CHEMICAL BIOLOGY
Artificial Metalloproteins, Design and Engineering of
Yi Lu, Dewain K. Garner and Jun-long Zhang, University of Illinois Urbana-Champaign,, Illinois
doi: 10.1002/9780470048672.wecb397
Engineering of artificial metalloproteins is an expanding field with potential impacts in many areas from fundamental understanding of protein structure and function to industrial production of specialty chemicals such as chiral drug intermediates. Incorporation of unnatural amino acids and non-native metal cofactors into proteins is an emerging field in the area of protein design, as it offers the tantalizing prospect of introducing new functionality and provides exquisite probes for and fine-tuning of native protein properties. Although it is a relatively young field, engineering of non-native metalloproteins boasts a myriad of techniques for design, construction, and/or expression of the desired artificial protein. Additionally, many groundbreaking studies exist in this field that have enhanced basic scientific understanding of protein function or generated promising artificial enzymes. Discussion of the techniques prominent for incorporation of unnatural amino acids and non-native cofactors is followed by some of the most interesting applications of these techniques reported to date.
Engineering of artificial metalloproteins is an expanding field with potential impact in many areas from fundamental understanding of protein structure and function to industrial production of specialty chemicals such as chiral drug intermediates (1-8). Native metalloproteins use 20 natural amino acids and physiologically available metal cofactors such as heme and cobalamin for their functions. Incorporation of unnatural amino acids and non-native metal cofactors into proteins is an emerging field in the area of protein design, as it offers the tantalizing prospect of introducing new functionality and provides exquisite probes for and fine-tuning of native protein properties beyond what is capable in the native proteins (4, 6, 8-10).
Although it is a relatively young field, design and engineering of artificial metalloproteins boasts a myriad of techniques for design, construction, and/or expression of the desired artificial protein. Additionally, many groundbreaking studies exist in this field that have enhanced basic scientific understanding of protein function or generated promising artificial enzymes. Much of this success has come because replacing a natural amino acid or cofactor with its unnatural analog, while altering a specific structural or electronic element, may be accomplished without gross perturbation of the protein site it replaces. As the title suggests, metal-containing proteins are the focus of this article, and although many wonderful examples of protein incorporation of unnatural amino acids and cofactors exist, only those for metalloproteins will be considered in this review. Because the elementary nature and function of amino acids and cofactors differ, it is sensible to discuss them separately. Furthermore, discussion of the techniques prominent for incorporation of unnatural amino acids and non-native cofactors will be followed by some of the most interesting applications of these techniques reported to date.
Approaches to Unnatural Amino Acid Incorporation into Proteins
Many methods have been developed to incorporate unnatural amino acids into metalloproteins, and these methods can be classified as either biological or synthetic in nature. Biological approaches rely on cellular machinery for placement of the unnatural amino acid within the protein sequence. Recent advances in molecular cloning and biology are the essence of these techniques. The simplest of these is cavity complementation in which a metal ligand is mutated to a Gly or Ala to create a cavity that can then be titrated by exogenous ligands
(11). The use of auxotrophs (bacteria lacking the ability to produce a specific amino acid) can be used for global replacement of a natural amino acid with a structurally similar unnatural analog (12). Finally, and more excitingly, it is now possible to use the amber stop codon and tRNAs that have been specially selected to charge with an unnatural amino acid to place the unnatural amino acid exactly at the coded position in the peptide sequence (13).
Synthetic approaches use chemical transformations on either native proteins, peptides obtained by solid-state synthesis, or both. Chemical modification is the simplest of these and requires the treatment of the selected peptide with a chemical reagent that alters reactive amino acid side chains such as Ser, Lys, or Cys (14). Because that particular amino acid at all positions of the protein is modified, it is difficult to make the chemical modification site-specific. Total solid phase peptide synthesis allows for site-specific incorporation of unnatural amino acids at any specific location (15), but it is limited in length, because the synthesis efficiency decreases with increased length of the peptide chain. This limitation has been eased by native chemical ligation which exploits the reactive properties of an N-terminal cysteine to connect two or more shorter synthetic peptides covalently via a peptide bond (16). The biological and synthetic approaches are complimentary and the combination of these two approaches has proven very useful. One excellent example is the semisynthetic technique of expressed protein ligation that couples a bacterially expressed peptide with a synthetic peptide in a fashion similar to that used for native chemical ligation (17). This hybrid of synthetic and biological techniques allows easy site-specific introduction of unnatural amino acids into much longer peptide sequences (18).
Design and engineering of metalloproteins that contain unnatural amino acids using the biological approach
The method of cavity complementation has been employed effectively to introduce unnatural amino acids as metal ligands into the oxygen storage protein myoglobin (11, 19) and into the electron transfer protein azurin (20). In the case of myoglobin, the proximal histidine that coordinates to the iron atom was removed, which generated a cavity that was titrated by neutral thiol and thioether ligands. Introduction of these unnatural amino acids demonstrated that ferrous heme iron proteins can be ligated by a neutral cysteine. These observations are important because they demonstrate that ferric heme proteins that apparently lose cysteinate ligation on reduction may in fact be ligated by neutral cysteine. Thus, ligation of the heme iron by unnatural amino acids has demonstrated the existence of a ligation state previously only hypothesized to exist and which significantly alters our understanding of the electronics and reactivity of the very important P450 class of enzymes. In the case of azurin, removal of one of the copper ligating histidines generated a cavity near the solvent exposed surface of the protein. It was shown that altering the type of unnatural amino acid titrated into the protein produced variations in the spectroscopic characteristics that encompass the known range of mononuclear copper-thiolate proteins. From this study, it became apparent that the protein scaffold displays preference for a few distinct geometries of the metal center. It is also important that the spectroscopic properties of the native protein could be restored by titration of the cavity with imidazole, which indicates that the introduction of the cavity had not altered the protein structure irreversibly. The use of unnatural amino acids has thus demonstrated that despite the wide variation in naturally occurring copper proteins, a few specific metal ligand geometries may be responsible for all variations in function.
The auxotrophic method has been used in the study of the monooxygenase P450 enzymes to replace all of the methionine residues with norleucine (Nle) (21). Although the Met to Nle substitution resulted in decreased thermal stability of the enzyme, a roughly two-fold increase in peroxygenase activity was observed. This was noteworthy because peroxygenase activity, which is the oxidation of organic substrates using hydrogen peroxide, is normally inefficient in the monooxygenase P450s. The increase in activity was attributed to the removal of the methionine residues, which are oxidized easily by hydrogen peroxide, resulting in enzyme inactivation. This study demonstrates the power of unnatural amino acids to tune the properties of native enzymes by introducing novel properties without sacrificing natural catalytic function.
The incorporation of unnatural amino acids into metallo- proteins via tRNA by using the amber stop codon has also contributed to the design of artificial metalloproteins. Using this technique, a para-cyano-L-phenylalanine was incorporated into myoglobin in place of the distal histidine (22). The cyano group on this unnatural amino acid proved to be a probe of ligand binding and local electronic environment, which distinguishes between linear and bent configurations of different diatomic heme ligands (i.e., O2, NO, and CO). This report demonstrated site-specific incorporation of a very useful probe via unnatural amino acids that might also find application in the study of protein folding and conformation, electronic fields within proteins, or biomolecule interactions. Site-specific modification of cytochrome c3 with a redox mediator was also accomplished using the amber stop codon encoding for unnatural amino acids (23). To achieve this goal, a para-propargyloxyphenylalanine unnatural amino acid introduced on the surface of the protein was reacted with an azido-viologen. The covalent anchoring of the redox active viologen near the hemes of cytochrome c3 was shown to increase the amount electron transfer between the solution and the protein cofactor by three fold. This report expands the number of modifications that can be made site specifically in a metalloprotein because the unnatural amino acid used can react with any azide-bearing molecule. The development of a system for introducing a selectively reactive unnatural amino acid near a c-type heme is important and applicable to the study of other heme proteins. These studies demonstrate the use of the tRNA method for incorporating unnatural amino acids, and an increase in the number of studies using this technique is anticipated.
Design and engineering of metalloproteins that contain unnatural amino acids using synthetic techniques
An excellent example of unnatural amino acid introduction via a synthetic technique may be found in work performed on the protease subtilisin. Chemical conversion of a catalytically active serine to a selenocysteine altered the catalytic activity significantly (24). Instead of native hydrolysis activity, which is greatly suppressed, selenosubtilisin promotes acyl transfer. Surprisingly, selenosubtilisin also catalyzes the reduction of peroxides in a highly enantioselective fashion, and it has even been used for resolution of functionalized racemic peroxides with up to 99% enantioselectivity in the alcohol produced (25). The methodology for incorporation of selenium on the catalytically active serine position has even been extended down the periodic table to incorporate tellurium (26) at this position. The new tellurosubtilisin displays similar reactivity to selenosubtilisin but with altered kinetic properties. The dramatic changes in reactivity observed in subtilisin on incorporation of selenium or tellurium demonstrate the range of protein properties accessible by incorporation of unnatural amino acids.
Although limited by the size of protein it can make, the total synthesis of proteins has contributed significantly to the study and the design of metalloproteins. Replacement of two histidine residues in a de novo designed four-α-helix bundle with 4-β-(pyridyl)-L-alanines resulted in ~6 x 104 weaker binding of the ferric heme and a near 300 mV increase in the reduction potential (Fig. 1a) (28). Changing this same position to 1-methyl-L-His causes >7 x 105-fold decrease in ferric heme affinity but only 125-fold decrease for ferrous heme (29). The methylated histidine unnatural amino acid also prevented the favored heme coordination geometry and resulted in a five-coordinate high-spin ferrous heme similar to deoxymyo- globin that could bind CO reversibly but not O2 (Fig. 1a). These substitutions of unnatural amino acids reveal the preferential binding of different redox states of the heme cofactor and provide insight into the importance of the protein environment for O2 transport. The use of unnatural amino acids with increased steric bulk to influence coordination geometry and to promote open binding sites on a metal center has also been used to control the properties of a ferredoxin model maquette. Replacement of the four Cys residues that ligate the iron atom with penicillamines (Pen) shifts the metal coordination environment from a symmetric (S-Cys)4 to an asymmetrical (S-Pen)3-(H2O) ligation (30). The shift is attributed to the increased steric bulk of the Pen, which has methyl groups in place of the β-methylene hydrogens of Cys. The increased bulk of Pen was also used to change the coordination geometry of Cd2+ in a helical peptide from four to three coordinate (Fig. 1b) (27). This successful control of the metal coordination geometry indicates clearly that small structural changes can alter the energy landscape of metalloproteins dramatically, and additional study of such systems should disclose information about metal protein interactions, particularly heavy metal insertion into protein frameworks.
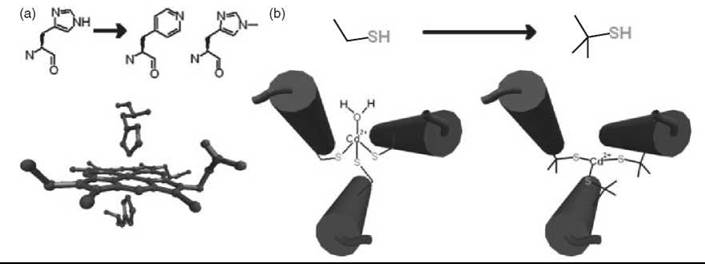
Figure 1. Controlling coordination geometry and metal binding properties using unnatural amino acids in de novo designed four-α-helix bundles. (a) Substitution of histidine with 4-β-(pyridyl)-L-alanines or 1-methyl-,L-histidine. (b) Substitution of cysteine with penicillamine. (Adapted from Reference 3, p. 120 copyright 2005 and Reference 27. Cover art copyright 2006 with permission from Elsevier and Wiley, respectively.)
Similar to total peptide synthesis, the use of native chemical ligation to study larger proteins has met with several successes. In rubredoxin, the replacement of a conserved Tyr near a Cys ligand with unnatural amino acids that substitute -F, -NO2, and -CN, for the para -OH group (Fig. 2a) (31) revealed a linear relationship between the reduction potential of the metal and the Hammett op values (a common measure of the electronic effects of the substituents on aromatic molecules) (31). This work confirmed the supposition that direct interaction of the rubredoxin active site and neighboring amino acids exists and influences the character of the metal site. The implications of this research reach beyond the electron transfer clusters and into nonheme iron or other metalloproteins with metal sulfur ligation and a nearby aromatic group and may provide a way to selectively tune the redox potential of some metalloproteins. In another study that relates reduction potential to secondary coordination environment, unnatural amino acids were used to remove a hydrogen bond from a backbone amide to a cysteine ligand in the (4Fe4S) cluster in high-potential iron proteins (33). A stabilizing effect of such a hydrogen bond on the reduced form of the protein had been predicted to increase the reduction potential. The replacement of the amide in question with an ester linkage lowered the reduction potential of the iron cluster by ~100 mV, in agreement with the predicted stabilizing effect for this hydrogen bond. Again, the use of unnatural amino acids confirmed the existence of subtle interactions between the protein environment and the metal center, and it emphasizes the necessity of including such interactions in protein design and engineering efforts. It is also important to note that an alteration such as exchanging an amide for an ester was impossible with conventional mutagenesis because all natural amino acids contain an amide linkage.

Figure 2. (a) Using native chemical ligation to introduce unnatural tyrosine analogs near the iron sulfur cluster in a rubredoxin. (b) Substituting the unnatural amino acid citruline for arginine alters the DNA recognition of a zinc finger protein. (Adapted from References 31, p. 11536 and References 32, p. 4960, copyright 1998 with permission from The American Chemical Society.)
Design and engineering of metalloproteins containing unnatural amino acids using semisynthetic techniques
Expressed protein ligation (EPL) has been used recently to introduce unnatural amino acids into metalloproteins to examine the effect of individual amino acids on fine-tuning the properties of the metal binding site. In the first report of EPL on a metalloprotein, the Cys in the type 1 blue copper electron transfer protein azurin has been replaced with selenocysteine, which resulted in marked changes in the electronic absorption and electron paramagnetic resonance (34, 35). However, little change was observed in the reduction potential. Conversely, when the axial methionine ligand in azurin is substituted by its structural analogs selenomethionine, norleucine, oxomethionine, trifluoromethionine, or difluoromethionine (Fig. 3), very little change is observed in the spectroscopic properties, but large changes in reduction potential are reported (>200 mV) (36, 37). A linear relationship between the reduction potential and the hydrophobicity of the axial ligand demonstrates that hydrophobicity is the dominant factor contributed by the axial ligand to tuning the reduction potential (36). Extrapolation of these results to studies with conventional mutagenesis demonstrate nearly identical effects of the axial ligand hydrophobicity on the reduction potential in all type 1 blue copper proteins (36). Type 1 blue copper proteins are one of the most common classes of electron transfer proteins, which play critical roles in processes such as photosynthesis and oxidative respiration. The role of the conserved axial methionine has been probed by mutagenesis using all other 19 natural amino acids, but the exact role remained undefined because conventional mutagenesis changes more than one factor, such as electronic and steric factors, at the same time. Using isostructural amino acids, the exact effect of the methionine is more readily defined, and observation of the above trend is only possible by introducing unnatural amino acids using EPL.
Although it is not used to study the metal binding site, EPL has been used to expand the DNA recognition abilities of a zinc-finger protein (32) by replacement of arginine with citrulline (Cit) (Fig. 2b). DNA-binding specificity of a zinc-finger is determined primarily by three amino acids. One such amino acid residue, Arg, recognizes the 5'-most guanosine (G) via donation of two hydrogen bonds. Replacement of the Arg with natural amino acids resulted in similar (as with Lys, which like Arg has only hydrogen bond donors) or decreased specificity (as with Gln, which is shorter than Arg). Replacing Arg with Cit alters the specificity from guanosine to adenosine because although Cit is the same length as Arg, it has one hydrogen bond donor and one acceptor (Fig. 2b). This work extends the application of unnatural amino acids for use in DNA recognition and overcomes an apparent “limitation” of the native system to provide recognition and specificity not achievable with natural amino acids.

Figure 3. Isostructural substitution of cysteine and methionine with unnatural analogs in the metal binding site of the electron transfer protein azurin. (Adapted from Reference 36, p. 15608, copyright 2006 with permission from The American Chemical Society.)
Approaches for Non-Native Cofactor Attachment
Naturally occurring proteins use a myriad of techniques to incorporate and to control metal cofactors. Many proteins such as myoglobins and cytochrome c peroxidase use a noncovalent approach, positioning cofactors such as heme through interactions including electrostatics, hydrogen bonding, amino acid side chain coordination of metals, and hydrophobicity. Some native proteins such as cytochrome c, mammalian peroxidases, and even hemoglobin, attach the heme cofactor to the protein via a covalent bond (or bonds) between amino acid side chains and substituents on the periphery of the heme (38). Inspired by nature’s way of positioning metal cofactors, non-native metal cofactors have been incorporated into proteins through both noncovalent and covalent approaches. Noncovalent introduction of non-native cofactors exploits interactions that are significantly weaker than a covalent bond between atoms. As such, the noncovalent approach is the most demanding in terms of careful design and modification of both the host protein and the prosthetic group. The mutation of bulky residues to less bulky residues may be required to create space for the new cofactor. The introduction of hydrophobic groups or salt bridges may be required to promote tight cofactor binding. One way to accomplish this task is to replace the native metal cofactor heme with a non-native cofactor of similar size and shape. Another general strategy for noncovalent attachment is to exploit strong native biological interactions such enzyme/substrate interactions. Covalent introduction of non-native cofactors relies on the strong interaction of an atom-atom bond to position the cofactor. Generally, this technique seeks a protein cavity and then tethers the cofactor by covalently bonding to a reactive amino acid side chain within the cavity.
Design and engineering of metalloproteins containing non-native cofactors through noncovalent attachment
The binding affinities of substrates and metal ligands have been exploited to attach ruthenium complexes to proteins noncovalently. Both the camphor substrate or an imidazole were covalently attached to a photoactive ruthenium complex via a covalent electron tunneling wire that allows direct interaction of a photogenerated radical with the active site of P450cam (39). Similar imidazole terminated rhenium molecular wires have also been employed with nitric oxide synthase (40). By allowing rapid electron transfer (nanosecond) between the heme and ruthenium complex, intermediates that are very difficult to see using the protein’s native redox partners were observed. Implementation of these unnatural cofactors may be far reaching, and it may allow detailed study of many biological electron transfer processes by removing the diffusion limits and time resolution restrictions of more conventional techniques.
An artificial catalyst that consists of a monoclonal antibody, 1G8, and the achiral rhodium complexes against which it was raised, exploits noncovalent interactions to promote enantioselective catalysis (41). Not only did the antibody bind tightly to the non-native cofactor, but it also provided a chiral environment. Hydrogenation of 2-acetamidoacrylic acid proceeded with 98% ee. The substrate specificity was also very high, and the antibody prevented the hydrogenation of larger substrates. Inclusion of a substrate mimic during antibody generation should improve the activity and substrate scope for the artificial catalyst. Additionally, rational design of the original antibody isolated is also possible. As the first report of asymmetric hydrogenation by a transition metalimmunoglobin complex, this approach holds promise for future development of tightly bound non-native cofactors as catalysts.
The noncovalent approach has been used to replace the native heme in heme proteins such as myoglobin, with non-native protoporphyrin IX modified at the propionate groups. Removal of the native heme followed by reconstitution with the propionate-modified protoporphyrin IX has led to myoglobins that display increased negative charge (42), flavin redox cofactors (43), and glycosylation (44) on the heme edge (Fig. 4). These modified hemes modulate a host of properties such as protein-protein interaction, oxygen activation, small molecule recognition, and electron transfer. Extensive reviews that contain the specifics of each heme modification are recommended to the reader (9). The propionate-substituted porphyrins allow much more direct contact between the heme and the solution outside the protein than occurs with only native heme. In a similar strategy, introduction of a porphycene in place of the native heme led to a myoglobin with dramatically enhanced dioxygen affinity (45). These authors show that direct alteration of the cofactor may be more effective than conventional mutagenesis for improving the properties of a native metalloprotein. The role of the formyl substituent on heme a has also been probed by comparing heme b and a heme a mimic inside a four-α-helix bundle (46). It was demonstrated that a major function of the formyl substituent is to raise the reduction potential of the heme, which it does by destabilizing the binding of the ferric state (47). This work with non-native cofactors is valuable because it is difficult to probe the thermodynamics of binding, oxidation reduction events, and variance of the heme type simultaneously in the native system. Lastly, a four-α-helix bundle that binds a nonbiological metalloporphyrin selectively over native heme has been reported, which demonstrates that proper design can actually favor the non-native cofactor (48, 49). The design of a peptide that can bind a non-native cofactor selectively takes us closer to the rational design of a metalloprotein from the ground up and provides an important testing ground for our knowledge of protein structure and function.
Reconstitution of heme proteins with modified hemes relies heavily on the strong protein-heme interaction and incorporation of non-native cofactors that bear less structural similarity to heme is more challenging. The metal ligand affinity of the proximal His in myoglobin for a Cu(II) N-salicylideneaminoacidato compound has been used for noncovalent attachment of this complex (50) demonstrating that noncovalent attachment to a protein is possible even if the new metal cofactor does not resemble heme. Although the metal Schiff base complexes Mn(III)salen and Cr(III)salophen are roughly planar and similar in dimension to heme, a structural-based design to modify both the metal complex (through substitution of the 5 and 5' positions) and protein (mutation of Ala71 with Gly) was required (Fig. 4) (51) to obtain cofactor incorporation. Crystal structures with an Fe(III)salophen derivative reveal disorder of the non-native cofactor induced by steric repulsion from Ala71 (52). In catalytic sulfoxidation, the modification of His64 to Asp allowed the Mn(III) derivative to produce enantioselectivity up to ~30% (53). Such detailed analysis of an artificial cofactor in a protein has allowed these authors to propose substrate-binding locations and to delineate substrate and cofactor interactions that may be responsible for the catalytic activity. Application of similar techniques allowed the heme oxygenase incorporation of Fe(III)salen (54). This non-native cofactor composite demonstrated the importance of a hydrogen bond to Arg177 for rapid electron transfer in heme oxygenase. Furthermore, the direct participation of the non-native cofactor in electron transfer reactions via the conserved interactions of the native system demonstrates the potential for regulation of non-native cofactors by the native protein environment.

Figure 4. Introduction of non-native metal Schiff base and substituted heme cofactors by noncovalent attachment in myoglobin. (Adapted from Reference 8, p. 6, 7, copyright 2007 with permission from Elsevier.)
A more general noncovalent attachment method is the use of biotin for attaching cofactors to avidin or streptavidin. This technique was first demonstrated by Wilson and Whitesides (55) who generated a biocatalyst capable of asymmetric hydrogenation of α-acetamidoacrylic acid with up to 44% ee using a biotinlated achiral diphosphinerhodium(I) complex and avidin. Lin et al. expanded this concept with a chiral Pyrphos-Rh(I) complex and hydrogenated itaconic acid with up to 48% ee (56). Recently, Letondor et al. (57) have demonstrated the use of this system by the noncovalent attachment of various diphosphine rhodium, ruthenium and iridium complexes to avidin and streptavidin (Fig. 5). The subsequent tuning of the linker and protein cavity has provided high enantiomeric selectivity for several reactions and good conversion. Examples include hydrogenation of acetamidoacrylic acid with 96% ee (58), transfer hydrogenation of p-methyl acetophenone with 94% ee (59), and oxidation of aromatic alcohols (60). A similar system has also been subjected to directed evolution for hydrogenation of acetamido-acrylic acid (6). The stepwise improvement of selectivity to 65% ee demonstrates that the noncovalent attachment of the metal cofactor can be coupled to biological techniques for improvement of the artificial metalloprotein.

Figure 5. Noncovalent attachment of non-native metal cofactors exploiting the strong interaction between biotin and streptavidin for generation of enantioselective hydrogenation biocatalysts. (Reproduced from Reference 57, p. 8321, copyright 2006 with permission from The American Chemical Society.)
Design and engineering of metalloproteins containing non-native cofactors through covalent attachment
The covalent attachment of non-native cofactors to a protein host has the advantage of site-specific incorporation and high yield and is usually achievable with minimal structural modification to the complex or protein host, even if the protein affinity for the cofactor is minimal. A Cu(II) 1,10 phenanthroline, an efficient DNA cleavage agent, was covalently attached via a cysteine to several DNA-binding proteins (Fig. 6a) (61-62). The resulting combination of efficient DNA cleavage by the cofactor and selective DNA positioning by the protein makes an excellent artificial nuclease. The covalent attachment of this same complex to an active site Cys in the adipocyte lipid-binding protein has yielded an artificial esterase that catalyzes enantioselective hydrolysis with 86% ee (62). Although the covalent attachment of a monodentate phosphate Rh(I) COD complex to this same protein yielded an active hydrogenation catalyst, no enantioselectivity was observed (64). Covalent attachment via a reactive Lys has been reported for a copper bis-imidazole ligand tethered to antibody 38 C2 via an anhydride linker (65). The artificial cofactor displays >3000 fold greater hydrolytic activity when complexed to the protein. Last, covalent anchoring via an active site Ser has been exploited to attach Pt-pincer type complexes to the lipase cutinase (66). A suicide inhibitor, a modified p-nitrophenol phosphonate, which covalently attaches to a lipase active site residue (Ser) on hydrolysis was used to form the covalent linkage. Because of the enzyme substrate specificity, this approach may provide chemoselective as well as stoichiometric ligand attachment, and the above examples show great potential for additional design and future site-specific artificial enzymes.
Covalent attachment has also been exploited for protein incorporation of non-native redox active cofactors. A photosensitive rhodium complex has been covalently attached to a cysteine near the heme of cytochrome c (67). The heme of these cytochrome c bioconjugates was photoreducible, which makes it possible for these artificial proteins to be potentially useful in electronic devices. The covalent anchoring, via a disulfide bond, of a redox active ferrocene cofactor has been demonstrated in the protein azurin (68). Not only did conjugation to the protein provide the cofactor with increased water stability and solubility, but it also provided, by means of mutagenesis, a means of tuning the reduction potential of the cofactor. The protein-aided transition of organometallic species into aqueous solution via increased solubility, stability and tuning are important benefits to the construction of artificial metalloproteins.
The above examples of covalent cofactor attachment all employ single-point covalent attachment. However, for some applications, single point attachment may be insufficient to produce the desired system properties. For example, when Mn(III)-salen is covalently attached to papain via a single maleimide linker, less than 10% ee was observed (69). Similarly, when Mn(III)-salen was covalently attached to myoglobin using a single disulfide bond, the resulting enantioselectivity in sulfoxidation was low, 12% ee. However, anchoring the Mn(III)-salen cofactor at two locations within myoglobin (Fig. 1b) increased the enantioselectivity of the artificial enzyme to 51% ee (63). Clearly, this work demonstrates the importance of controlling catalyst positioning within the protein cavity, and dual covalent anchoring is a promising approach if high chemoselectivity and enantioselectivity is the desirable goal of artificial metalloprotein design.
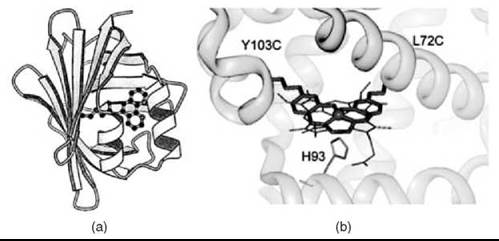
Figure 6. Introduction of non-native metal cofactors by covalent attachment. (a) Computer model of a phenanthroline complex bound to adipocyte lipid binding protein. (b) Computer model of a dual covalently attached Mn Salen. (Reproduced from Reference 62, p. 11644, copyright 1997 and Reference 63, p. 10812, copyright 2004 with permission from The American Chemical Society.)
Conclusions
Great progress has been made in the design and engineering of artificial metalloproteins. The introduction of unnatural amino acids has created new probes of physiologic activity and provided methods for fine-tuning protein properties not available with conventional biochemical techniques. Such techniques have also been used to elucidate the subtle role of key residues in protein metal binding sites as was never possible before. The incorporation of non-native cofactors has enhanced our understanding of protein design by revealing factors, both covalent and noncovalent, that govern cofactor binding and enzyme enantio- and chemoselectivity. Last, novel function has been introduced in metalloproteins. A combination of directed evolutionary, rational design, and combinatorial techniques for the design of non-native metalloproteins has a bright future in biotechnological and pharmaceutical applications.
References
1. Lu Y, Berry SM, Pfister TD. Engineering novel metalloproteins: design of metal-binding sites into native protein scaffolds. Chem. Rev. 2001; 101:3047-3080.
2. Reedy CJ, Gibney BR. Heme protein assemblies. Chem. Rev. 2004; 104:617-649.
3. Lu Y. Design and engineering of metalloproteins containing unnatural amino acids or non-native metal-containing cofactors. Curr. Opin. Chem. Biol. 2005; 9:118-126.
4. Letondor C, Ward TR. Artificial metalloenzymes for enantioselective catalysis: recent advances. ChemBioChem. 2006; 7:1845-1852.
5. Lu Y. Metalloprotein and metallo-DNA/RNAzyme design: current approaches, success measures, and future challenges. Inorg. Chem. 2006; 45:9930-9940.
6. Reetz MT, Peyralans JJP, Maichele A, Fu Y, Maywald M. Directed evolution of hybrid enzymes: evolving enantioselectivity of an achiral Rh-complex anchored to a protein. Chem. Commun. 2006; 41:4318-4320.
7. Goodman CM, Choi S, Shandler S, DeGrado WF. Foldamers as versatile frameworks for the design and evolution of function. Nat. Chem. Biol. 2007; 3:252-262.
8. Ueno T, Abe S, Yokoi N, Watanabe Y. Coordination design of artificial metalloproteins utilizing protein vacant space. Coord. Chem. Rev. 2007; 251:2717-2731.
9. Hayashi T, Hisaeda Y. New functionalization of myoglobin by chemical modification of heme-propionates. Acc. Chem. Res. 2002; 35:35-43.
10. Lu Y. Biosynthethic inorganic chemistry. Angew. Chem. Int. Ed. 2006; 45:5588-5601.
11. Barrick D. Depletion and replacement of protein metal ligands. Curr. Opin. Biotechnol. 1995; 6:411-418.
12. Ikeda Y, Kawahara S-i, Taki M, Kuno A, Hasegawa T, Taira K. Synthesis of a novel histidine analog and its efficient incorporation into a protein in vivo. Protein Eng. 2003; 16:699-706.
13. Xie J, Schultz PG. A chemical toolkit for proteins—an expanded genetic code. Nat. Rev. Mol. Cell Biol. 2006; 7:775-782.
14. Qi D, Tann C-M, Haring D, Distefano MD. Generation of new enzymes via covalent modification of existing proteins. Chem. Rev. 2001; 101:3081-3111.
15. Merrifield B. Concept and early development of solid-phase peptide synthesis. Methods Enzymol. 1997; 289:3-13.
16. Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Synthesis of proteins by native chemical ligation. Science 1994; 266:776-779.
17. Muir, TW. Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 2003; 72:249-289.
18. Muir TW, Sondhi D, Cole PA. Expressed protein ligation: a general method for protein engineering. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:6705-6710.
19. Perera R, Sono M, Sigman JA, Pfister TD, Lu Y, Dawson JH. Neutral thiol as a proximal ligand to ferrous heme iron: Implications for heme proteins that lose cysteine thiolate ligation on reduction. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:3641-3646.
20. den Blaauwen T, Canters GW. Creation of type-1 and type-2 copper sites by addition of exogenous ligands to the Pseudomonas aeruginosa azurin His117Gly mutant. J. Am. Chem. Soc. 1993; 115:1121-1129.
21. Cirino PC, Tang Y, Takahashi K, Tirrell DA, Arnold FH. Global incorporation of norleucine in place of methionine in cytochrome P 450 BM-3 heme domain increases peroxygenase activity. Biotechnol. Bioeng. 2003; 83:729-734.
22. Schultz KC, Supekova L, Ryu Y, Xie J, Perera R, Schultz PG. A genetically encoded infrared probe. J. Am. Chem. Soc. 2006; 128:13984-13985.
23. Iida S, Asakura N, Tabata K, Okura I, Kamachi T. Incorporation of unnatural amino acids into cytochrome c3 and specific viologen binding to the unnatural amino acid. ChemBioChem. 2006; 7:1853-1855.
24. Wu ZP, Hilvert D. Conversion of a protease into an acyl transferase: selenolsubtilisin. J. Am. Chem. Soc. 1989; 111:4513-4514.
25. Haring D, Schueler E, Adam W, Saha-Moeller CR, Schreier P. Semisynthetic enzymes in asymmetric synthesis: enantioselective reduction of racemic hydroperoxides catalyzed by selenosubtilisin. J. Org. Chem. 1999; 64:832-835.
26. Mao S, Dong Z, Liu J, Li X, Liu X, Luo G, Shen J. Semisynthetic tellurosubtilisin with glutathione peroxidase Activity. J. Am. Chem. Soc. 2005; 127:11588-11589.
27. Lee K-H, Cabello C, Hemmingsen L, Marsh ENG, Pecoraro VL. Using nonnatural amino acids to control metal-coordination number in three-stranded coiled coils. Angew. Chem. Int. Ed. 2006; 45:2864-2868.
28. Privett HK, Reedy CJ, Kennedy ML, Gibney BR. Nonnatural amino acid ligands in heme protein design. J. Am. Chem. Soc. 2002; 124:6828-6829.
29. Zhuang J, Amoroso JH, Kinloch R, Dawson JH, Baldwin MJ, Gibney BR. Design of a five-coordinate heme protein maquette: a spectroscopic model of deoxymyoglobin. Inorg. Chem. 2004; 43:8218-8220.
30. Petros AK, Shaner SE, Costello AL, Tierney DL, Gibney BR. Comparison of cysteine and penicillamine ligands in a Co(II) maquette. Inorg. Chem. 2004; 43:4793-4795.
31. Low DW, Hill MG. Rational fine-tuning of the redox potentials in chemically synthesized rubredoxins. J. Am. Chem. Soc. 1998; 120:11536-11537.
32. Jantz D, Berg JM. Expanding the DNA-recognition repertoire for zinc finger proteins beyond 20 amino acids. J. Am. Chem. Soc. 2003; 125:4960-4961.
33. Low DW, Hill MG. Backbone-engineered high-potential iron proteins: effects of active-site hydrogen bonding on reduction potential. J. Am. Chem. Soc. 2000; 122:11039-11040.
34. Berry SM, Gieselman MD, Nilges MJ, Van der Donk WA, Lu Y. An engineered azurin variant containing a selenocysteine copper ligand. J. Am. Chem. Soc. 2002; 124:2084-2085.
35. Ralle M, Berry SM, Nilges MJ, Gieselman MD, Van der Donk WA, Lu Y, Blackburn NJ. The selenocysteine-substituted blue copper center: spectroscopic investigations of Cys112SeCys pseudomonas aeruginosa azurin. J. Am. Chem. Soc. 2004; 126:7244-7256.
36. Garner DK, Vaughan MD, Hwang HJ, Savelieff MG, Berry SM, Honek JF, Lu Y. Reduction potential tuning of the blue copper center in pseudomonas aeruginosa azurin by the axial methionine as probed by unnatural amino acids. J. Am. Chem. Soc. 2006; 128:15608-15617.
37. Berry SM, Ralle M, Low DW, Blackburn NJ, Lu Y. Probing the role of axial methionine in the blue copper center of azurin with unnatural amino acids. J. Am. Chem. Soc. 2003; 125:8760-8768.
38. Stevens JM, Daltrop O, Allen JWA, Ferguson SJ. C-type cytochrome formation: chemical and biological enigmas. Acc. Chem. Res. 2004; 37:999-1007.
39. Dunn AR, Dmochowski IJ, Winkler JR, Gray HB. Nanosecond photoreduction of cytochrome P450cam by channel-specific ru-diimine electron tunneling wires. J. Am. Chem. Soc. 2003; 125:12450-12456.
40. Belliston-Bittner W, Dunn AR, Nguyen YHL, Stuehr DJ, Winkler JR, Gray HB. Picosecond photoreduction of inducible nitric oxide synthase by rhenium(I)-diimine wires. J. Am. Chem. Soc. 2005; 127:15907-15915.
41. Yamaguchi H, Hirano T, Kiminami H, Taura D, Harada A. Asymmetric hydrogenation with antibody-achiral rhodium complex. Org. Biomol. Chem. 2006; 4:3571-3573.
42. Hitomi Y, Hayashi T, Wada K, Mizutani T, Hisaeda Y, Ogoshi H. Interprotein electron transfer reaction regulated by an artificial interface. Angew. Chem., Int. Ed. 2001; 40:1098-1101.
43. Matsuo T, Hayashi T, Hisaeda Y. Reductive Activation of dioxygen by a myoglobin reconstituted with a flavohemin. J. Am. Chem. Soc. 2002; 124:11234-11235.
44. Matsuo T, Nagai H, Hisaeda Y, Hayashi T. Construction of glycosylated myoglobin by reconstitutional method. Chem. Commun. 2006; 29:3131-3133.
45. Hayashi T, Dejima H, Matsuo T, Sato H, Murata D, Hisaeda Y. Blue myoglobin reconstituted with an iron porphycene shows extremely high oxygen affinity. J. Am. Chem. Soc. 2002; 124:11226-11227.
46. Zhuang J, Amoroso JH, Kinloch R, Dawson JH, Baldwin MJ, Gibney BR. Evaluation of electron-withdrawing group effects on heme binding in designed proteins: implications for heme a in cytochrome c oxidase. Inorg. Chem. 2006; 45:4685-4694.
47. Zhuang J, Reddi AR, Wang Z, Khodaverdian B, Hegg EL, Gibney BR. Evaluating the roles of the heme a side chains in cytochrome c oxidase using designed heme proteins. Biochemistry 2006; 45:12530-12538.
48. Cochran FV, Wu SP, Wang W, Nanda V, Saven JG, Therien MJ, DeGrado WF. Computational de novo design and characterization of a four-helix bundle protein that selectively binds a nonbiological cofactor. J. Am. Chem. Soc. 2005; 127:1346-1347.
49. Bender GM, Lehmann A, Zou H, Cheng H, Fry HC, Engel D, Therien MJ, Blasie JK, Roder H, Saven JG, DeGrado WF. De novo design of a single-chain diphenylporphyrin metalloprotein. J. Am. Chem. Soc. 2007; 129:10732-10740.
50. Abe S, Ueno T, Reddy PAN, Okazaki S, Hikage T, Suzuki A, Yamane T, Nakajima H, Watanabe Y. Design and structure analysis of artificial metalloproteins: selective coordination of His64 to copper complexes with square-planar structure in the apo-myoglobin scaffold. Inorg. Chem. 2007; 46:5137-5139.
51. Ohashi M, Koshiyama T, Ueno T, Yanase M, Fujii H, Watanabe Y. Preparation of artificial metalloenzymes by insertion of chromium Schiff base complexes into apomyoglobin mutants. Angew. Chem. Int. Ed. 2003; 42:1005-1008.
52. Ueno T, Ohashi M, Kono M, Kondo K, Suzuki A, Yamane T, Watanabe Y. Crystal structures of artificial metalloproteins: tight binding of FeIII(Schiff-Base. by mutation of Ala71 to Gly in apo-myoglobin. Inorg. Chem. 2004; 43:2852-2858.
53. Ueno T, Koshiyama T, Ohashi M, Kondo K, Kono M, Suzuki A, Yamane T, Watanabe Y. Coordinated design of cofactor and active site structures in development of new protein catalysts. J. Am. Chem. Soc. 2005; 127:6556-6562.
54. Ueno T, Yokoi N, Unno M, Matsui T, Tokita Y, Yamada M, Ikeda-Saito M, Nakajima H, Watanabe Y. Design of metal cofactors activated by a protein-protein electron transfer system. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:9416-9421.
55. Wilson ME, Whitesides GM. Conversion of a protein to a homogeneous asymmetric hydrogenation catalyst by site-specific modification with a diphosphinerhodium(I) moiety. J. Am. Chem. Soc. 1978; 100:306-307.
56. Lin C-C, Lin C-W, Chan ASC. Catalytic hydrogenation of itaconic acid in a biotinylated Pyrphos-rhodium(I) system in a protein cavity. Tetrahedron: Asymmetry 1999; 10:1887-1893.
57. Letondor C, Pordea A, Humbert N, Ivanova A, Mazurek S, Novic M, Ward TR. Artificial transfer hydrogenases based on the biotin-(strept)avidin technology: fine tuning the selectivity by saturation mutagenesis of the host protein. J. Am. Chem. Soc. 2006; 128:8320-8328.
58. Collot J, Gradinaru J, Humbert N, Skander M, Zocchi A, Ward TR. Artificial metalloenzymes for enantioselective catalysis based on biotin-avidin. J. Am. Chem. Soc. 2003; 125:9030-9031.
59. Letondor C, Humbert N, Ward TR. Artificial metalloenzymes based on biotin-avidin technology for the enantioselective reduction of ketones by transfer hydrogenation. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:4683-4687.
60. Thomas CM, Letondor C, Humbert N, Ward TR. Aqueous oxidation of alcohols catalyzed by artificial metalloenzymes based on the biotin-avidin technology. J. Organomet. Chem. 2005; 690:4488-4491.
61. Chen C-hB, Milne L, Landgraf R, Perrin DM, Sigman DS. Artificial nucleases. ChemBioChem. 2001; 2:735-740.
62. Davies RR, Distefano MD. A semisynthetic metalloenzyme based on a protein cavity that catalyzes the enantioselective hydrolysis of ester and amide substrates. J. Am. Chem. Soc. 1997; 119:11643-11652.
63. Carey JR, Ma SK, Pfister TD, Garner DK, Kim HK, Abramite JA, Wang Z, Guo Z, Lu Y. A site-selective dual anchoring strategy for artificial metalloprotein design. J. Am. Chem. Soc. 2004; 126:10812-10813.
64. Panella L, Broos J, Jin JF, Fraaije MW, Janssen DB, Jeronimus- Stratingh M, Feringa BL, Minnaard AJ, de Vries JG. Merging homogeneous catalysis with biocatalysis; papain as hydrogenation catalyst. Chem. Commun. 2005; 45:5656-5658.
65. Nicholas KM, Wentworth P, Harwig CW, Wentworth AD, Shafton A, Janda KD. A cofactor approach to copper-dependent catalytic antibodies. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:2648-2653.
66. Kruithof CA, Casado MA, Guillena G, Egmond MR, van der Kerk-van Hoof A, Heck AJR, Gebbink RJMK, van Koten G. Lipase active-site-directed anchoring of organometallics: metallopincer. Chem. Eur. J. 2005; 11:6869-6877.
67. Peterson JR, Smith TA, Thordarson P. Photoinduced reduction of catalytically and biologically active Ru(II)bisterpyridine-cytochrome c bioconjugates. Chem. Commun. 2007; 19:1899- 1901.
68. Hwang HJ, Carey JR, Brower ET, Gengenbach AJ, Abramite JA, Lu Y. Blue ferrocenium azurin: an organometalloprotein with tunable redox properties. J. Am. Chem. Soc. 2005; 127:15356-15357.
69. Reetz MT, Rentzsch M, Pletsch A, Maywald M. Towards the directed evolution of hybrid catalysts. Chimia 2002; 56:721-723.
Further Reading
Benson DE, Wisz MS, Hellinga HW. The development of new biotechnologies using metalloprotein design. Curr. Opin. Biotechnol. 1998; 9:370-376.
Forum on Biomolecular Design. Inorg. Chem. 2006; 45:9927-10012.
Lu Y. Metalloprotein Design and Engineering, Volume V. In: Encyclopedia of Inorganic Chemistry, 2nd ed. King RB, ed. 2006. Wiley, Chichester. pp. 3159-3192. http://www.mrw.interscience.wiley.com/eic/.
Lu Y, Berry SM. Protein structure design and engineering. In: Encyclopedia of Life Sciences. 2005. John Wiley & Sons, Ltd: Chichester. http://www.els.net/.
Watanabe Y. Construction of heme enzymes: four approaches. Curr. Opin. Chem. Biol. 2002; 6:208-216.
See Also
Expanding the Genetic Code Through Chemical Biology
Metallo-enzymes and Metallo-Proteins, Chemistry of
Proteins, in vivo Chemical Modification of
Protein Engineering: Overview of Applications in Chemical Biology