CHEMICAL BIOLOGY
Nuclear Receptors, Chemistry of
Lioudmila A. Campbell, Graduate Program in Biological Sciences, Department of Physiology, University of California, San Francisco, California
Holly A. Ingraham, Departments of Cellular and Molecular Pharmacology and Physiology, University of California, San Francisco, California
doi: 10.1002/9780470048672.wecb402
Nuclear hormone receptors are integral players in endocrine networks that lie at the interface between biology and chemistry. Unlike most other classes of transcription factors, these proteins are designed uniquely to bind small molecules and, thus, affect gene expression in response to the cellular and organismal chemical environment. After several decades of research, it is now appreciated that nuclear receptors bind very diverse lipophilic small molecules with a wide range of specificity and affinities. Recent nuclear receptor structures coupled with large-scale screening efforts challenge the dogma that all nuclear receptors, especially the large subset of constitutively active receptors, will have ligands and will represent tractable drug targets. As such, the ''pharmacologic future'' for such orphan nuclear receptors may reside outside of the ligand-binding pocket.
Nuclear hormone receptors are classically defined as ligand- regulated transcription factors. The transcriptional programs affected by these proteins are linked to metabolic pathways, endocrine homeostasis, and organ development; thus, both the loss and the gain of function of these receptors are associated closely with a variety of human diseases that include developmental and metabolic defects, cardiovascular disease, diabetes, reproductive failure, and cancer. Forty-eight nuclear receptors have been identified in the human genome and are subclassified into seven distinct subfamilies that consist of NR1, NR2, NR3, NR4, NR5, NR6, and NR0 based largely on sequence similarity in their two signature domains (1). These two domains are present in almost all nuclear receptors and consist of the N-terminal DNA-binding domain (DBD) and the C-terminal ligand-binding domain (LBD). The DBD interacts with specific DNA elements located in promoters of target genes, whereas the LBD binds hormones or other lipophilic molecules (2). Additionally, receptors include two highly variable domains: the N-terminal domain preceding the DBD and the flexible hinge region between the DBD and the LBD. Currently, no pharmaceutical compound is targeted directly to the DBD or the flexible domains of any nuclear receptor.
Here we will focus on current progress in structural analyses of nuclear hormone receptors, and how these proteins interact with their ligands, both natural and pharmaceutical. We will provide first a general overview of nuclear receptors and then using several nuclear receptors as examples, we will discuss the receptor-ligand specificity throughout the nuclear receptor superfamily and its implications for successful, rational drug design to target the activity of these proteins. Additionally, we will review the emerging drug strategies that target regions outside of the ligand-binding pocket that might potentially provide new therapeutics aimed at this large family of receptors.
Overview
Nuclear receptors are sophisticated homeostatic sensors that function in the endocrine network of vertebrate organisms and allow for communication between or within different tissues and organs, often over large distances. These receptors can detect a constantly changing environment by binding small lipophilic hormones and metabolic intermediates. The ligand dependent feature of some nuclear receptors has been exploited successfully for therapeutic intervention against diseases such as breast cancer, type 2 diabetes, and hypertension (Table 1) (3-39). The use of nuclear receptors to mediate hormone signaling seems to have developed late during metazoan evolution. Indeed, genome-wide comparisons reveal that nuclear receptors are absent in some eukaryotic genomes. However, in those organisms, other signaling pathways have been adapted to meet their endocrine needs and respond to small lipophilic molecules. For instance, no nuclear receptors have been identified in the yeast genome. Interestingly, a protein fold similar to the nuclear receptor LBD was identified by structural prediction in two transcription factors Oaf1 and Pip2 in the budding yeast S. cerevisiae. These transcription factors heterodimerize and bind the fatty acid oleate, reminiscent of the mammalian retinoic X receptor (RXR, NR2B)/peroxisome proliferators-activated receptor (PPAR, NR1 C) signaling pathway (40). Similarly, hormone signaling in multicellular plants is not mediated by nuclear receptors despite the fact that sterols mediate many analogous functions in plant biology. Instead, plants seem to use other ligand binding motifs. For example, the growth promoting plant phytohormone brassinosteroid binds a cell surface receptor that activates downstream kinases and ultimately Myc family transcription factors (41, 42). Another large family of homeodomain-START (star-related lipid-transfer) proteins is hypothesized to affect gene expression directly after binding sterols and lipids selectively via the START domain (43, 44). Collectively, these examples suggest conserved signaling by lipophilic molecules using evolutionarily distinct binding proteins.
Table 1. Nuclear hormone receptors and their ligands
|
Subfamily |
Examples of members |
Endogenous ligands |
Examples of synthetic ligands |
Reference |
|
NR1 |
TR |
Thyroid hormone |
GC-1 |
(3-5) |
|
|
PPAR |
Fatty acids |
GW6471(PPARa), Rosiglitazone (PPARy) |
(6-9) |
|
|
LXR |
Oxysterols |
GW3965, T0901317 |
(10, 11) |
|
|
PXR |
Not known |
Rifampicin, SR12813, Hyperforin |
(12-14) |
|
|
CAR |
Not known |
Androstanol, Phenobarbital, CITCO |
(15-19) |
|
NR2 |
RXR |
Retinoic acid |
GW0791 (RXRa) |
(17, 20, 21) |
|
|
HNF4 |
Fatty acids (?) |
None to date |
(22, 23) |
|
NR3 |
ER |
Estradiol |
Tamoxifen, ICI164,384 |
(24-26) |
|
|
ERR |
Not known |
GSK4716 (ERRy) |
(27, 28) |
|
NR4 |
NGFI-B |
Not known |
None to date |
(29) |
|
|
Nurr1 |
Not known |
None to date |
(29, 30) |
|
NR5 |
SF-1 |
Phospholipids (?) |
GSK8470 |
(31-34) |
|
|
LRH-1 |
Phospholipids (?) |
GSK8470 |
(31, 33-35) |
|
NR6 |
GCNF |
Not known |
None to date |
(1) |
|
NR0 |
DAX-1 |
Not known |
None to date |
(36) |
Ligand Activation of Nuclear Receptors
To carry out the transcriptional programs that require both activation and repression of target genes, nuclear receptors interact with numerous coregulators, which nucleate the assembly of macromolecular protein complexes that remodel chromatin and modulate transcription initiation or silencing (45, 46). For ligand-dependent receptors, the presence or absence of ligand determines the nature of the assembled protein complex. Given the importance of the LBD in binding ligand and interacting with coregulators, collective efforts of academia and industry have now elucidated LBD crystal structures for nearly all seven subfamilies (47). The nuclear receptor LBD structure is conserved and consists of an a-helical bundle (α1-α12), one to five P-strands, with three to four antiparallel layers and a hydrophobic ligand-binding pocket that occupies the core of the bundle (20). The volume of this pocket varies greatly among the receptors, which enables these proteins to accommodate ligands of varying shapes and sizes (Fig. 1) (47, 48). In addition, the LBD contains a dimerization interface that allows receptors to bind DNA as homodimers or heterodimers (49). And, for some receptors, it is also a key site for interaction with the heat-shock proteins (50).
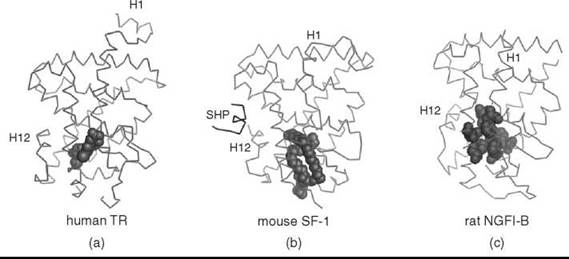
Figure 1. Nuclear receptors can accommodate ligands of various sizes (a) LBD structure of human TR bound to its ligand, triiodothyronine (shown as spheres) (PDB 1XZX) (3). (b) LBD structure of mouse SF-1 bound to a bacterial phospholipid (shown as spheres) and the mouse SHP peptide (PDB 1YMT) (31). (c) LBD structure of rat NGFI-B with an empty ligand-binding pocket; hydrophobic amino acids occluding the ligand-binding pocket are highlighted as spheres (PDB 1YJE) (29). Helix 1 (H1) and helix 12 (H12) for each structure are indicated.
Based on the first crystal structures of liganded nuclear receptors, the “mousetrap model” was proposed to account for ligand-initiated activation (51). Ligand was proposed to complete and stabilize the hydrophobic core of the receptor in an active conformation of the LBD. Concomitant with binding of the ligand, helix H12 that contains the Activation Function 2 (AF2) undergoes a dramatic rearrangement, docking across the ligand-binding pocket and trapping the ligand inside (51). This repositioning of helix H12 creates a new hydrophobic surface (52, 53) that is bound by the LXXLL motif within coactivator proteins (Fig. 2a) (54, 55). Interestingly, corepressor proteins compete with coactivators for binding to the same hydrophobic groove but form a slightly extended surface that eliminates the need for ligand (6, 56). This mechanism allows the ligand to dictate nuclear receptor action by repositioning AF2 and thus shifting the equilibrium between coactivator and corepressor binding (Fig. 2b) (57). However, many nuclear receptors, especially orphan receptors, are constitutively active in the apparent absence of a ligand. Moreover, structural studies point to seemingly small receptor-specific differences within the LBDs that must underlie the diversity of receptor action in controlling distinct biologic processes (47).

Figure 2. Helix 12 position differs in agonist- and antagonist-bound receptors. (a) LBD structures of human PPAR bound by an agonist ligand and a coactivator peptide (SRC-1) (PDB 1K7L) or an antagonist ligand and a corepressor peptide (SMRT) (PDB 1KKQ) (6, 7). (b) Helix 12 structure from agonist- and antagonist-bound receptor shown in A. (c) LBD structures of human ER bound by an agonist, 17β-estradiol (PDB 1ERE), and rat ER bound by an antagonist, ICI 164,384 (PDB 1HJ1) (24, 26). Helix 1 (H1) and helix 12 (H12) for each structure are indicated; ligands are shown as spheres.
Thus far, about half of all nuclear receptors have been paired with physiologic ligands. The other half remain orphaned, and either await identification of their native ligands or alternatively will never be bound by a ligand. For the most part, matching ligands with their cognate receptors has followed traditional drug discovery approaches using both cell-based assays and biologic clues. Although nuclear receptors are found readily in tractable genetic model organisms, such as flies and worms (58), hunting for ligands by standard genetic screens has proven difficult and may reflect an overrepresentation of receptors that belong to the so-called “orphan receptor” subfamilies in these invertebrate species. Exceptions include the discovery of the ecdysone hormone receptors and heme receptors in Drosophila (59), and 3-keto-sterols as ligands for the C. elegans nuclear hormone receptor DAF-12 involved in regulating lifespan (60, 61).
Despite the collective efforts of academia and pharmaceutical enterprises, ligands have remained elusive for many receptors. A feature of these so-called “orphan receptors” is their constitutive activity as evident by the robust, induced activity after overexpressing these receptors in a cellular reporter assay. In the absence of ligands, the best insights into the role of most of these receptors in vertebrate development and physiology comes from engineered mutants in mice or naturally occurring human mutations. Whether obligate ligands exist for the divergent but conserved ligand-binding pockets of all nuclear receptors is debated. Moreover, no clear consensus exists on the evolution of ligand dependence; two opposing hypotheses have been proposed. The first hypothesis suggests that primordial nuclear hormone receptors were ligand-independent, and regulation by specific, high affinity ligands evolved later, several times during the evolution of the nuclear receptor superfamily (62). Consistent with this notion, many orphan receptor LBD structures reveal an active conformation with the AF2 containing helix H12 packed against the LBD, and with ligand-binding pockets that are either small or absent because of the presence of bulky hydrophobic amino acids (Fig. 1c) (63). These active but empty orphan receptors may represent an intermediate state as receptors were transitioning from ligand independence to ligand dependence (62).
The second hypothesis suggests that the ancestral nuclear receptors were ligand dependent, and throughout evolution particular receptors lost the need for ligand activation. The idea that primordial nuclear receptors were responsive to estrogens is consistent with this notion (64). Moreover, the rodent lineage of the NR5A subfamily that includes Steroidogenic factor 1 and Liver receptor homolog 1 (SF-1, NR5A1 and LRH-1, NR5A2) exhibits specific features that diminish ligand binding, which suggests that loss of ligand dependency occurred late in evolution. Structures of human and mouse NR5A subfamily LBDs revealed bacterial phospholipid ligands (Fig. 1b) (31-33, 35), except for the rodent LRH-1 where a key glycine residue has been replaced with a glutamate; the resulting salt bridge at the mouth of the ligand-binding pocket stabilizes the rodent LBD without a need for ligand. Taken together, it suggests that the ancestral NR5A receptor was regulated by a ligand. Clearly, the debate on whether nuclear receptors evolved to bind ligand or not is currently unresolved. Nonetheless, the collective structural and cellular data establish definitively that the binding capacity and ligand requirement vary drastically among the LBDs within all seven receptor subfamilies. More importantly, newly available structures of orphan nuclear receptor LBDs beg the question as to how tractable all nuclear receptors will be as drug targets? Below we will illustrate both the successes and the challenges of ligand discovery for different nuclear receptor proteins.
ER: The Drug Target Darling
Given the significant role of steroid receptors in human biology and disease, especially breast and prostate cancer, it is not surprising that some of the first LBD crystal structures were those of the steroid receptors (52). The steroid receptors were also the first to be targeted by pharmaceutical compounds, even before the availability of the high-resolution LBD structures that paved the way for structure-based drug design. The estrogen receptor (ER, NR3A) is the best example of successful manipulation of a nuclear receptor with synthetic ligands. Crystal structures of the ER LBD bound by several distinct ligands reveal the exquisite specificity with which these ligands manipulate ER into active and inactive conformations. Each ER-ligand complex presents a distinct set of structural changes in the position of the AF2 relative to the core LBD, which suggests that standard approaches can be used to design specific agonists or antagonists for this receptor. When bound by the natural ligand estradiol (E2), ER possesses a relatively small and well-defined ligand-binding pocket, and multiple contacts between the receptor and the ligand result in high specificity of interaction (24). These features allow one to design ER modulatory ligands that range from selective ER modulators (SERMs) such as tamoxifen, which exhibit mixed agonist/antagonist properties depending on the tissue or promoter, to complete antagonists such as ICI 164,384 (25, 26). In the latter case, the ER/ICI structure revealed how addition of bulky constituents to an agonist scaffold results in a protrusion from the ligand-binding pocket and movement of the AF2 helix into nonproductive conformation, which provides a paradigm for designing steroid nuclear receptor antagonists (Fig. 2c) (26). Regrettably, this approach has not worked for other ligand dependent receptors. Indeed, in a search for a thyroid hormone receptor (TR, NR1A, Fig. 1a) antagonist for treatment of hyperthyroidism, adding bulky constituents onto the endogenous TR ligand triiodothyronine (T3) does not create a true antagonist as would be predicted from studies on synthetic ER ligands (3, 4). On the other hand, novel synthetic TR agonists have emerged based on the structure of T3 complexed with the LBD (5).
The existence of SERMs raises some intriguing questions: What does the inactive LBD structure mean at a cellular level? Do only active nuclear receptors interact with the genome? Based on the ER LBD structures with tamoxifen and raloxifene, no productive interactions with coactivator proteins are possible because the AF2 helix adopts an inactive conformation (24, 25); however, paradoxically, SERM-bound ER receptors retain transcriptional activity in certain tissues and on certain promoters. Thus, the small overlap in tamoxifen and raloxifene regulated genes when profiled in an osteosarcoma cell line (65) illustrates how diverse the transcriptional outcomes can be for different SERMs. Similarly, an extremely small overlap was noted between groups of genes regulated by tamoxifen and E2 in a uterine cell line, despite the fact that tamoxifen is thought to be a partial agonist in this tissue (66). Although additional studies are needed, these results illustrate how ligands can alter gene expression dramatically. With the onset of new genome-wide technologies, one can begin to examine how promoter occupancy is affected by ligands. Recent studies that use chromatin immunoprecipitation combined with microarray analyses (ChIP/CHIP) reveal that many ER binding sites are located at a great distance from the proximal promoters and that some sites could be bound by the receptor even in the absence of E2 (67). For receptors fortunate enough to have high affinity [ligands, as found for steroid receptors (NR3A, NR3C)], the collective information gathered from these genome-wide approaches is likely to shed new insights into the physiologic consequences of drug and provide for additional refinement of drug structure.
PPAR and LXR: Orphans Adopted by Pharmaceuticals
Peroxisome Proliferator-Activated Receptor (PPAR, NR1C) and Liver X Receptor (LXR, NR1h3) represent two clear examples in which the lack of structural information did not hinder the development of efficacious high affinity pharmaceutical compounds. PPAR and LXR are responsive to glucose and lipid levels, and play important roles in inflammation, cholesterol and lipid metabolism, and energy balance (68-70). Despite the fact that natural ligands for PPAR remain controversial, with fatty acids and ecosinoids as the proposed low affinity endogenous ligands for PPARa (71), highly specific synthetic agonists and antagonists have been developed (Fig. 2a) (6, 7). Indeed, thiazolidinediones and the structurally related fibrates are used widely to treat diabetes and cardiovascular disease (8, 9).
Oxysterols are the proposed endogenous LXR ligands and can bind the ligand-binding pocket of LXR and activate its transcription in cellular assays (10, 72). Additionally, genetic disruption of oxysterol biosynthesis in mice attenuates LXR function greatly (73). Existing synthetic LXR agonists show potential to treat cardiovascular disease, although their collective role in controlling liver and gut metabolism may impose unwanted, off-target effects (10, 11). Oxysterols may not be the only endogenous ligands for LXR. Remarkably, a recent study reports that LXR also acts as a glucose sensor, in which high concentrations of glucose (2 mM) displace oxysterols from the ligand-binding pocket, bind directly to the LBD, and also seem to act synergistically with the synthetic LXR ligand to affect endogenous target gene expression in the liver (74). If true, LXR would be the first intracellular glucose sensor to be discovered and could provide a molecular explanation for the prominent linkage of diabetes with cardiovascular disease. Mechanistically, the authors suggest that glucose binds directly to the LXR LBD, perhaps in combination with oxysterol, or alternatively binds elsewhere in the pocket or on the solvent exposed surface of the LBD to modulate LXR activity allosterically (74, 75). If structural analysis upholds the latter, it would raise an interesting dilemma—how would a hydrophilic molecule, such as glucose or the cellular glucose-6-phosphate, bind tightly into the hydrophobic pocket of LXR? Nonetheless, this finding is provocative and potentially provides a new paradigm for targeting nuclear receptors.
NR5A Receptors: Large Pockets in Search of Large Ligands
The NR5A subfamily of nuclear hormone receptors includes LRH-1 and SF-1, as well as the Drosophila nuclear receptor Ftz-F1. SF-1 is required for endocrine tissue development and sexual differentiation, and it is a major regulator of steroid biosynthesis (76). LRH-1 is essential in embryonic development, and, in adults, it regulates bile acid production, cholesterol transport, and ovarian function (77). All LBD crystal structures of murine and human members of this subfamily revealed large ligand-binding pockets and structural inflexibility as evidenced by the minimal changes observed with or without ligand or coactivator peptide (31-3335, 78). The overall stability of the NR5A subfamily can be explained partially by the presence of an additional stabilizing layer caused by a well-formed and elongated helix H2.
Phospholipids were found in the ligand-binding pockets of mouse and human SF-1 and human LRH-1 and are relatively large (~750Da) compared with other ligands such as steroid derivatives. The lipid tails fit exceptionally well into the ligand-binding pocket and make several specific contacts with helix H12 and the hydrophobic cavity (Fig. 1b) (31, 32). In addition to being integral membrane components, phospholipids also bind in the ligand pocket of START domain and in phosphatidyl inositol transport proteins (79). All NR5A receptors exhibit constitutive activity in cells, thus it is unclear whether these ligands serve simply to stabilize the LBD helical bundle or whether they act as regulatory ligands. Notably, filling the pocket with bulky residues diminishes ligand uptake in biochemical assays (31) and also attenuates transcriptional activity in cells (32, 33, 35). The challenge in designing synthetic ligands for NR5A receptors is two-fold. First, finding a ligand that recapitulates the positioning of the acyl chains and the phosphate head group might be problematic; and second, whereas the SF-1 LBD protein readily exchanges the bacterial phosphatidyl glycerol with PIP3 or PIP2 (HAI, unpublished data), and might be bound naturally by phosphatidic acid (80), displacing the endogenous phospholipid with a small molecule in a cellular environment might prove difficult. However, a recent report describes a small molecule that at nanomolar concentrations promotes coactivator peptide recruitment to SF-1 and LRH-1, displaces the phospholipid ligand, and evokes a modest increase in endogenous target gene activation in human hepatocytes (34). These studies suggest that, perhaps, this family of nuclear receptors is still tractable for drug discovery.
PXR and CAR: Too Much Receptor for a Single Ligand
Pregnane X receptor (PXR, NR1I2) and the constitutive androstane receptor (CAR, NR1I3) are highly promiscuous nuclear receptors that bind a variety of structurally diverse compounds. Thus, no difficulty exists in identifying ligands for these receptors—finding highly specific ligands seems to be the challenge; which is true especially for PXR and most likely reflects its role in the xenobiotic response. Several different compounds that range from small hydrophobic drugs to the large antibiotic rifampicin are accommodated in its ligand-binding pocket (12-14). Five expandable β-sheets, unique to PXR, allow for this dramatic increase in the size of the ligand-binding pocket (12). Interestingly, a similar structural feature is also found in START domain proteins and may represent a critical structural arrangement for binding a wide variety of lipophilic molecules (79). CAR exhibits a large but empty ligand-binding pocket and high constitutive activity that results from two structural features: an aX helix that precedes helix H12 that stabilizes AF2 in an active conformation and an extended helix H2, similar to the NR5A receptors (15-18, 21). For an organism, the promiscuity of PXR and CAR activation is an indispensable feature because it assures protection from a variety of harmful xenobiotics and metabolites. However, this characteristic also presents a formidable challenge to rational drug design. Once again, and as found with TR, bulky constituents added onto existing PXR agonist scaffolds fail to yield suitable antagonists (81). For CAR, it appears that a significant mode of regulation occurs by shuttling between the nucleus and cytoplasm rather than by ligand activation (82). Interestingly, inverse agonists or ligands that reduce the constitutive activity of CAR have been reported (15, 19). Whether natural ligands exist for PXR and CAR remains unclear, and it may be more likely that these receptors are designed to sample their chemical environment constantly, and protect the organism from harmful cellular metabolites or from environmental toxins.
True Orphans Without Pockets
Finally, structural information on other receptor subfamilies reveals some receptors either to be complexed with “structural non-exchangeable ligands” or to simply have inadequate capacity in their pockets to accommodate the smallest of ligands. To date, hepatocyte nuclear factor 4 (HNF4, NR2A) is an example of a receptor with a structural ligand. Structures of the rat HNF4a (NR2A1) and human HNF4γ (NR2A2) LBDs showed a mixture of bacterial fatty acids that occupy the ligand-binding pocket (22, 23). Although HNF4 is found complexed with only a small selection of fatty acids among an assortment of many, this fatty acid ligand is entrenched completely in the ligand-binding pocket and is dislodged only after complete denaturation of the protein, which suggests that in vitro approaches to ligand identification may not be feasible. Similar to this finding, the phospholipid ligand in human LRH-1 is also resistant to in vitro exchange with other phospholipids perhaps, presenting another case of a structural ligand for a nuclear receptor (35).
As mentioned above, the lack of a conventional hydrophobic cavity makes the ligand hunt extremely difficult. Two receptor subfamilies appear to be “pocketless,” including members of NR4A ([NGFI-B/Nurr77, NR4A1], [Nurr1, NR4A2], [NOR1, NR4A3]) and their fly ortholog DHR38, and members of the NR0 subfamily including Dax-1 (NR0B1). All three structures of NR4 LBDs adopt a canonical protein fold but lack any ligand-binding pocket due to obstruction by bulky side chains (Fig. 1c). These LBDs also lack a hydrophobic coactivator cleft that is instead replaced with a charged surface (29, 30, 83). Another case of an empty pocket is the new structure of the atypical orphan nuclear receptor Dax-1 complexed with LRH-1. Both Dax-1 and SHP (NR0B2) lack a DBD altogether and thus rely on interactions with other NRs and transcription factors to be recruited to the DNA, but both are potent repressors in cellular reporter assays (36, 84). From the crystal structure, it is evident that the ligand-binding pocket of Dax-1 (80 A3) cannot accommodate even the smallest ligand (E. Sablin and R. J. Fletterick, personal communication). Based on this structure and given the high identity with Dax-1, SHP is also predicted to be refractory to ligand regulation.
Finding pharmaceutical ligands for receptors with very small pockets still remains a feasible option as illustrated by recent discovery of a synthetic agonist for the estrogen-related receptor y (ERRγ, NR3B3). ERRγ is a constitutively active nuclear receptor with no known natural ligand, and the crystal structure of the ERRγ LBD revealed an extremely small ligand-binding pocket (220 A3) (27). Remarkably, in a new crystal structure of ERRγ LBD with a synthetic agonist, GSK4716, the ligand-binding pocket expanded to a notable 610 A3 (28). This result underscores the ability of the LBD to accommodate ligands of varying size, and suggests that continuing the hunt for ligands might yield some future surprises.
Alternative Surfaces for Regulation
Despite the fact that ligand discovery has focused historically on the LBD; emerging data suggest that alternative surfaces might be targeted to regulate receptor activity. Alternative binding surfaces have been suggested by structural studies on the NR4A subfamily member, Nurr1. Nuclear magnetic resonance footprinting studies of Nurr1 LBD with peptides derived from the nuclear receptor corepressor (NCoR) and a related corepressor SMRT identified a hydrophobic binding site on the surface of the LBD between helices H11 and H12 (85). Mutational disruption of this interaction surface abolished transcriptional activity of Nurr1 underscoring its importance in Nurr1 function. Because the canonical coactivator groove is absent in the NR4A subfamily, this additional LBD surface is possibly the major site for interaction with the coregulators. On that note, it is of interest that crystal structures of the rat farnesoid X receptor LBD (FXR, NR1H4) and the human LRH-1 LBD revealed two coactivator LXXLL peptides bound to the receptor (31, 33, 86); in these cases, the relevance of this additional bound peptide remains to be determined.
New pharmaceuticals might act by covalent modification of a key protein-protein interaction surface, by blocking an interaction surface, or by affecting the ligand-binding pocket allosterically, as suggested for glucose binding to LXR. Presumably, for most interactions, one would disrupt the assembly of receptor-coregulator complexes and in essence mimic conventional antagonists (87, 88). Rodriguez et al. (89) synthesized a small molecule inhibitor of coactivator binding that structurally mimics key contacts of a coactivator LXXLL motif with the hydrophobic binding groove of the nuclear receptor. The authors used a crystal structure of agonist-bound ERα in a complex with a coactivator peptide to guide small molecule design followed by a screen to identify molecules that abolish peptide recruitment but do not directly compete with ligand binding. A similar high-throughput approach was used to identify novel covalent inhibitors of TRP (NR1A2), β-aminoketones. These inhibitors irreversibly react with a cysteine residue located in the coactivator groove of TRβ LBD thus disrupting the interaction between TRP and an LXXLL-containing coactivator peptide (90). TRP has multiple solvent-exposed cysteines on the LBD, yet these compounds show high selectivity towards a single residue, unique to the TR family of receptors. Additionally, some of the tested compounds appear to be isoform-specific, demonstrating vastly different affinities for TRα and TRβ. Similar to these findings, 4-hydroxytamoxifen (OHT) was found to inhibit coactivator recruitment to ERβ and surprisingly, the crystal structure of the ERβ LBD revealed two bound OHT molecules (91). One molecule was bound in the ligand-binding pocket, and another molecule was revealed in the coactivator groove, displacing the AF2 away from the LBD, into inactive conformation (25). While the exact contribution of this external OHT binding site to the antagonistic effects of OHT on ERβ function is unclear, this binding event could be uncovering a subtle structural difference between the two ER isoforms. Finally, another allosteric inhibitor compound has been identified for the androgen receptor (AR, NR3 C4). It shows reversible binding at a novel hydrophobic LBD surface, conserved in other steroid receptors, and this binding allosterically moves the AF2 helix into an inactive conformation (E. Estebanez-Perpina and R. J. Fletterick, personal communication). Collectively, these studies raise the possibility that new drugs may emerge that target additional surfaces other than the hydrophobic ligand-binding pocket.
Summary and Future Directions
The ability of nuclear hormone receptors to bind small molecules with high affinity and high specificity places them squarely at the interface between biology and chemistry. As such, the nuclear receptor field has historically been focused primarily on the identification of regulatory ligands. Now, an alternative approach is needed for those receptors that fail to exhibit classic ligand dependency, but instead appear to be ligand-independent. Domains outside the DBD and the LBD, especially the Activation Function 1 (AF1) offer a regulatory platform for multiple posttranslational events and coregulator interactions (92, 93). Positioning of the AF1 varies among nuclear receptors, suggesting that it has hopped around throughout evolution, and can be found in the variable N-terminal extension preceding the DBD (94-97) for steroid receptors or in the hinge region close to the LBD for the NR4 and NR5 subfamilies (98-101). Both the N-terminal extension and the hinge region are highly variable in length and sequence, and most likely are disordered and flexible, thus making their structural determination elusive. Multiple sites of posttranslational modifications are found in these variable regions, such as phosphorylation, sumoylation, acetylation, and ubiquitination, and these sites of modification often cluster closely together. Additionally, the AF1 appears to be a major surface for interaction with numerous coregulator proteins (102-106). Considering the importance of AF1 in nuclear receptor activity, its structure, function, and interaction with the DBD and the LBD are still poorly understood, and there are no pharmaceuticals available for direct manipulation of AF1 function. For the ligand-independent receptors, such as the NR4 subfamily, posttranslational modifications might be crucial in regulating their activity (107, 108).
The interplay between posttranslational modifications and the ligand potentially leads to a myriad of functional outcomes for the nuclear receptors. We are only beginning to map out the relationships between individual posttranslational events and to understand the specific effects of their combinations on receptor activity. Numerous studies highlight the importance of “the histone code” or how posttranslational modifications of histone proteins affect transcriptional state of the chromatin and dictate transcriptional competency of genes. The abundance of posttranslational modifications on nuclear receptors suggests a similar idea of regulation.
From the extensive cellular, biochemical and structural studies carried out on nuclear hormone receptors it is now appreciated that their ability to be “classically” regulated by ligands is no longer taken for granted. Indeed, we now know that over half of these receptors are not regulated by ligands as discovered for the steroid receptors many decades ago. In the last ten years, intensive research has focused on the “orphan receptors” with the goal of finding their high affinity ligands. Now, it is realized that many receptors cannot be bound by a ligand or have a non-exchangeable “structural” ligand embedded in their pockets. For ligand-dependent receptors, the challenge for the next decade will be to refine the specificity of the existing known ligands or identify allosteric modulatory ligands. For ligand-independent receptors, research will have to take a new direction to identify other regulatory sites that can then be targeted by small molecules. Given the importance of nuclear receptors in human biology and disease, they are likely to remain a primary focus for both academia and industry for years to come.
References
1. Germain P, et al. Overview of nomenclature of nuclear receptors. Pharmacol. Rev. 2006; 58:685-704.
2. Bain DL, et al. Nuclear receptor structure: implications for function. Annu. Rev. Physiol. 2006.
3. Sandler B, et al. Thyroxine-thyroid hormone receptor interactions. J. Biol. Chem. 2004; 279:55801-55808.
4. Borngraeber S, et al. Ligand selectivity by seeking hydrophobicity in thyroid hormone receptor. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:15358-15363.
5. Chiellini G, et al. A high-affinity subtype-selective agonist ligand for the thyroid hormone receptor. Chem. Biol. 1998; 5:299-306.
6. Xu HE, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature 2002; 415:813-817.
7. Xu HE, et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. 2001; 98:13919-13924.
8. Nolte RT, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998; 395:137-143.
9. Xu HE, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell. 1999; 3:397-403.
10. Williams S, et al. X-ray crystal structure of the liver X receptor beta ligand binding domain: regulation by a histidine-tryptophan switch. J. Biol. Chem. 2003; 278:27138-27143.
11. Farnegardh M, et al. The three-dimensional structure of the liver X receptor beta reveals a flexible ligand-binding pocket that can accommodate fundamentally different ligands. J. Biol. Chem. 2003; 278:38821-38828.
12. Watkins RE, et al. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science 2001; 292:2329-2333.
13. Chrencik JE, et al. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol. Endocrinol. 2005; 19:1125-1134.
14. Watkins RE, et al. 2.1 A crystal structure of human PXR in complex with the St. John’s wort compound hyperforin. Biochemistry 2003; 42:1430-1438.
15. Shan L, et al. Structure of the murine constitutive androstane receptor complexed to androstenol: a molecular basis for inverse agonism. Mol. Cell 2004; 16:907-917.
16. Suino K, et al. The nuclear xenobiotic receptor CAR: structural determinants of constitutive activation and heterodimerization. Mol. Cell 2004; 16:893-905.
17. Xu RX, et al. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol. Cell 2004; 16:919-928.
18. Maglich JM, et al. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J. Biol. Chem. 2003; 278:17277-17283.
19. Honkakoski P, et al. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol. Cell Biol. 1998; 18:5652-5658.
20. Bourguet W, et al. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature 1995; 375:377-382.
21. Haffner CD, et al. Structure-based design of potent retinoid X receptor alpha agonists. J. Med. Chem. 2004; 47:2010-2029.
22. Dhe-Paganon S, et al. Crystal structure of the HNF4 alpha ligand binding domain in complex with endogenous fatty acid ligand. J. Biol. Chem. 2002; 277:37973-37976.
23. Wisely GB, et al. Hepatocyte nuclear factor 4 is a transcription factor that constitutively binds fatty acids. Structure 2002; 10:1225-1234.
24. Brzozowski AM, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature, 1997; 389:753-758.
25. Shiau AK, et al. The structural basis of estrogen receptor/ coactivator recognition and the antagonism of this interaction by tamoxifen. Cell, 1998; 95:927-937.
26. Pike AC, et al. Structural insights into the mode of action of a pure antiestrogen. Structure, 2001; 9:145-153.
27. Greschik H, et al. Structural and functional evidence for ligand-independent transcriptional activation by the estrogen-related receptor 3. Mol. Cell 2002; 9:303-313.
28. Wang L, et al. X-ray crystal structures of the estrogen-related receptor-gamma ligand binding domain in three functional states reveal the molecular basis of small molecule regulation. J. Biol. Chem. 2006; 281:37773-37781.
29. Flaig R, et al. Structural basis for the cell-specific activities of the NGFI-B and the Nurr1 ligand-binding domain. J. Biol. Chem. 2005; 280:19250-19258.
30. Wang Z, et al. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature 2003; 423:555-560.
31. Krylova IN, et al. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell 2005; 120:343-55.
32. Li Y, et al. Crystallographic identification and functional characterization of phospholipids as ligands for the orphan nuclear receptor steroidogenic factor-1. Mol. Cell, 2005; 17:491-502.
33. Wang W, et al. The crystal structures of human steroidogenic factor-1 and liver receptor homologue-1. Proc. Natl. Acad. Sci. U.S.A, 2005; 102:7505-7510.
34. Whitby RJ, et al. Identification of small molecule agonists of the orphan nuclear receptors liver receptor homolog-1 and steroidogenic factor-1. J. Med. Chem. 2006; 49:6652-6655.
35. Ortlund EA, et al. Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nat. Struct. Mol. Biol. 2005; 12:357-363.
36. Niakan KK, McCabe ER. DAX1 origin, function, and novel role. Mol. Genet. Metab. 2005; 86:70-83.
37. Jordan VC. Antiestrogenic action of raloxifene and tamoxifen: today and tomorrow. J. Natl. Cancer Inst. 1998; 90:967-971.
38. Day C. Thiazolidinediones: a new class of antidiabetic drugs. Diabet. Med. 1999; 16:179-192.
39. Baxter JD, et al. Towards selectively modulating mineralocorticoid receptor function: lessons from other systems. Mol. Cell Endocrinol. 2004; 217:151-165.
40. Phelps C, et al. Fungi and animals may share a common ancestor to nuclear receptors. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:7077-7081.
41. Belkhadir Y, Wang X, Chory J. Brassinosteroid signaling pathway. Sci. STKE. 2006:cm4.
42. Vert G, et al. Molecular mechanisms of steroid hormone signaling in plants. Annu. Rev. Cell Dev. Biol. 2005; 21:177-201.
43. Ponting CP, Aravind L. START: a lipid-binding domain in StAR, HD-ZIP and signalling proteins. Trends Biochem. Sci. 1999; 24:130-132.
44. Schrick K, et al. START lipid/sterol-binding domains are amplified in plants and are predominantly associated with homeodomain transcription factors. Genome Biol. 2004; 5:R41.
45. 45. Freedman LP. Increasing the complexity of coactivation in nuclear receptor signaling. Cell 1999; 97:5-8.
46. Nettles KW, Greene GL. Ligand control of coregulator recruitment to nuclear receptors. Annu. Rev. Physiol. 2005; 67:309-333.
47. Ingraham HA, Redinbo MR. Orphan nuclear receptors adopted by crystallography. Curr. Opin. Struct. Biol. 2005; 15:708-715.
48. DeLano WL. MacPyMOL: a PyMOL-based Molecular Graphics Application for MacOS X. 2007. DeLano Scientific LLC, Palo Alto, CA.
49. Glass CK. Differential recognition of target genes by nuclear receptor monomers, dimers, and heterodimers. Endocr Rev. 1994; 15:391-407.
50. Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997; 18:306-360.
51. Renaud JP, et al. Crystal structure of the RAR-gamma ligandbinding domain bound to all-trans retinoic acid. Nature 1995; 378:681-689.
52. Weatherman RV, Fletterick RJ, Scanlan TS. Nuclear-receptor ligands and ligand-binding domains. Annu. Rev. Biochem. 1999; 68:559-581.
53. Darimont BD, et al. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998; 12:3343-3356.
54. Heery DM, et al. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 1997; 387:733-736.
55. Glass CK, Rose DW, Rosenfeld MG. Nuclear receptor coactivators. Curr. Opin. Cell Biol. 1997; 9:222-2232.
56. Baniahmad A. Nuclear hormone receptor co-repressors. J. Steroid Biochem. Mol. Biol. 2005; 93:89-97.
57. Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000; 14:121-141.
58. King-Jones K, Thummel CS. Nuclear receptors--a perspective from Drosophila. Nat. Rev. Genet. 2005; 6:311-323.
59. Billas IM, Moras D. Ligand-binding pocket of the ecdysone receptor. Vitam. Horm. 2005; 73:101-129.
60. Motola DL, et al. Identification of ligands for DAF-12 that govern dauer formation and reproduction in C. elegans. Cell 2006; 124:1209-1223.
61. Beckstead RB, Thummel CS. Indicted: worms caught using steroids. Cell 2006; 124:1137-1140.
62. Escriva H, Delaunay F, Laudet V. Ligand binding and nuclear receptor evolution. Bioessays 2000; 22:717-727.
63. Privalsky ML. Activation incarnate. Dev. Cell 2003; 5:1-2.
64. Thornton JW, Need E, Crews D. Resurrecting the ancestral steroid receptor: ancient origin of estrogen signaling. Science 2003; 301:1714-1717.
65. Kian Tee M., et al. Estradiol and selective estrogen receptor modulators differentially regulate target genes with estrogen receptors alpha and beta. Mol. Biol. Cell, 2004; 15:1262-1272.
66. Wu H, et al. Hypomethylation-linked activation of PAX2 mediates tamoxifen-stimulated endometrial carcinogenesis. Nature 2005; 438:981-987.
67. Carroll JS, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005; 122:33-43.
68. Willson TM, et al. The PPARs: from orphan receptors to drug discovery. J Med. Chem. 2000; 43:527-550.
69. Michalik L, et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006; 58:726-741.
70. Li AC, Glass CK. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J. Lipid Res. 2004; 45:2161-2173.
71. Kliewer SA, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator- activated receptors alpha and gamma. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:4318-4323.
72. Janowski BA, et al. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 1996; 383:728-731.
73. Chen W, et al. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab. 2007; 5:73-79.
74. Mitro N, et al. The nuclear receptor LXR is a glucose sensor. Nature 2007; 445:219-223.
75. Lazar MA, Willson TM. Sweet Dreams for LXR. Cell Metab. 2007; 5:159-161.
76. Val P, et al. SF-1 a key player in the development and differentiation of steroidogenic tissues. Nucl. Recept. 2003; 1:8.
77. Fayard E, Auwerx J, Schoonjans K. LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004; 14:250-260.
78. Sablin EP, et al. Structural basis for ligand-independent activation of the orphan nuclear receptor LRH-1. Mol. Cell 2003; 11:1575-1585.
79. Tsujishita Y, Hurley JH. Structure and lipid transport mechanism of a StAR-related domain. Nat. Struct. Biol. 2000; 7:408-414.
80. Li D, et al. cAMP-Stimulated Interaction Between Steroidogenic Factor-1 and Diacylglycerol Kinase-theta Facilitates Induction of CYP17. Mol. Cell Biol. 2007.
81. Xue Y, et al. Crystal structure of the PXR-T1317 complex provides a scaffold to examine the potential for receptor antagonism. Bioorg. Med. Chem. 2007; 15:2156-2166.
82. Swales K, Negishi M. CAR, driving into the future. Mol. Endocrinol. 2004; 18:1589-1598.
83. Baker KD, et al. The Drosophila orphan nuclear receptor DHR38 mediates an atypical ecdysteroid signaling pathway. Cell 2003; 113:731-742.
84. Bavner A, et al. Transcriptional corepression by SHP: molecular mechanisms and physiological consequences. Trends Endocrinol. Metab. 2005; 16:478-488.
85. Codina A, et al. Identification of a novel co-regulator interaction surface on the ligand binding domain of Nurr1 using NMR footprinting. J. Biol. Chem. 2004; 279:53338-53345.
86. Mi LZ, et al. Structural basis for bile acid binding and activation of the nuclear receptor FXR. Mol. Cell 2003; 11:1093-1100.
87. Leduc AM, et al. Helix-stabilized cyclic peptides as selective inhibitors of steroid receptor-coactivator interactions. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:11273-11278.
88. Geistlinger TR, Guy RK. Novel selective inhibitors of the interaction of individual nuclear hormone receptors with a mutually shared steroid receptor coactivator 2. J. Am. Chem. Soc. 2003; 125:6852-6853.
89. Rodriguez AL, et al. Design, synthesis, and in vitro biological evaluation of small molecule inhibitors of estrogen receptor alpha coactivator binding. J. Med. Chem. 2004; 47:600-611.
90. Arnold LA, et al. Discovery of small molecule inhibitors of the interaction of the thyroid hormone receptor with transcriptional coregulators. J. Biol. Chem. 2005; 280:43048-43055.
91. Wang Y, et al. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor beta. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:9908-9911.
92. Warnmark A, et al. Activation functions 1 and 2 of nuclear receptors: molecular strategies for transcriptional activation. Mol. Endocrinol. 2003; 17:1901-1909.
93. McEwan IJ. Molecular mechanisms of androgen receptor-mediated gene regulation: structure-function analysis of the AF-1 domain. Endocr. Relat. Cancer 2004; 11:281-293.
94. Miesfeld R, et al. Glucocorticoid receptor mutants that define a small region sufficient for enhancer activation. Science 1987; 236:423-427.
95. Dahlman-Wright K, et al. Delineation of a small region within the major transactivation domain of the human glucocorticoid receptor that mediates transactivation of gene expression. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:1619-1623.
96. Hadzopoulou-Cladaras M, et al. Functional domains of the nuclear receptor hepatocyte nuclear factor 4. J. Biol. Chem. 1997; 272:539-550.
97. He B, Wilson EM. The NH(2)-terminal and carboxyl-terminal interaction in the human androgen receptor. Mol. Genet. Metab. 2002; 75:293-298.
98. Desclozeaux M, et al. Phosphorylation and intramolecular stabilization of the ligand binding domain in the nuclear receptor steroidogenic factor 1. Mol. Cell Biol. 2002; 22:7193-7203.
99. Wansa KD, Harris JM, Muscat GE. The activation function-1 domain of Nur77/NR4A1 mediates trans-activation, cell specificity, and coactivator recruitment. J. Biol. Chem. 2002; 277:33001-33011.
100. Wansa KD, et al. The AF-1 domain of the orphan nuclear receptor NOR-1 mediates trans-activation, coactivator recruitment, and activation by the purine anti-metabolite 6-mercaptopurine. J. Biol. Chem. 2003; 278:24776-247790.
101. Maira M, et al. Dimer-specific potentiation of NGFI-B (Nur77) transcriptional activity by the protein kinase A pathway and AF-1-dependent coactivator recruitment. Mol. Cell Biol. 2003; 23:763-776.
102. Faus H, Haendler B. Post-translational modifications of steroid receptors. Biomed. Pharmacother. 2006;60:520-528.
103. Jackson TA, et al. The partial agonist activity of antagonist- occupied steroid receptors is controlled by a novel hinge domainbinding coactivator L7/SPA and the corepressors N-CoR or SMRT. Mol. Endocrinol. 1997; 11:693-705.
104. Chen M, et al. Phosphorylation of the liver X receptors. FEBS Lett. 2006; 580:4835-4841.
105. Garcia-Pedrero JM, et al. The SWI/SNF chromatin remodeling subunit BAF57 is a critical regulator of estrogen receptor function in breast cancer cells. J. Biol. Chem. 2006; 281:22656-22664.
106. Ou Q, et al. The DEAD box protein DP103 is a regulator of steroidogenic factor-1. Mol. Endocrinol. 2001; 15:69-79.
107. Galleguillos D, et al. PIASgamma represses the transcriptional activation induced by the nuclear receptor Nurr1. J. Biol. Chem. 2004; 279:2005-2011.
108. Wingate AD, et al. Nur77 is phosphorylated in cells by RSK in response to mitogenic stimulation. Biochem. J. 2006; 393:715-724.
See Also
Crystallization of Proteins: Overview of Applications in Chemical Biology
Liver X Receptors (LXR), Chemistry of
Orphan Nuclear Hormone Receptors
Peroxisome Proliferator-Activated Receptor (PPAR), chemistry of
Steroid Hormones, Biology and Biochemistry of