apo state contains cross peaks from 12 of 13 Phe residues. The missing peak corresponds to the Phe residue in the DFG-loop and only appears during addition of a DFG-out ligand as shown in the second spectrum, which proves that the conformational equilibrium of the free kinase is disrupted by this allosteric ligand.
This DFG-in/out phenomenon was first observed for the Abelson kinase where the ligand gleevec (78, 79) was found to bind both the ATP binding site and the allosteric binding pocket in the DFG-loop. This mechanism is observed not only for the Abelson and p38 kinases (74, 80, 81) but also for Raf (82) and KDR (83).
The lack of signals caused by the flexible nature of the kinase in the free state is observed for most of the activation loop as well as other highly conserved domains (84). The same pattern of missing peaks from these domains is also seen for the protein kinase A (PKA) (85).

Figure 8. 1H-15N TROSY spectra of p38a(a) In the free state and (b) After binding a DFG-out ligand. The ligand structure is depicted in the upper left corner of the bound spectrum. The phenylalanine residue 169 situated in the DFG loop appears during binding. (c) Schematic representation of the effect of DFG-in ligand (SB203580) versus DFG-out ligand on the conformational exchange of p38 (1P38), the activation loop is indicated by an arrow.
SAR by NMR
To reduce the effort of chemical synthesis in drug discovery, the structure-activity relationship (SAR) method uses results from the chemical shift perturbation experiments (SAR by NMR). Optimizing the binding properties of initial hits from a high-throughput screening process without additional knowledge of the exact binding site and orientation is challenging. The SAR by NMR approach, which was developed by Stephen W. Fesik of Abbott Labs, is based on enhancing binding properties of small molecules to the protein surface by linking two active compounds together. First, smaller fragments are screened for binding, and initial hits are optimized toward stronger binding. Second, two compounds that bind to close, but not overlapping, sites on the protein surface are connected by a linker of appropriate length (Fig. 9). Strong dual-site binders can be generated from two initial relatively weak single-site binders with interaction sites close together on the protein surface by connecting them with a linker of suitable length.
Optimizing the linker is important because the free energy of binding for the composite ligand ∆G°AB is not just the sum of the binding enthalpies for the individual ligands A and B (∆H°A and ∆H°B, respectively), but it includes an intrinsic entropic penalty term ∆S° depending on rigidity and orientation of the linker. The origin of this entropic loss is the conformational adaptation the ligand undergoes during binding.
Mechanistically, inhibition must not necessarily block the active site itself, but it can exert allosteric effects on the substrate-binding pocket, which thereby enhances or suppresses enzymatic activity. Additional considerations regarding enzymatic reactions are discussed in Reference 86. SAR by NMR has been successfully applied to various systems [i.e., for disrupting intracellular protein-protein binding (87) as well as cytokine-receptor interaction (88)]. High-affinity enzyme inhibitors have been developed by this technique [e.g., for the metalloproteinase Stromelysin (89) and the protein tyrosine phosphatase 1B (90)].

Figure 9. Flow diagram of the SAR by NMR procedure. In case of completely independent binding of two ligands Left: primary screening can be performed individually for each ligand. Ligands binding to two independent sites are linked, which enhances ligand-binding affinity. Right: for allosteric binders, screening for a second ligand should be done in the presence of a primary ligand.
Hydrogen/deuterium (H/D) exchange
Protein hydrogen atoms bound to N, O, or S are in constant exchange with solvent protons, and therefore they can be exchanged readily by deuterium. The exchange reaction can be followed by measuring a series of 1H-15N correlation spectra (e.g., HSQC, HMQC, or TROSY) after the solvent exchange. Residues that are buried in the core of the protein are not as solvent exposed as residues on the surface of the protein and will therefore exchange more slowly. Equally, protons that are involved in hydrogen bonding are not as prone to exchange as non-hydrogen-bonded protons.
One of the most elegant experiments that uses the H/D exchange phenomenon is the characterization of protein folding by Miranker et al. (91). In addition, useful information can be obtained about protein-protein interactions, in which residues at the interaction surface will be solvent accessible before the complexation but not after. Domain reorientation of a protein during binding of a ligand can result in some amino acid residues being buried in the interior of the protein and therefore showing significantly different exchange times before and after binding.
Changes in secondary structure of the target protein during ligand binding can also be extracted from H/D exchange measurements as secondary structure elements, such as α-helices and β-sheets, which are highly stabilized by hydrogen bonds. This is not the case for the more flexible loop regions or domains.
Experimentally, H/D exchange for a stable globular protein sample can normally be performed by lyophilizing the sample and redissolving it in the same amount of D2O as there was H2O before lyophilizing to ensure that the buffer concentration is maintained. (Furthermore, the different pH values of H2O and D2O have to be taken into account.) Enzymes, such as kinases, may lose activity during lyophilization, which renders buffer exchange more tedious and time consuming, and only protons that are not exchanged within the first hour will be measurable.
Fast exchange reactions can be monitored by the SEA-TROSY (Solvent Exposed Amides with TROSY) experiment. Initial filtration of all amide signals and subsequent transfer of magnetization from water allows observation of solvent-exposed amides that are in fast exchange with bulk water (92).
Dynamic information from line shape analysis
The simple exchange reaction of a protein and a ligand (below) is described by an equilibrium constant K = kon/koff
![]()
The effect of this exchange reaction on a protein proton in the binding pocket will depend on the difference in Larmor frequency (chemical shift) between the free and bound form compared with the exchange rate. When exchange rate and difference in Larmor frequency are comparable, the reaction is in the intermediate exchange regime, and useful information can be obtained from line shape analysis. Slow intermediate exchange in which the exchange rate is slower than the difference in Larmor frequencies leads to cross-peaks from both the free and bound state. Fast intermediate exchange, however, produces only one single peak at the average chemical shift value. When the exchange rate and frequency difference are equal (the crossover point), the signal is broadened in between the two states.
Proteins at ambient temperature exhibit conformational dynamics on a wide range of time-scales (Fig. 10), and studies of such dynamics have been shown to be particularly important to understand the pharmaceutical properties of kinases (vide infra).
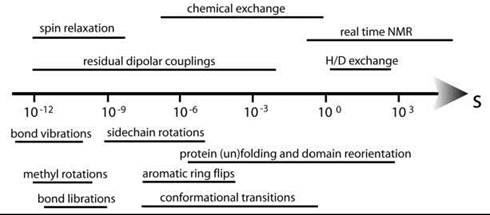
Figure 10. The different processes amenable to NMR spectroscopy are indicated above the time arrow, below are the typical time windows for different molecular motions and events.
Line shape analysis is used to study exchange processes as described by the relative population of the different states (mole fractions), line widths of the signals, and their frequency separation Av (93). It is assumed that the lifetimes are significantly longer than the rotational correlation time Tc, and that spin-spin couplings do not interfere.
Starting from the simplest case of slow exchange between the free and ligand-bound form of an NMR signal from a biomacromolecule, this exchange situation will be indicated by two distinct peaks that do not change position but vary only in intensity. More complex exchange mechanisms may lead to overlaying changes in line shapes and can be simulated by models that are more complex. An example is the introduction of an additional fast exchange step between the free form and an intermediate form that subsequently is in slow exchange with the ligand-bound form. In this case, the peak that corresponds to the ligand-bound form will only increase in intensity during titration with the ligand. The peak of the free form, however, will not only decrease in intensity, but also change position toward the averaged peak position of free form and intermediate. Even more elaborate peak patterns will develop if additional intermediates are in fast exchange with the free form or if alternative ligand-bound forms are present.
The simulated 1H-line shapes of a 1H-15N HSQC spectrum of a protein signal during titration with an inhibitor are shown in (Fig. 11). The expected line shapes for three different binding mechanisms of increasing complexity have been simulated for a protein concentration of 0.1 mM. The ligand concentration increases from 0 to 0.1 mM (blue through red lines), and the ligand has a binding affinity of 1 μM. The line shapes for a direct key/lock mechanism are shown in (Fig. 11a). This mechanism is the simplest conceivable mechanism and the first model to use when fitting experimental NMR line shapes. Even when a simple key/lock mechanism is presumed, the line shape can look different depending on the off-rate of the reaction. A slow reaction on a high ms time scale or slower leads the sharp peak of the free protein to disappear at the same time as a sharp peak of the complex appears. An off-rate in the ps time regime or faster will result in a line that shifts from its original position to the position of the bound form (Fig. 11a). The peak evolves from its free form to a broadened line shape that sharpens again at the end of the titration. Failure to reproduce the measured line shape by any parameter set of a certain model means that the complexity of the model has to be increased. Figure 11b shows the behavior expected from a single intermediate mechanism, whereas (Fig. 11c) presumes binding of the ligand in two different ways, of which only one species reacts to the complex structure. For each model, the input parameters, such as off-rates, are varied until the resulting line shapes fit the experimental data. In case of line broadening of the amide protons because of exchange with solvent water the line shapes of the nitrogen may also be simulated (93-96).
Thus, it is possible to calculate the off-rates of protein-ligand interactions observed for individual amino acid residues of the protein of a verified reaction mechanism. A nice example that shows various cases was recently published for the Apo-Cellular Retinol Binding Protein (97).
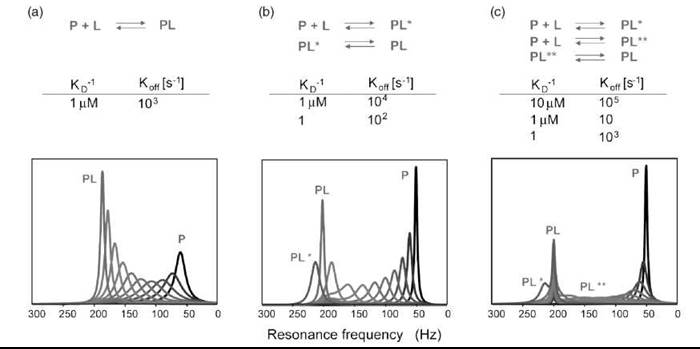
Figure 11. Simulation of line shapes of a reaction according to the binding model: (a) Two-state reaction. (b) Two-state reaction with one intermediate and (c) Two-state reaction with two possible intermediates. Protein concentration is 0.1 mM. Titration in equal steps until an equimolar amount of ligand is reached (0, 0.01, 0.02 ... 0.1 mM). The signals of P and PL are separated by ∆v = 150 Hz. The sequential steps in the titration change color from black to gray.
An example of dramatic change of line shapes is observed when the p38α kinase is bound to the previously mentioned DFG-out ligand. p38α is activated by dual phosphorylation of two conserved residues in the activation loop. The 31P spectrum of the apo state (Fig. 12) shows two broad peaks; during addition of a DFG-out ligand, one of these is shifted. Both peaks show significant narrowing, which indicates that ligand binding hinders the flexibility not only of the DFG motif but also of the entire activation loop, as the phosphorylation sites are situated 11 and 13 residues down the amino acid chain from the DFG loop.
Line shape analysis may be complemented or verified by measuring chemical exchange by NMR by relaxation dispersion experiments. Relaxation dispersion is based on measuring a series of CPMG-based relaxation rates at different temperatures. Excited state intermediates in folding reactions or ligand-accessible intermediates can thereby be probed, even when they constitute as little as 1% of the entire population (34, 98).

Figure 12. 31P spectrum of activated p38 in apo and ligand bound state, which shows significant line-narrowing and shift of the two phosphorous signals during ligand binding.
Dynamic information from relaxation studies
The heteronuclear relaxation properties as measured by T1, T2, T1p and hetNOEs contain valuable information about molecular dynamics of biomolecules.
The relaxation data shown in (Fig. 13a) (73) describe the dynamics of the ribosomal protein L11 before (blue) and after (red) complexation with the GTPase region of 23S rRNA and of the subsequent binding of the antibiotic thiostrepton to the protein-RNA complex (in green). L11 consists of an N- and a C-terminal domain. The latter shows a flexible loop region in the free form around residues 86-96, which is evident by small values of {1H}-15N hetNOEs, large T2 values, and correspondingly low order parameters. During binding to 23S rRNA, this loop region shows a significant reduction of both hetNOEs and T2 relaxation times, which indicates a rigidification of the region. This finding is shown in (Fig. 13a) with blue arrows. The overall correlation times that describe random tumbling of the C- and N-terminal domains are similar in the free form of the protein, which indicates that the protein domains tumble together more or less like a rigid body. During addition of RNA, both domains show a reduction of the correlation times, which indicates binding of the RNA to the protein; however, the effect is much more pronounced in the C-terminal domain, which indicates that this region is most tightly bound. When adding thiostrepton to the complex, the T1 relaxation times of the N-terminal domain match the ones of the C-terminal domain, which indicates that the thiostrepton locks the N-terminal domain to the RNA, thus creating a stable protein-RNA-ligand complex. The highly flexible parts of the protein are shown on the structures below the relaxation data (Fig. 13b). The color coding goes from yellow for highly flexible over gray to blue for “nonflexible” regions.
Obtaining such detailed information about the dynamics of a complexation reaction is labor intensive. To obtain these data, it was necessary first to assign the protein in its free form as well as in complex with RNA and RNA-antibiotic by a series of 3D NMR spectra of 2H, 15N, 13C triple-labeled samples. The labor intensity of dynamic studies like this is, however, offset by the invaluable insights into the detailed dynamic driving forces behind biological processes uniquely obtainable by NMR.

Figure 13. Relaxation parameters of protein-RNA-ligand interactions of the L11 system. Free L11 (blue), L11, and RNA (red) and L11, RNA, and Thiostrepton (green). RNA binding shows effects on all parameters, pronounced for certain residues in the C-terminal domain (arrows), whereas the additional Thiostrepton binding leads to an increase in T of the N-terminal domain. Binding of RNA locks the two domains in a fixed orientation relative to each other and strongly restricts the motion of the C-terminal domain, Thiostrepton then locks the N-terminal in place. See color insert.
From dynamics to thermodynamics
For a comprehensive understanding of ligand binding, the thermodynamics of the association process must be taken into account. On this topic, we follow the line of discussion of Steve Homans (99). The binding constant is related to the Gibbs free energy of binding in the following manner: ∆G°b = —RT ln Ka = RT ln Kd.
∆G°b is composed of an enthalpic and an entropic term (∆G°b = ∆H°b — T∆S°b), the former representing the structural features and the latter constituting the changes of dynamic properties of ligand, protein, and solvent. Although the binding event is mainly enthalpy driven, the entropic component should not be underestimated. Using only the enthalpic term as a measure of binding strength will lead to incorrect evaluation of the data. In some cases, the entropic term accounts for a difference in Gibbs energy that will distinguish between a mediocre and a tight binder (about 4 kcal mol-1). Macroscopic techniques such as isothermal calorimetry can determine the overall thermodynamic parameters of binding events, whereas a microscopic view on the mechanism can only be obtained by NMR. Using the relaxation methods described above, a per-residue analysis of protein dynamics is possible. Especially for ligand binding, no other technique will provide such detailed account of the binding mechanism. For the major urinary protein, an interesting effect of ligand binding has been observed (100). The increase in protein backbone flexibility during ligand binding results in an entropic contribution to the Gibbs free energy of binding of a magnitude comparable with other driving components. The elucidation of such processes in microscopic detail that NMR can provide may lead to a reassessment of the governing factors of ligand affinity, as this might well be a general mechanism of small ligand binding.
Not only are protein backbone dynamics accessible to relaxation studies, but also side-chain dynamics can be measured. Different techniques that quantify the relaxation of 13C or 2H nuclei are also available (101-104). The focus usually lies on the dynamics of methyl groups that require elaborate isotopic labeling schemes to obtain either 13CHD2 or 13CH2D isotopomers (105). This finding has enabled determination of the order parameters of methyl group rotation axes. In the case of calmodulin, which is a calcium binding protein, it has been shown that complex formation with a peptide leads to nonuniform entropy changes along the protein backbone and side chains. Whereas the backbone only shows small changes in its motional characteristics, some side chains close to the binding site are restricted in their motion. Simultaneously, the motional entropy of more remote side chains increases, which thereby compensates for the entropy losses at the binding site (106, 107).
A nice example of the power of dynamic NMR spectroscopy is the work by Kern and coworkers (108, 109) on the correlation between enzymatic activity and the kinetics of structural dynamics in the coupled network of proteins.
These NMR methods provide greater insight into the complex thermodynamic mechanisms of molecular recognition, which thereby supplies the field of Chemical Biology with unique and invaluable information.
Ligand-observed experiments
Analysis of binding events by observing the NMR signals of the ligand rather than the biomolecular target is an alternative to protein-observed experiments. This approach is attractive to screen large libraries of compounds in a cheap and efficient manner, because expensive isotopic labeling of target or ligand is not required. It is even possible to screen mixtures of ligands using distinct spectral features of each ligand to identify the biologically active compound. Another advantage of ligand-observed drug screening is that it allows all biological targets, regardless of size, to be studied by NMR spectroscopy. This approach is especially useful when dealing with cells, viruses, or membrane proteins. G-coupled receptors and ion channels are membrane proteins that are highly interesting as drug targets but are difficult to express in high yields, which renders isotopic enrichment of such proteins costly. Furthermore, the required presence of phospholipids in samples of membrane proteins increases the size of the complex, which restricts the use of protein-observed measurements even more because of fast T2 relaxation (63). Contrarily, size restrictions do apply to the ligand, as the distinction between ligand and target is based on their difference in molecular weight. Therefore, ligands must be much smaller than the target. The binding event of a ligand to a biological target will transfer magnetization from target to ligand, which alters the NMR spectrum of the ligand. To observe binding of a ligand, it should usually be in medium to fast exchange with the target that corresponds to moderate binders with KD values in the mM to μM range.
Saturation transfer difference NMR (STD-NMR)
STD-NMR (110) observes magnetization transfer from protein to ligand molecules that are in fast exchange between the free and bound state. The protein is excited selectively by irradiating at a frequency specific to protein signals, typically the protein side-chain protons. A large excess of ligands ensures that the fraction of ligand molecules bound to the target at any one time is negligible. The transfer of magnetization from a large saturated protein to a bound ligand is, however, very efficient, and a fast koff rate of the ligand ensures that a measurable fraction of the free ligand has been bound to the protein during the course of the experiment. This fraction remains saturated throughout the experiment and does not contribute to the ligand NMR spectrum, which reduces the signal intensity. The fraction of ligand molecules that have been bound to the protein during the experiment can be calculated from the difference of two 1D spectra with and without protein saturation. The large excess of ligands also ensures that the amount of ligand molecules that will bind to the target more than once is negligible. The STD effect of a ligand-binding event is strong when the protein is large, even if the ligand only transiently binds to the protein, as the magnetization transfer depends on the molecular weight of the macromolecular complex and is very efficient for large complexes but inefficient for small molecules.
STD-NMR cannot readily distinguish between specific and nonspecific binding events. Therefore, an extension of STD-NMR involves the titration of a protein-inhibitor complex with a test ligand that will compete with the inhibitor in fast exchange for the same receptor-binding site on the protein. This situation is described by Equation (2) (111)
![]()
where KI and KD are the inhibition and dissociation constants of the inhibitor and the added ligand, cI and cL are the concentrations of inhibitor and ligand, and i is the extent of inhibition measured by the decrease of the STD signals of the known inhibitor. Assuming that both compounds have koff values that are relatively fast on the NMR timescale, it is possible to calculate the KD value of the competing ligand [Equation (3)].
![]()
In a fast-exchange situation with a large excess of ligands compared to receptors, the KD value derived from NMR experiments is comparable with the IC50 value (i.e., the concentration of the inhibitor required to reduce the target activity by 50%). The IC50 values are usually derived from biochemical assays; from an inhibitor of known IC50 value, it is therefore possible to derive relative IC50 values for compounds in competition with this inhibitor by NMR spectroscopy.
The competition between an inhibitor of known binding properties, which is also called a reporter-ligand, and a ligand for the same binding site is used in NMR reporter assays as illustrated in (Fig. 14). The reporter ligand must be highly soluble and have strong, quantifiable NMR resonances. Competing ligands are titrated to the NMR sample that contains a target protein and reporter ligand, and the reduction of the reporter signals corresponds to the fraction that has been displaced from the binding site by the competing ligand. By selecting a reporter-ligand of low affinity toward the target protein, it is even possible to detect binding of high-affinity or poorly soluble ligands (112-114), as well as ligands in slow exchange with the target (115).
Other methods similar to the STD experiment are Water-LOGSY (Water-Ligand Observed via Gradient Spectroscopy) (116) and NOE pumping (117). WaterLOGSY is based on the transfer of magnetization from biomolecule to ligand via bulk water, whereas NOE pumping relies on first suppressing all ligand signals and then transferring magnetization via an NOE experiment from bio-macromolecule to the fraction of ligand molecules that have been bound.
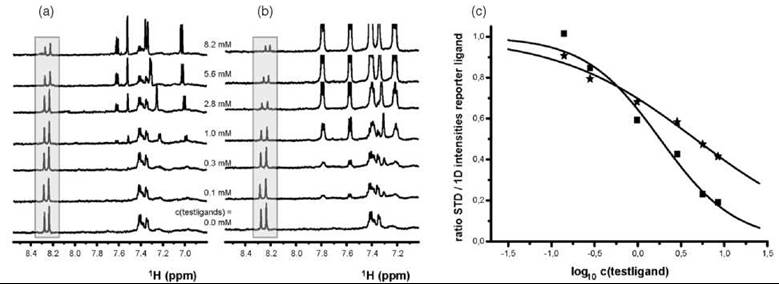
Figure 14. STD-NMR reporter assay as a competition experiment. The resonances of the highly soluble reporter ligand of known binding properties to the target protein are accented by a grey box. The test ligand competes with the reporter ligand for the binding site. (a) Stepwise addition of the test ligand to the kinase-reporter-ligand sample decreases the STD reporter signals in comparison with the reference spectrum in the absence of any competitor. (b) Analogous procedure with a more potent ligand. (c) The resonances of the reporter ligand deplete faster when titrated with the more potent ligand B as observed from the fitted curve of the titration data (Ligand A: stars, Ligand B: squares).
Exchange-transferred NOE spectroscopy (et-NOESY)
Similar to the STD experiment, exchange-transferred NOE spectroscopy (et-NOESY) provides information about a binding event between a small molecule and a high molecular weight biomolecule, even when the biomolecule itself is not amenable to NMR studies because of broad line widths, low solubility, or fast T2 relaxation.
The principle of et-NOESY (118-120) is applied to low-affinity protein-ligand complexes where exchange is fast on the NMR timescale. High molar excess of the ligand is used (typical ligand to receptor site ratios range from 10 to 50); therefore, the resonance line shapes are governed by the properties of the unbound ligand in solution (i.e., sharp signals caused by slow T2 relaxation). The et-NOEs observed, however, are caused by the conformation of the ligand in the binding pocket. The efficiency of magnetization transfer caused by NOE effects depends on the overall correlation time of the molecular complex and becomes more efficient for larger complexes. For small molecules, the NOE is positive and weak, whereas it is negative and strong for large biomolecular complexes. Therefore, the cross-relaxation measured by the NOE transfer within the ligand when it is bound to the target will dominate the spectrum.
The NOEs from the bound state are transferred into the free state of the ligand when it leaves the binding pocket. To observe NOEs from the bound state, the off-rate (Koff) of the ligand must be much faster than the cross-relaxation rate, only then is the initial build up of the NOE proportional to the cross-relaxation rate (121). Based on the internuclear distances obtainable from the NOEs it is possible to deduce the conformation of the ligand when bound to the target.
The inversion of sign between NOEs from small and large molecules simplifies measurement of et-NOEs for binding studies of small ligands <1 kDa and large targets. For larger ligands, such as ligand-peptides, two et-NOESY spectra are recorded in the presence and absence of target protein. By subtracting the two spectra, it is possible to distinguish between NOEs that develop from target-bound ligands and ligands that have not been bound.
The combination of STD-NMR and et-NOESY experiments provides information on ligand protons in direct contact with the binding pocket as well as the overall ligand structure. The two experiments thus complement each other in elucidating the binding surface of a target of otherwise unknown structure.
NOEs and isotope-filtered NMR experiments
In the strong binding regime, the measurable NOEs between the biomolecule and the bound ligand can be exploited to derive the 3D complex structure. A common approach involves uniform labeling of the biomolecule with 15N and 13C isotopes, whereas the ligand remains unlabeled (122). To distinguish between the intra-molecular NOEs of the biomolecule, the intra-molecular NOEs of the ligand and the intermolecular NOEs that occur at the binding interface, isotope-filtered NOESY experiments are used. The proton magnetization that originates from either 1H-13C or 1H-15N atom pairs is either filtered out or selected (123, 124).
The 3D 13C- and 15N-filtered NOESY experiments are used to identify NOE restraints within the biomolecule itself. Intra-molecular NOE restraints of the ligand can be assigned from the 12C,14N-filtered NOESY experiment, which suppresses the signal of 13C- and 15N-attached protons of the biomolecule. The intermolecular NOEs between the ligand and biomolecule can be detected by a combination of selection and filtering out of proton magnetization in the 3D 12C,14N ω1-filtered NOESY 1H,13C HSQC experiment, which observes the interface between the two molecules as observed in the 2D projection in (Fig. 15).
The information derived from the analysis of this example was used as distance restraints for calculation of the 3D structure of the complex of calmodulin, a calcium binding protein, and a peptide ligand. The amino acid sequence of the peptide ligand, C20W, corresponds to the N-terminal part of the calmodulin-binding domain of the plasma membrane calcium pump (125).

Figure 15. 2D 1H,1H projection of the 12C/14Nω1-filtered 3D NOESY 1H,13C HSQC spectrum that contains only intermolecular NOEs between the double-labeled protein (Calmodulin, resonance assignment in black) and a bound peptide ligand (in light grey). Only contacts between the protein and the peptide ligand are observed. Assigned cross-peaks are indicated in the spectrum.
Structure calculation based on NMR data
To provide new insights in the biological context of biomolecules, obtaining structural information is frequently essential. The tertiary structure of molecules with accuracy at the atom level can be calculated using NMR-derived data on intra-molecular atom distances and angles. Many biomolecules form complexes with each other, and the nature of these complex formations can be deduced from intermolecular distances and determination of the interaction surfaces in combination with computational docking procedures to determine their global structure.
Simulated annealing
Traditionally, the structure of biomolecules and macromolecular complexes are solved by NMR using proton-proton NOEs that are translated into distances. In addition to NOEs, the structures may be refined using additional NMR data, such as coupling constants, RDCs, chemical shifts, and hydrogen bonds. Many programs and protocols are available for calculation of NMR structures [e.g., XPLOR-NIH (126), CNS (127), CNX (Accelrys, San Diego, USA), CYANA (128, 129), and ARIA (130)]. Structural calculations using simulated annealing (SA) and molecular dynamics (MD) require a forcefield with the molecular topology and parameters to define the atomic masses, bond lengths, charges, angles, planarity, and connectivity information of the entire macromolecule (131). Most biomolecular NMR structures are calculated by SA with MD in either Cartesian or torsion angle coordinate space. The torsion angle dynamics protocol allows simulation of longer time intervals and higher temperatures than dynamics in Cartesian space and is thus very efficient for the calculation of NMR structures (128, 132). The SA algorithm generally starts from an extended conformation of the macromolecule and proceeds from a high temperature stage over slow cooling-down stages to a final energy minimization at low temperature. This protocol is commonly repeated multiple times in an iterative way to recalibrate the NOE-based distances. The final NMR structures are often refined in explicit solvent to improve the structure quality significantly (133).
HADDOCK
The conventional NMR approach for determining biomolecular complex structures is based on a time-consuming process of collecting intermolecular NOEs and complementing RDCs. Another approach uses the easily obtainable chemical shift perturbations from NMR titration experiments in combination with computational docking. The HADDOCK (High Ambiguity Driven Docking) approach uses the experimental NMR data of the intermolecular interactions as Ambiguous Interaction Restraints (AIRs) to drive the docking process (134). Residues that show significant chemical shift changes during interaction are designated as active. Residues that show less significant changes and/or are surface neighbors of the active residues are called passive. The AIR is accordingly defined as an ambiguous intermolecular distance between any atom of an active residue and any atom of an active or passive residue of the partner molecule in the complex. The HADDOCK approach uses CNS for structural calculations and protocols derived from ARIA for automation. Generally, the docking protocol starts with calculating many random orientations in which the partner molecules are positioned far away from each other. After a rigid-body docking, the best scoring solutions in terms of intermolecular energies are refined in torsion angle space using a multistage, semi-rigid simulated annealing protocol. Subsequently, the structures are refined in explicit water in Cartesian space. Next to protein-protein docking, HADDOCK has also been applied to model protein-DNA, protein-RNA, protein-oligosaccharide, and protein-ligand complexes (135, 136). The HADDOCK protocols allow the usage of additional NMR data, such as RDCs (137) and diffusion anisotropy data (138). Furthermore, HADDOCK is able to perform solvated docking (139), as water molecules in the interaction interface play an important role in mediating biomolecular interactions. These implementations lead to improved accuracy and quality of the final complex structures. The HADDOCK method can calculate models of multicomponent complexes and can easily be applied to drug design.
RDC-based structure refinement
RDCs are especially useful for determining the relative orientation of different partner molecules in a complex or of different domains within a single macromolecule in a complex. When two domains of a macromolecule are connected by a flexible linker and NOE data between those domains is scarce or lacking, RDCs can be used to determine the exact orientation of the domains with respect to each other. In the case of ribosomal L11 protein, these additional orientational restraints have been used to solve the structure of the protein in its free form as well as in complex with its RNA binding partner and an antibiotic (vide supra) (73,140). The relative orientation of the two domains that are connected by a short linker region is rigid and well defined in solution (Fig. 16). Using 3668 distance restraints (26.4 per residue), 77 TALOS-derived dihedral angle restraints, and 102 J-couplings derived from the backbone HN-Ha, the structure of each domain could be determined accurately. The relative orientation between the N- and C-terminal, however, could not be determined unambiguously because of the limited amount of interdomain NOEs. To calculate the overall structure of the full-length protein, an additional 429 RDCs were measured and incorporated in the structural calculation. This step allowed the exact determination of the relative orientation of both domains (73). This result is in good agreement with the fact that the two domains tumble together as a rigid body, based on the dynamic measurements (Fig. 13).
The same method has been applied to measure the L11 domain orientation when the protein is in complex with its RNA partner or both RNA and thiostrepton antibiotic. The additional RDCs revealed a rearrangement of the N-terminal domain of L11 placing it closer to the RNA after binding of thiostrepton. HADDOCK has been used to calculate a model of the ternary structure of the L11 protein in complex with RNA and antibiotic. Based on the orientational data, the dynamics and the docking model, it seems that thiostrepton locks the domain conformation of L11 in a rigid (inhibitory) state. The antibiotic thiostrepton interferes with the interaction of elongation factors to this L11-RNA region, which has a dramatic effect on the level of protein synthesis by the ribosome.
Orientational restraints, which are derived by paramagnetic labeling of a single domain of a protein, are especially important for the analysis of domain-domain interactions (141). The paramagnetic tag induces anisotropic alignment, which is scaled for the two domains of the protein according to their relative domain mobility.
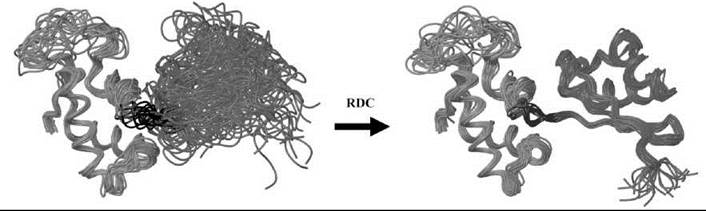
Figure 16. Structure calculation purely based on NOE and dihedral angle restrictions often fails to determine proper domain orientations. By incorporating the long-range orientation parameters that RDCs provide, it is possible to overcome this problem and to define relative orientation of the two domains of the ribosomal L11 protein.
LIGDOCK and SOS-NMR
In the weak binding regime, intermolecular NOEs between biomolecule and ligand are usually not exploitable in protein- observed experiments. Instead, other NMR-derived information is used as an experimental restraint for molecular docking procedures to solve the protein-ligand structure. The readily obtainable 15N chemical shift perturbations (CSPs) of the protein amide resonances caused by ligand binding are often used as experimental restraints. During the molecular dynamics simulation, penalty energies calculated from these experimental restraints together with conventional intermolecular van der Waals energies or electrostatic interactions guide the protein-ligand complex toward an energy minimum. This minimum represents a physically reasonable structure that fulfills a maximum number of restraints introduced by NMR-derived information.
Accuracy of the NMR structure, as judged by comparison with X-ray crystallographic structures, depends on the nature and completeness of the NMR data. Figure 17 shows two different outcomes of calculation of complex structures from CSPs with the LIGDOCK procedure (142) (an extension of HADDOCK). The first example, which includes phosphotyrosine phosphatase 1b (PTP1b) and 5-iodo-2-(oxalylamino) benzoic acid (143), gives straightforward results, and the calculated complex of lowest energy shows excellent correlation with the reference X-ray structure. The second example, which involves mitogen-activated protein kinase p38 and the small molecule ligand SB203580 (144), does not lead to accurate structures by the introduction of CSPs alone. First, the binding site is less defined because the extent of assignment is lower compared to that of PTP1b. Second, the specific shape of SB203580 with a twofold and a threefold rotation symmetry axis implies that the ligand can occupy the binding site in more than one symmetry-related orientation. It is concluded that the ambiguity of the ligand binding can only be solved by inclusion of additional experimental data.
To find a unique solution of the p38α-SB203580 complex structure, additional NMR information was obtained from the SOS-NMR method (Structural information using Overhauser effects and Selective labeling) (145). The SOS concept is based on STD measurements of selectively 1H labeled and otherwise deuterated protein. The selectively 1H-labeled amino acids lead to patches of STD-active surface that also results in ambiguous information, but in combination with CSPs, the number of possible solutions is reduced. In this case, inspection of the binding site, as defined by the CSPs, leads to the conclusion that p38α should be selectively 1H-labeled on all isoleucines. The subsequent STD experiment confirmed that the pyridinium moiety of the ligand is close to Ile-84, which yields a pronounced STD effect of its protons as observed in (Fig. 18). The fluoro-phenyl group shows a weak STD effect and is therefore concluded to be situated farther away from Ile-84.
Figure 17. Molecular docking guided by NMR-derived information. (a) The complex structure of PTP1b with a ligand (modeled structure in green/yellow) calculated from CSPs only corresponds well with the X-ray reference structure (blue). (b) The cluster of lowest energy structures calculated for p38α and ligand SB203580 shows two possible orientations (c) that differ in their ability to form a hydrogen bond to the Met109 amide group. Additional information is needed to clarify the binding mode. See color insert.

Figure 18. SOS-NMR using deuterated and selectively protonated protein can resolve ambiguity of ligand orientation. (a) Bottom: 1D 1H NMR reference spectrum that shows the resonances of the ligand SB203580. Middle: 1D 1H STD spectrum recorded in the presence of p38a. Top: 1D 1H STD spectrum recorded of a sample prepared according to the SOS procedure of perdeuterated kinase and unlabeled isoleucine residues. A pronounced STD effect for the H1 and H4 protons and only weak effects for the H6 and H5 protons resolves ambiguity of ligand orientation in the binding pocket. (b) Distance analysis of the reference X-ray structure.
Outlook
In addition to the in vitro studies of well-defined systems that have been discussed here, NMR spectroscopy can also be applied to living systems or complex substance mixtures (146). This broad applicability is an advantage of NMR spectroscopy over X-ray crystallography. Early in vivo NMR studies that mostly identify small metabolite molecules by 31P or *H 1D experiments have led to different applications. In-cell NMR uses isotopic labeling combined with two or higher dimensional NMR experiments for structural studies of the phosphorylation state of a given protein in the cellular environment or of intrinsically unstructured proteins. In-cell NMR applications in prokaryotic systems cover structural and functional studies (i.e., protein-protein interactions, protein dynamics, automated structure determination, and de novo resonance assignments (147-149)).
In eukaryotic systems, challenges intrinsic to NMR (e.g., low sensitivity or proper discrimination between target and background signals) become even more pronounced. Interesting applications of NMR to eukaryotic systems are developing that study important cellular features like posttranslational protein modification or directed transport that are not observed in prokaryotic cells (150).
Whereas the above-mentioned NMR techniques are used to define individual interactions of proteins in a more or less artificial setting, NMR is also used for metabonomic analysis that focuses on identification of small molecules in biofluids and tissues, tracking changes that occur during variation of the local environment. A chemical biologist is not only interested in defining the properties of a protein-ligand interaction but also in the outcome. Therefore, systematic changes of bio-fluid or tissue extract composition are tracked during administration of enzyme inhibitors or activators. In combination with other techniques, especially mass spectrometry, NMR is a very valuable tool for this task. The role of NMR has been evaluated for analytical strategies in metabonomic research (151) and pharmaceutical discovery (152, 153).
Magnetic resonance imaging has long been used for clinical diagnostic purposes, often in combination with relaxationenhancing contrast agents. Recent development of lipid nanoparticle systems as carriers of relaxation enhancers to specific targets (154) makes imaging methods applicable to the field of chemical biology.
References
1. Levitt M. Spin dynamics: Basics of Nuclear Magnetic Resonance. 2001. John Wiley & Sons, New York.
2. Hore PJ, Jones JA, Wimperis, S. NMR: The toolkit. 2000. Oxford University Press, New York.
3. Wishart DS, Sykes BD, Richards FM. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 1992; 31:1647-1651.
4. Wang Y, Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 2002; 11:852-861.
5. Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 1999; 13:289-302.
6. Xu XP, Case DA. Automated prediction of 15N, 13Calpha, 13Cbeta and 13C’ chemical shifts in proteins using a density functional database. J. Biomol. NMR 2001; 21:321-333.
7. Neal S, et al. Rapid and accurate calculation of protein 1H, 13C and 15N chemical shifts. J. Biomol. NMR 2003; 26:215-240.
8. Shen Y, Bax A. Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology. J. Biomol. NMR 2007; 38:289-302.
9. Wishart DS, et al. Automated 1H and 13C chemical shift prediction using the BioMagResBank. J. Biomol. NMR 1997; 10:329-336.
10. Cavalli A, et al. Protein structure determination from NMR chemical shifts. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:9615-9620.
11. Kontaxis G, Delaglio F, Bax A. Molecular fragment replacement approach to protein structure determination by chemical shift and dipolar homology database mining. Methods Enzymol. 2005; 394:42-78.
12. Karplus M. Contact electron-spin coupling of nuclear magnetic moments. J. Chem. Phys. 1959; 30:11-15.
13. Cordier F, et al. Direct detection of N-H[...]O = C hydrogen bonds in biomolecules by NMR spectroscopy. Nat. Protoc. 2008; 3:235-241.
14. Cornilescu G, Hu JS, Bax A. Identification of the hydrogen bonding network in a protein by scalar couplings. J. Am. Chem. Soc. 1999; 121:2949-2950.
15. Wohnert J, et al. Direct identification of NH...N hydrogen bonds in non-canonical base pairs of RNA by NMR spectroscopy. Nucleic Acids Res, 1999; 27:3104-3110.
16. Overhauser AW. Polarization of nuclei in metals. Phys. Rev. 1953; 92:411.
17. Prestegard JH, Bougault CM, Kishore AI. Residual dipolar couplings in structure determination of biomolecules. 2004; 3519-3540.
18. Rodriguez-Castaneda F, et al. Paramagnetic tagging of diamagnetic proteins for solution NMR. Magn. Reson. Chem. 2006; 44:S10-6.
19. Deschamps ML, et al. Probing protein-peptide binding surfaces using charged stable free radicals and transverse paramagnetic relaxation enhancement (PRE). J. Biomol. NMR 2005; 31:155-160.
20. Martin LJ, et al. Double-lanthanide-binding tags: design, photophysical properties, and NMR applications. J. Am. Chem. Soc. 2007; 129:7106-7113.
21. Tolman JR. Dipolar couplings as a probe of molecular dynamics and structure in solution. Curr. Opin. Struct. Biol. 2001; 11:532-539.
22. Lipsitz RS, Tjandra N. Residual dipolar couplings in NMR structure analysis. Annu. Rev. Biophys. Biomol. Struct. 2004; 33:387-413.
23. Mulder FAA, et al. An off-resonance rotating frame relaxation experiment for the investigation of macromolecular dynamics using adiabatic rotations. J. Magnet. Res. 1998; 131:351-357.
24. Mulder FA, et al. Microsecond time scale dynamics in the RXR DNA-binding domain from a combination of spin-echo and off-resonance rotating frame relaxation measurements. J. Biomol. NMR 1999; 13:275-288.
25. Carr HY, Purcell EM. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys. Rev. 1954; 94:630.
26. Meiboom S, Gill D. Modified spin-echo method for measuring nuclear relaxation times. Rev. Scient. Inst. 1958; 29:688-691.
27. Millet O, et al. The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. J. Am. Chem. Soc. 2000; 122:2867-2877.
28. Ishima R, Torchia DA. Estimating the time scale of chemical exchange of proteins from measurements of transverse relaxation rates in solution. J. Biomol. NMR 1999; 14:369-372.
29. Meekhof AE, Freund SMV. Separating the contributions to 15N transverse relaxation in a fibronectin type III domain. J. Biomol. NMR 1999; 14:13-22.
30. Cavanagh J, Venters RA. Protein dynamic studies move to a new time slot. Nat. Struct. Biol. 2001; 8:912-914.
31. Ishima R, et al. Carbonyl carbon transverse relaxation dispersion measurements and ms-micros timescale motion in a protein hydrogen bond network. J. Biomol. NMR 2004; 29:187-198.
32. Ishima R, Torchia DA. Extending the range of amide proton relaxation dispersion experiments in proteins using a constant-time relaxation-compensated CPMG approach. J. Biomol. NMR 2003; 25:243-248.
33. Mulder FA, et al. Measurement of slow (micros-ms) time scale dynamics in protein side chains by (15)N relaxation dispersion NMR spectroscopy: application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 2001; 123:967-975.
34. Mulder FA, et al. Studying excited states of proteins by NMR spectroscopy. Nat. Struct. Biol. 2001; 8:932-935.
35. Tollinger M, et al. Slow dynamics in folded and unfolded states of an SH3 domain. J. Am. Chem. Soc. 2001; 123:11341-11352.
36. Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. 1982: 4559-4570.
37. Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. 1982; 104:4546-4559.
38. Mandel AM, Akke M, Palmer AG 3rd. Dynamics of ribonuclease H: temperature dependence of motions on multiple time scales. Biochemistry 1996; 35:16009-16023.
39. Clore GM, et al. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 1990; 112:4989-4991.
40. Clore GM, et al. Analysis of the backbone dynamics of interleukin-1 beta using two-dimensional inverse detected heteronuclear 15N-1H NMR spectroscopy. Biochemistry 1990; 29:7387-7401.
41. Aue WP, Bartholdi E, Ernst RR. Two-dimensional spectroscopy. Application to nuclear magnetic resonance. J. Chem. Phys. 1976; 64:p.2229-2246.
42. Braunschweiler L, Ernst RR. Coherence transfer by isotropic mixing: application to proton correlation spectroscop. J. Magnet. Reson. 1983; 53:521-528.
43. Jeener J, et al. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979; 71:4546-4553.
44. Bax A, Davis DG. Practical aspects of two-dimensional transverse NOE spectroscopy. J. Magnet. Reson. (1969) 1985; 63:207-213.
45. Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Progress Nuclear Magnet. Reson. Spectros. 1999; 34:93-158.
46. Strauss A, et al. Efficient uniform isotope labeling of Abl kinase expressed in Baculovirus-infected insect cells. J. Biomol. NMR 2005; 31:343-349.
47. Werner K, et al. Isotope labeling of mammalian GPCRs in HEK293 cells and characterization of the C-terminus of bovine rhodopsin by high resolution liquid NMR spectroscopy. J. Biomolec. NMR 2008; 40:49-53.
48. Otomo T, et al. NMR observation of selected segments in a larger protein: central-segment isotope labeling through intein-mediated ligation. Biochemistry 1999; 38:16040-16044.
49. Valiyaveetil FI, MacKinnon R, Muir TW. Semisynthesis and folding of the potassium channel KcsA. J. Am. Chem. Soc. 2002; 124:9113-9120.
50. Tzakos AG, Easton LE, Lukavsky PJ. Preparation of large RNA oligonucleotides with complementary isotope-labeled segments for NMR structural studies. Nat. Protoc. 2007; 2:2139-2147.
51. Bertini I, et al. Mapping protein-protein interaction by 13C’-detected heteronuclear NMR spectroscopy. J. Biomol. NMR 2006; 36:111-122.
52. Fares C, Amata I, Carlomagno T. 13C-detection in RNA bases: revealing structure-chemical shift relationships. J. Am. Chem. Soc. 2007; 129:15814-15823.
53. Fiala R, Sklenar V. 13C-detected NMR experiments for measuring chemical shifts and coupling constants in nucleic acid bases. J. Biomol. NMR. 2007; 39:153-163.
54. Bothner-By AA, et al. Structure determination of a tetrasaccharide: transient nuclear Overhauser effects in the rotating frame. 1984; 811-813.
55. Bodenhausen G, Ruben DJ. Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 1980; 69:185-189.
56. Bax A, Griffey RH, Hawkins BL. Correlation of proton and nitrogen-15 chemical shifts by multiple quantum NMR. J. Magnet. Reson. (1969) 1983; 55:301-315.
57. Bax A, Summers MF. Proton and carbon-13 assignments from sensitivity-enhanced detection of heteronuclear multiple-bond connectivity by 2D multiple quantum NMR. 1986; 2093-2094.
58. Cicero DO, Barbato G, Bazzo R. Sensitivity enhancement of a two-dimensional experiment for the measurement of heteronuclear long-range coupling constants, by a new scheme of coherence selection by gradients. J. Magnet. Reson. 2001; 148:209-213.
59. Claridge TD, Perez-Victoria I. Enhanced 13C resolution in semi-selective HMbC: a band-selective, constant-time HMBC for complex organic structure elucidation by NMR. Org. Biomol. Chem. 2003; 1:3632-3634.
60. Pervushin K, et al. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:12366-12371.
61. Morris GA, Freeman R. Enhancement of nuclear magnetic resonance signals by polarization transfer. 1979; 760-762.
62. Furtig B, et al. NMR spectroscopy of RNA. ChemBioChem. 2003; 4:936-962.
63. Meyer B, Peters T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew. Chem. Internat. Ed. 2003; 42:864-890.
64. Stockman BJ, Dalvit C. NMR screening techniques in drug discovery and drug design. Prog. Nucl. Magnet. Reson. Spectros. 2002; 41:187-231.
65. Diercks T, Coles M, Kessler H. Applications of NMR in drug discovery. Curr. Opin. Chem. Biol. 2001; 5:285-291.
66. Pellecchia M, Sem DS, Wuthrich K. NMR in drug discovery. Nat. Rev. Drug Discov, 2002; 1:211-219.
67. Salvatella X, Giralt E. NMR-based methods and strategies for drug discovery. Chem. Soc. Rev. 2003; 32:365-372.
68. LeMaster DM. Deuteration in protein proton magnetic resonance. Methods Enzymol. 1989; 177:23-43.
69. LeMaster DM. Deuterium labelling in NMR structural analysis of larger proteins. Quarter. Rev. Biophys. 1990; 23:133-174.
70. LeMaster DM. Uniform and selective deuteration in two-dimensional NMR of proteins. Annual review of biophysics and biophysical chemistry. 1990; 19:243-266.
71. Rao NS, et al. NMR Pulse schemes for the sequential assignment of arginine side-chain h[epsilon]protons. J. Magnet. Reson. Series B 1996; 113:272-276.
72. Medek A, et al. The use of differential chemical shifts for determining the binding site location and orientation of protein-bound ligands. 2000; 1241-1242.
73. Jonker HR, et al. L11 domain rearrangement upon binding to RNA and thiostrepton studied by NMR spectroscopy. Nucleic Acids Research, 2007; 35:441-454.
74. Pargellis C, et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002; 9:268-272.
75. Wang Z, et al. The structure of mitogen-activated protein kinase p38 at 2.1-A resolution. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:2327-2732.
76. Wang Z, et al. Structural basis of inhibitor selectivity in MAP kinases. Structure 1998; 6:1117-1128.
77. Vogtherr M, et al. NMR characterization of kinase p38 dynamics in free and ligand-bound forms. Angew. Chem. Internat. Ed. 2006; 45:993-997.
78. Schindler T, et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 2000; 289:1938-1942.
79. Nagar B, et al. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002; 62:4236-4243.
80. Regan J, et al. Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate. J. Med. Chem. 2002; 45:2994-3008.
81. Branger J, et al. Anti-inflammatory effects of a p38 mitogen-activated protein kinase inhibitor during human endotoxemia. J. Immunol. 2002; 168:4070-4077.
82. Wan PT, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004; 116:855-867.
83. Manley PW, et al. Advances in the structural biology, design and clinical development of VEGF-R kinase inhibitors for the treatment of angiogenesis. Biochim. Biophys. Acta 2004; 1697:17-27.
84. Vogtherr M, et al. NMR backbone assignment of the mitogen- activated protein (MAP) kinase p38. J. Biomol. NMR 2005; 32:175.
85. Langer T, et al. NMR backbone assignment of a protein kinase catalytic domain by a combination of several approaches: application to the catalytic subunit of cAMP-dependent protein kinase. ChemBioChem. 2004; 5:1508-1516.
86. Jencks WP. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. U.S.A. 1981; 78:4046-4050.
87. Shuker SB, et al. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996; 274:1531-1534.
88. Braisted AC, et al. Discovery of a potent small molecule IL-2 inhibitor through fragment assembly. J. Am. Chem. Soc. 2003; 125:3714-3715.
89. Hajduk PJ, et al. Discovery of potent nonpeptide inhibitors of stromelysin using SAR by NMR. J. Am. Chem. Soc. 1997; 119:5818-5827.
90. Szczepankiewicz BG, et al. Discovery of a potent, selective protein tyrosine phosphatase 1B inhibitor using a linked-fragment strategy. J. Am. Chem. Soc. 2003; 125:4087-4096.
91. Miranker A, et al. Demonstration by NMR of folding domains in lysozyme. Nature 1991; 349:633-636.
92. Pellecchia M, et al. SEA-TROSY (Solvent Exposed Amides with TROSY): a method to resolve the problem of spectral overlap in very large proteins. 2001; 4633-4634.
93. Gunther UL, Schaffhausen B. NMRKIN: simulating line shapes from two-dimensional spectra of proteins upon ligand binding. J. Biomol. NMR 2002; 22:201-209.
94. Mittag T, Schaffhausen B, Gunther UL. Tracing kinetic intermediates during ligand binding. J. Am. Chem. Soc. 2004; 126:9017-9023.
95. Gunther U, Mittag T, Schaffhausen B. Probing Src homology 2 domain ligand interactions by differential line broadening. Biochemistry 2002; 41:11658-11669.
96. Mittag T, Schaffhausen B, Gunther UL. Direct observation of protein-ligand interaction kinetics. Biochemistry 2003; 42:11128-11136.
97. Mittag T, et al. Retinol modulates site-specific mobility of apo-cellular retinol-binding protein to promote ligand binding. J. Am. Chem. Soc. 2006; 128:9844-9848.
98. Korzhnev DM, et al. Low-populated folding intermediates of Fyn SH3 characterized by relaxation dispersion NMR. Nature 2004; 430:586-590.
99. Homans SW. NMR spectroscopy tools for structure-aided drug design. Angew. Chem. Internat. Ed. 2004; 43:290-300.
100. Zidek L, Novotny MV, Stone MJ. Increased protein backbone conformational entropy upon hydrophobic ligand binding. Nat. Struct. Biol. 1999; 6:1118-1121.
101. Millet O, et al. Deuterium spin probes of side-chain dynamics in proteins. 1. Measurement of five relaxation rates per deuteron in 3C-labeled and fractionally 2H-enriched proteins in solution. J. Am. Chem. Soc. 2002; 124:6439-6448.
102. Muhandiram DR, et al. Measurement of 2H T1 and T1.rho. Relaxation times in uniformly 13C-labeled and fractionally 2H-labeled proteins in solution. J. Am. Chem. Soc. 1995; 117:11536-11544.
103. Ishima R, Louis JM, Torchia DA. Optimized labeling of 13CHD2 methyl isotopomers in perdeuterated proteins: potential advantages for 13C relaxation studies of methyl dynamics of larger proteins. J. Biomol. NMR 2001; 21:167-171.
104. Ishima R, et al. Comparison of methyl rotation axis order parameters derived from model-free analyses of 2H and 13C longitudinal and transverse relaxation rates measured in the same protein sample. J. Am. Chem. Soc. 2001; 123:6164-6171.
105. Kainosho M, et al. Optimal isotope labelling for NMR protein structure determinations. Nature 2006; 440:52-57.
106. Lee AL, Kinnear SA, Wand AJ. Redistribution and loss of side chain entropy upon formation of a calmodulin-peptide complex. Nature Struct. Biol. 2000; 7:72-77.
107. Cavanagh J, Akke M. May the driving force be with you—whatever it is. Nat. Struct. Molec. Biol. 2000; 7:11-13.
108. Eisenmesser EZ, et al. Intrinsic dynamics of an enzyme underlies catalysis. Nature 2005; 438:117-121.
109. Kern D, Eisenmesser EZ, Wolf-Watz M. Enzyme dynamics during catalysis measured by NMR spectroscopy. Methods Enzymol. 2005; 394:507-524.
110. Moriz Mayer BM. Characterization of ligand binding by saturation transfer difference. NMR Spectros. 1999; 1784-1788.
111. Meinecke R, Meyer B. Determination of the binding specificity of an integral membrane protein by saturation transfer difference NMR: RGD peptide ligands binding to integrin alphaIIbbeta3. J. Med. Chem. 2001; 44:3059-3065.
112. Dalvit C, et al. NMR-Based screening with competition water- ligand observed via gradient spectroscopy experiments: detection of high-affinity ligands. J. Med. Chem. 2002; 45:2610-2614.
113. Siriwardena AH, et al. A straightforward NMR-spectroscopy- based method for rapid library screening. Angew. Chem. Int. Ed. Engl. 2002; 41:3454-3457.
114. Jahnke W, et al. NMR reporter screening for the detection of high-affinity ligands. Angew. Chem. Int. Ed. Engl. 2002; 41:3420-3423.
115. Wang YS, Liu D, Wyss DF. Competition STD NMR for the detection of high-affinity ligands and NMR-based screening. Magn. Reson. Chem. 2004; 42:485-489.
116. Dalvit C, et al. Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J. Biomol. NMR 2000; 18:65-68.
117. Chen A, Shapiro MJ. NOE pumping: a novel NMR technique for identification of compounds with binding affinity to macromolecules. 1998; 10258-10259.
118. Balaram P, Bothner-By AA, Dadok J. Negative nuclear Over-hauser effects as probes of macromolecular structure. J. Am. Chem. Soc. 1972; 94:4015-4017.
119. Clore GM, Gronenborn AM. Theory and applications of the transferred nuclear overhauser effect to the study of the conformations of small ligands bound to proteins. J. Magnet. Reson. (1969) 1982; 48:402-417.
120. Balaram P, Bothner-By AA, Breslow E. Nuclear magnetic resonance studies of the interaction of peptides and hormones with bovine neurophysin. Biochemistry 1973; 12:4695-4704.
121. Post CB. Exchange-transferred NOE spectroscopy and bound ligand structure determination. Curr. Opin. Struct. Biol. 2003; 13:581-588.
122. Zwahlen C, et al. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: application to a bacteriophage λ N-peptide/boxB. RNA Complex 1997; 6711-6721.
123. Kogler H, et al. Low-pass J filters. Suppression of neighbor peaks in heteronuclear relayed correlation spectra. J. Magnet. Reson. (1969) 1983; 55:157-163.
124. Otting G, et al. Editing of 2D 1H NMR spectra using X half-filters. combined use with residue-selective 15N labeling of proteins. J. Magnet. Reson. (1969) 1986; 70:500-505.
125. Elshorst B, et al. NMR solution structure of a complex of calmodulin with a binding peptide of the Ca2+ pump. Biochemistry 1999; 38:12320-12332.
126. Schwieters CD, et al. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003; 160:65-73.
127. Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. Biol. Crystallogr. 1998; 54:905-921.
128. Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997; 273:283-298.
129. Guntert P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004; 278:353-378.
130. Linge JP, O’Donoghue SI, Nilges M. Automated assignment of ambiguous nuclear overhauser effects with ARIA. Methods Enzymol. 2001; 339:71-90.
131. Linge JP, Nilges M. Influence of non-bonded parameters on the quality of NMR structures: a new force field for NMR structure calculation. J. Biomol. NMR 1999; 13:51-59.
132. Stein EG, Rice LM, Brunger AT. Torsion-angle molecular dynamics as a new efficient tool for NMR structure calculation. J. Magn. Reson. 1997; 124:154-164.
133. Linge JP, et al. Refinement of protein structures in explicit solvent. Proteins 2003; 50:496-506.
134. Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003; 125:1731-1737.
135. van Dijk M, et al. Information-driven protein-DNA docking using HADDOCK: it is a matter of flexibility. Nucleic Acids Res. 2006; 34:3317-3325.
136. van Dijk AD, Boelens R, Bonvin AM. Data-driven docking for the study of biomolecular complexes. FEBS J. 2005; 272:293-312.
137. van Dijk AD, Fushman D, Bonvin AM. Various strategies of using residual dipolar couplings in NMR-driven protein docking: application to Lys48-linked di-ubiquitin and validation against 15N-relaxation data. Proteins 2005; 60:367-381.
138. van Dijk AD, et al. Combining NMR relaxation with chemical shift perturbation data to drive protein-protein docking. J. Biomol. NMR. 2006; 34:237-244.
139. van Dijk AD Bonvin AM. Solvated docking: introducing water into the modelling of biomolecular complexes. Bioinformatics 2006; 22:2340-2347.
140. Ilin S, et al. Domain reorientation and induced fit upon RNA binding: solution structure and dynamics of ribosomal protein L11 from Thermotoga maritima. ChemBioChem. 2005; 6:1611- 1618.
141. Babini E, et al. A structural and dynamic characterization of the EF-hand protein CLSP. Structure 2006; 14:1029-1038.
142. Schieborr U, et al. How much NMR data is required to determine a protein-ligand complex structure? ChemBioChem. 2005; 6:1891-1898.
143. Andersen HS, et al. 2-(oxalylamino)-benzoic acid is a general, competitive inhibitor of protein-tyrosine phosphatases. J. Biol. Chem. 2000; 275:7101-7108.
144. Wang Z, et al. Structural basis of inhibitor selectivity in MAP kinases. Structure 1998; 6:1117-1128.
145. Hajduk PJ, et al. SOS-NMR: a saturation transfer NMR-based method for determining the structures of protein-ligand complexes. J. Am. Chem. Soc. 2004; 126:2390-2398.
146. Blaise BJ, et al. Metabotyping of Caenorhabditis elegans reveals latent phenotypes. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:19808-18912.
147. Reckel S, Lohr F, Dotsch V. In-cell NMR spectroscopy. Chem-BioChem. 2005; 6:1601-1606.
148. Serber Z, et al. In-cell NMR spectroscopy. Methods Enzymol. 2005; 394:17-41.
149. Serber Z, et al. Investigating macromolecules inside cultured and injected cells by in-cell NMR spectroscopy. Nat. Protoc. 2006; 1:2701-2709.
150. Selenko P, Wagner G. Looking into live cells with in-cell NMR spectroscopy. J. Struct. Biol. 2007; 158:244-253.
151. Lenz EM, Wilson ID. Analytical strategies in metabonomics. J. Proteome Res. 2007; 6:443-458.
152. Robertson DG, Reily MD, Baker JD. Metabonomics in pharmaceutical discovery and development. J. Proteome Res. 2007; 6:526-539.
153. Lindon JC, Holmes E, Nicholson JK. Metabonomics in pharmaceutical R&D. FEBS J. 2007; 274:1140-1151.
154. Mulder WJM, et al. Lipid-based nanoparticles for contrast- enhanced MRI and molecular imaging. 2006; 142-164.