CHEMICAL BIOLOGY
Chemical Damage to Nucleic Acids
Errol C. Friedberg, Laboratory of Molecular Pathology, Department of Pathology and University of Texas Southwestern Medical Center, Dallas, Texas
Thomas J. Kodadek, Division of Translational Research, Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas
doi: 10.1002/9780470048672.wecb412
Nucleic acids are highly reactive macromolecules that can undergo a variety of chemical changes that may be deleterious to their function. In particular, deoxyribonucleic acid (DNA) undergoes multiple spontaneous chemical changes and is reactive with reactive oxygen species generated during oxidative metabolism and with naturally occurring chemicals, ultraviolet radiation, and a myriad of environmental chemicals. This review highlights many of these chemical changes.
Alterations in the chemistry of DNA, particularly those that affect the nitrogenous bases, the coding elements of the genome, are an incontrovertible fact of life as we know it on planet Earth. This fact of life derives from the intrinsic relative instability of nucleic acids, as well as their chemical reactivity with naturally occurring physical and chemical agents (in particular, free radicals generated during normal oxidative metabolism), other naturally occurring reactive chemicals, and ultraviolet (UV) radiation from the Sun. Indeed, life on Earth might have been very different if the genetic blueprint of cells had evolved as a chemical entity that was not as reactive, or if the generation of energy in cells was not oxygen-based. Some nucleic acids, DNA in particular, are reactive with other products of normal cellular metabolism, such as steroids and alkylating agents, compounds that can crosslink the two polynucleotide chains of DNA, and those that can crosslink DNA to proteins.
Biologic Background
The fact that nucleic acids are highly reactive and are thus subject to frequent chemical change, i.e., DNA damage, necessitated the evolutionary selection of multiple cellular protective mechanisms in order to sustain life. These mechanisms include multiple ones designed to (1) dispose of free radicals, (2) restore the normal chemistry and nucleotide sequence of damaged DNA (DNA repair), and (3) tolerate persistent DNA damage that threatens the viability of cells as a result of arrested DNA replication or transcription, or because of high levels of mutation.
This review is largely restricted to a consideration of chemical alterations of the nitrogenous bases, the informational elements of DNA. However, consideration is also given to the formation of strand breaks in DNA. The article does not consider interactions of nucleic acids with synthetic compounds, of which a very large number is known to react with both DNA and RNA. Nor do we consider the mispairings of bases that arises from the occasional errors associated with normal DNA replication. Finally, we do not address chemical alterations in RNA, since most of the RNA in cells turns over rapidly, and the biological significance (if any) of RNA damage is apparently limited.
The nature and mechanism of the numerous biologic responses that cells can mount to cope with the consequences of DNA damage is also beyond the scope of this limited review. We refer interested readers to a recent comprehensive textbook on these topics and to numerous references quoted therein (1).
Spontaneous Alterations in DNA Chemistry
Tautomeric shifts
All of the bases commonly found in DNA [adenine (A), cytosine (C), guanine (G), and thymine (T)] can undergo spontaneous, pH-dependent chemical changes called tautomeric shifts (2). The base T normally exists in the keto form (C=O) in the C-4 position, but it can occasionally exist in the rare enol form (C-OH) (Fig. 1). Under such circumstances it can anomalously pair with G in the usual keto state. Conversely, the C-6 position of G in the rare enol form can pair with T in the usual keto form (Fig. 1). The same is true of the N-6 position of A, usually in the amino form (NH2), switching to the imino form (NH) tautomer; in which case, it can mispair with cytosine in the amino form (Fig. 1). Reciprocally C can mispair with A when the C-4 position switches to the rare imino form (Fig. 1).

Figure 1. Anomalous base pairing involving rare tautomeric forms of the bases in DNA. When either T or G are in the rare enol form, they can pair. Similarly, when A or C are in the rate imino form, they can pair. (Reproduced from Reference 1 with permission.)
Tautomerism in the bases commands interesting prominence in the history of the elaboration of the structure of DNA by James Watson and Francis Crick. When in the early 1950s Watson was attempting to identify structurally compatible base pairing schemes using simple two-dimensional cardboard models of the bases, he inadvertently deployed the rare tautomeric states until the chemist Jerry Donahue pointed out this error to him.
The tautomeric forms I had copied out of Davidson’s book were, in Jerry’s opinion, incorrectly assigned. My immediate retort that several other texts also pictured guanine and thymine in the enol form cut no ice with Jerry. Happily he let out that for years organic chemists had been arbitrarily favoring particular tautomeric forms over their alternatives on only the flimsiest of grounds (3).
Spontaneous Deamination of Nitrogenous Bases in DNA
The common DNA bases A, C, and G, as well as the less common base 5-methylC, are endowed with exocyclic amino groups. These bases can be spontaneously lost as a function of the ambient pH and/or temperature (deamination), thus altering their ability to correctly pair with other bases (4).
Deamination of cytosine (C) to generate uracil (U) in DNA
Perhaps the most extensively studied example of spontaneous deamination, certainly in terms of its biologic outcome, is that of C to yield uracil (U) in DNA (Table 1). U, which is chemically identical to T except for the methyl group in the C-5 position of the latter, most often pairs with A, resulting in permanent G:C → A:T transition mutations during subsequent rounds of DNA replication (5). Additionally, the presence of U instead of T in the promoter regions of coding genes may affect the binding of regulatory proteins, thereby altering gene expression (1).
Table 1. Endogenous DNA lesions arising and repaired in a mammalian cella in 24 h
|
|
|
98% double-stranded DNA |
|
Endogenous source |
100% double-stranded DNA |
2% single-stranded DNA |
|
Hydrolysis depurination depyrimidination cytosine deamination 5-methylcytosine deamination
Oxidation 8-hydroxyguanine (8-oxoG) Ring-saturated pyrimidines (thymine glycol, cytosine hydrates) Lipid peroxidation products (MiG, etheno-A, etheno-C)
Nonenzymatic methylation by S-adenosylmethionine 7-methylguanine 3-methyladenine 1-methyladenine & 3-methylcytosine
Nonenzymatic methylation by nitrosated polyamines & peptides O6-methylguanine |
9000 300 50 5
500 1000 500
3000 600 n.d.
10 - 50 |
9000 300 250 25
500 1000 500
3000 600 100
10 - 50 |
aEstimates are for a 3 x 109 bp genome. (Reproduced from Reference 1 with permission.)
The chemistry of C deamination has been extensively studied (6, 7). It has been suggested that at neutral pH hydroxyl ions can directly attack the C-4 position of C (Fig. 2), especially in single-stranded DNA. An alternative explanation offered is an addition-elimination reaction during which dihydrocytosine is generated as an intermediate. This reaction may involve the formation of dihydrocytosine and dihydrouracil as intermediates (Fig. 2).
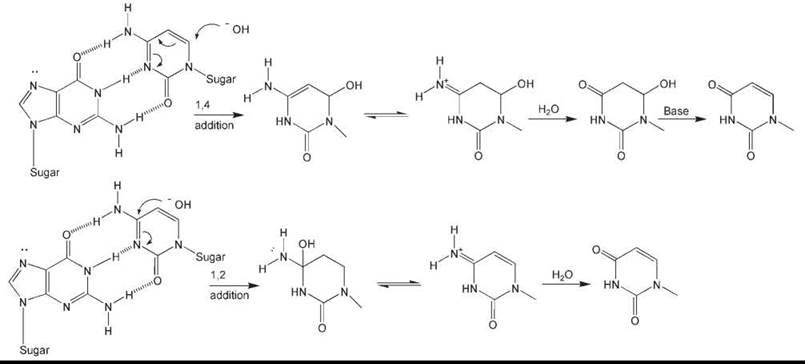
Figure 2. Plausible chemical mechanisms for the base-mediated deamination of cytosine. Top: Deamination resulting from 1,4-addition. Bottom: Deamination resulting from 1,2-addition. In both cases, only the cytosine ring is shown subsequent to hydroxide addition.
Cytosine in DNA is stable, with an estimated half-life of about 200 years (4). This value derives for a calculated rate constant for cytosine deamination in single-stranded DNA of ~2 x 10-10/s based on the direct measurement of deamination in nucleotides and polynucleotides in vitro at various temperatures and pH (8). Evaluating the biologic significance of these values is problematic because they derive from in vitro measurements using single-stranded polynucleotides, whereas in living cells, most DNA is, of course, in the duplex configuration and the rate of spontaneous deamination of cytosine in duplex DNA in vitro is believed to be <1% of that in single-stranded DNA (8). However, limited regions of single-stranded DNA are transiently generated in the course of various DNA metabolic transactions, in particular replication, transcription, and recombination. Additionally, considerable evidence exists that duplex DNA undergoes spontaneous localized denaturation, or “breathing,” that could further promote this process (9). (On the other hand, single-stranded DNA can interact with specific DNA-binding proteins that may protect these regions of the genome from chemical change.)
The literature documents various situations in which C deamination is enhanced. These situations include the presence of C in cyclobutane pyrimidine dimers (a form of DNA damage caused by exposure to UV radiation; see later discussion) or in mispairings with other bases or with alkylated bases (1). Cytosine deamination is also promoted in the presence of nitrous acid, a reaction that although not considered in this review, has lent much to our understanding of possible chemical mechanisms of spontaneous deamination (1).
While considering the presence of U in DNA, it is relevant to point out that this base can also arise in DNA by its misincorporation (instead of T) during normal DNA replication as a consequence of the existence of a small pool of dUTP (1). Not unexpectedly, perturbations that increase the pool size of dUTP relative to that of TTP promote this misincorporation (1). However, the biologic consequences of A:U “mispairs” in DNA are limited because such base pairs have the same coding potential as A:T pairs (1).
Essentially all life forms studied are endowed with enzymes that recognize and remove U from DNA. These enzymes, called uracil-DNA glycosylases (UDGs), are historically noteworthy because their discovery identified a new class of DNA repair enzymes that collectively embrace many more substrates than uracil in DNA. The several DNA glycosylases that selectively remove uracil from DNA comprise a superfamily of proteins that are represented in all life forms (1).
Deamination of 5-methylcytosine
Cytosine that is methylated at the 5 position (5-methylC) is naturally encountered in the genomes of some organisms, both prokaryotic and eukaryotic (10) (Table 1). It has been estimated that the rate of deamination of 5-methylC is significantly faster than that of C in single-stranded polynucleotides, and that it could account for as much as 10% of the spontaneous deamination events in the genome of mammalian cells under physiologic conditions (4). Since deamination of 5-methylC generates T in DNA, this spontaneous reaction is considered to be an important source of T:G mispairs that can lead to G:C → A:T transition mutations, especially in CpG-rich promoter regions of many genes, so-called CpG islands (1).
For many years it was considered that T:G mispairs generated by the deamination of 5-methylC would not be repaired by mismatch repair mechanisms because of the intuitive notion that conventional mismatch repair would not distinguish T in T:G base pairs from that in normal T:A base pairs, potentially leading to wholesale loss of TMP from DNA. However, the discovery of DNA glycosylases as a general class of base excision repair enzymes led to the detection of a DNA glycosylase that specifically recognizes T in T:G mispairs (1). Additionally, some bacteria are endowed with a specialized form of mismatch repair that distinguishes between T:G and T:A base pairs (1).
Deamination of adenine and guanine
The deamination of A and G in DNA to hypoxanthine and xanthine, respectively, transpires at rates about 50-fold less than that of C (9). The biologic significance of such deamination reactions is also overshadowed by data indicating that they transpire about 10,000 times less frequently than their complete loss from the genome during spontaneous depurination (see later discussion).
Spontaneous Loss of Bases from DNA (Depurination and Depyrimidination)
The spontaneous loss of bases from DNA after hydrolysis of the N-glycosyl bond linking them to the sugar-phosphate backbone is a frequent source of spontaneous DNA damage, with potentially important biologic consequences (1) (Table 1). It is estimated that the genome of the average mammalian cell sustains the loss of about 10,000 purines a day, generating that many apurinic/apyrimidinic (AP) sites (9). AP sites are very unstable, often leading to hydrolysis of the sugar phosphate backbone and the formation of strand breaks (1, 4, 9).
The kinetics of base loss has been extensively studied in vitro using polynucleotide substrates incubated at varying pH and temperatures. Such studies indicate that G is lost from DNA about 1.5 times more rapidly than A (11). Extrapolation of Arrhenius plots generated by direct measurements at varying temperatures yields a rate constant for the depurination of duplex DNA of k = 3 x 10-11/s at physiologic ionic strength and temperature (11). This result translates to the loss of ~1 purine/E. coli genome equivalent. However, thermophilic bacteria that thrive at high temperatures and often at very acidic pH may sustain the loss of several hundreds of purines each generation absent specific biologic mechanisms to offset this loss. As just mentioned, mammalian cells may sustain the loss of ~ 10,000 purines/day. The chemistry of depurination at acidic pH is believed to involve protonation of bases followed by elimination of the base in an SN1-like process that probably involves a ribose-derived oxonium ion (Fig. 3). Pyrimidines are spontaneously lost from polynucleotides at about 1/20th the rate of the loss of purines, but this process may translate into the loss of hundreds of pyrimidines during each mammalian cell cycle (12).
The deoxyribose residues that are generated at sites of base loss in DNA exist in equilibrium between the closed furanose form and the open aldehyde form (13) (Fig. 3). The 3' phosphodiester bonds associated with the latter are labile and are readily hydrolyzed by β-elimination during which the pentose carbon tothe aldehyde is activated at alkaline pH and at increased temperatures (13). The same reaction proceeds at a reduced rate at neutral pH. At physiologic pH and temperature, a site of base loss in DNA has an estimated average lifetime of ~400 h (14).

Figure 3. Plausible chemical mechanism for acid-catalyzed depurination and subsequent base-mediated strand cleavage, shown in the context of an A-T base pair. Only the adenine-containing segment of the molecule is shown subsequent to the initial reaction. The depurination reaction proceeds through an oxonium ion intermediate (top right) that is quenched by water. The resultant hemiacetal exists in equilibrium with the open-chain aldehyde, thus making the 2'-hydrogen relatively acidic and facilitating base-catalyzed elimination that leads to strand cleavage.
Spontaneous Oxidative Damage to DNA
Products of oxygen breakdown, such as superoxide radicals, hydroxyl radicals (the most reactive oxygen radical known to chemistry) and hydrogen peroxide, collectively referred to as reactive oxygen species (ROS) (15, 16), are continuously generated as byproducts of mitochondrial respiration in all aerobic organisms, as well as from lipid peroxidation and from the metabolic activity of phagocytic cells. ROS can also be generated when cells are exposed to various exogenous agents, notably ionizing radiation and numerous chemicals (especially redox-cycling drugs) (1). ROS are highly reactive with DNA, as well as with proteins and lipids. Indeed, the so-called oxygen paradox points out that although oxygen is fundamental to aerobic life forms, ROS pose what is perhaps the most pervasive natural threat to the integrity of both the nuclear and the mitochondrial genomes of cells (15). Numerous disease states (in particular cancer) as well as the process of aging have been implicated as consequences of oxidative damage to DNA (1).
To cope with this fundamental problem, cells have evolved a myriad of defense mechanisms designed to both reduce the burden of free ROS and to repair damage generated in DNA by ROS (1). The former defense mechanisms primarily comprise multiple cellular antioxidants; antioxidant enzymes such as superoxide dismutase, glutathione peroxidase, and catalase (strategically located in or near the mitochondrial membrane), as well as naturally occurring antioxidants such as vitamins A, E, and C; glutathione; and thioredoxin. Cellular oxidative stress, a state characterized by (but not limited to) extensive oxidative damage to DNA, results when levels of ROS exceed these various antioxidant defense mechanisms. The following discussion focuses on the chemistry and to a lesser extent the functional consequences of oxidative base damage to DNA. Details of the repair of such damage and of the regulation of various antioxidant enzymes are reviewed elsewhere (1).
Reactive oxygen species (ROS)
Molecular oxygen [dioxygen (O2)] exists in a triplet ground state and hence is relatively unreactive with the singlet state of biologic molecules. But the unpaired orbitals of dioxygen can sequentially accommodate single electrons, yielding superoxide radicals (∙O2-), hydrogen peroxide (H2O2), hydroxyl radicals (∙OH), and water as shown in the following equation (15, 17):
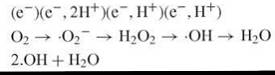
The parameters that most significantly influence the potential of ROS to interact with and damage DNA are their reactivity, their half-life, and their diffusability in cells. Being highly reactive, ∙OH can readily oxidize organic molecules. However, their extreme reactivity dictates a high probability that they will rapidly encounter a reactive cellular component other than DNA. Thus, they have limited diffusability, and ∙OH generated directly from leaky mitochondrial respiration is probably not a major source of DNA damage.
The generation of ∙OH in the very close proximity required for its interaction with DNA is believed to transpire by the Fenton reaction (1), first described around the turn of the nineteenth century by Henry John Horstman Fenton, who discovered that hydrogen peroxide is a much more potent oxidant in the presence of various metals, notably the ferrous ion (FeII) than in their absence. Subsequently, Fritz Haber and coworkers proposed that the potent oxidant generated during the Fenton reaction is in fact ∙OH, produced according to the following reaction (18):
![]()
Even trace concentrations of iron support a robust Fenton reaction if reducing agents such as NADH (17), ascorbate (19), or superoxide are available to recycle Fe3+ to the active Fe2+ form. Although the Fenton reaction typically involves Fe2+, it is also supported in the presence of other transitional metals such as copper.
Like hydrogen peroxide, the free superoxide radical (∙O2-) is not highly reactive. However, this chemical moiety can readily generate H2O2, especially in the presence of the enzyme superoxide dismutase, by the reaction:
![]()
Superoxide radicals can also reduce and liberate Fe3+ from ferritin, or liberate Fe2+ from iron-sulfur clusters (17, 20), thereby generating free iron that facilitates the formation of highly reactive oxygen species from H2O2 and ∙O2- by way of the Fenton reaction.
Hydroxyl radicals can abstract electrons from residues of organic macromolecules (RH) by the following general reaction:
![]()
This reaction can initiate chain reactions that may generate DNA damage at considerable distances from the initial chemical event (21). Peroxidation of unsaturated lipids initiated by reactive free radicals such as ∙OH represents a prominent example of such a chain reaction. The phospholipids of all membranes contain high concentrations of polyunsaturated fatty acids, and chain reactions involving hundreds of phospholipids can react in each oxidation event (22). The initial products of such fatty acid oxidation are lipid hydroperoxides, but these are relatively short-lived; they are either reduced by glutathione peroxidases to unreactive fatty acid alcohols or they react with metals to produce products such as epoxides and aldehydes, which are themselves reactive.
The major aldehyde products of lipid peroxidation are malondialdehyde and 4-hydroxynonenal (Table 1, Fig. 4). Malondialdehyde can react with DNA to generate adducts at the bases A, C, and G. The mutagenic adduct M1G (pyrimido(1,2-a)purin-10(3H)one) has been detected at levels as high as 1 adduct per 106 nucleosides in human tissues. M1G is a reactive electrophile that can undergo further modification, leading to crosslinking of an adducted DNA strand to the opposite strand, or to some protein (22). Exocyclic etheno adducts can also arise from lipid peroxidation, possibly by reaction of an epoxide of 4-hydroxynonenal with A, C, or G in DNA.
In addition to oxidants that are generated by the Fenton reaction, superoxide radicals (∙O2-) readily react with nitric oxide (NO∙), generating peroxynitrite anion (ONOO-) in the following reaction:
![]()
The protonated form of peroxynitrite anion, peroxynitrous acid, is highly reactive with biologic molecules. Hence, the production of nitric oxide from nitric oxide synthase (a complex enzyme containing several cofactors, and a heme group that is part of the catalytic site), which catalyzes the formation of NO from oxygen and arginine, can render cellular components such as DNA susceptible to superoxide-mediated damage (1).

Figure 4. (a) Malonaldehyde is a major product of lipid peroxidation that is reactive with A, G, and C in DNA. (b) 4-Hydroxynonenal, another major product of lipid peroxidation, can generate exocyclic etheno adducts of A, C, and G in DNA. (Reproduced from Reference 1 with permission.)
DNA damage by ROS
Hydroxyl radicals can add to the DNA bases or abstract hydrogen atoms, generating multiple types of DNA damage. The C4-C5 double bond of the pyrimidines C and T is particularly sensitive to attack by hydroxyl radicals, generating a large spectrum of oxidized pyrimidines, notably thymine glycol, urea residues, 5-OHdU, 5-OHdC, and hydantoin (Fig. 5, Tables 1 and 2). Similarly, interaction of hydroxyl radicals with the purines A and G generates multiple purine oxidative products. The altered base 7,8-dihydro-8-oxo-guanine (23, 24), sometimes referred to as 8-oxoguanine (8-oxoG or as its isomeric form 8-hydroxyguanine), is a biologically important form of base damage caused by oxygen free radicals (1) (Table 1). When present in DNA this residue readily assumes a syn conformation that can base pair with A, resulting in transversion mutations after DNA replication (25).
Table 2. Radiation yields (nmol J-1) of DNA base products under oxygen-free (N2O) and oxygenated conditions (N2O + O2)
|
|
|
Chromatin |
Naked DNA |
||
|
Parent base |
Product formed |
N2O |
N2O + O2 |
N2O |
N2O + O2 |
|
T |
5,6-dihydrothymine |
0.26 |
nil |
5.6 |
nil |
|
|
5-hydroxy-5,6-dihydrothymine |
0.30 |
nil |
7.7 |
<0.1 |
|
|
thymine glycol |
0.094 |
0.40 |
10.2 |
43.4 |
|
|
5-hydroxymethyluracil |
0.064 |
0.052 |
n.d. |
n.d. |
|
|
5-hydroxy-5-methylhydantoin |
b |
0.042 |
n.d. |
n.d. |
|
C |
5-hydroxy-5,6-dihydrocytosine |
0.17 |
nil |
2.6 |
<0.1 |
|
|
cytosine glycol |
0.95 |
2.30 |
10.7 |
25.6 |
|
|
5,6-dihydroxycytosine |
b |
0.33 |
n.d. |
n.d. |
|
|
5-hydroxyhydantoin |
b |
0.22 |
n.d. |
n.d. |
|
A |
FaPy-A (4,6-diamino-5-fomamidopyrimidine) |
0.96 |
1.02 |
8.7 |
5.9 |
|
|
8-oxoA (7,8-dihydro-8-oxoadenine) |
0.80 |
3.50 |
5.3 |
15.8 |
|
G |
FaPy-G (2,6-diamino-4-hydroxy-5-fomamidopyrimidine) |
1.81 |
1.81 |
12.4 |
3.6 |
|
|
8-oxoG (7,8-dihydro-8-oxoguanine) |
1.35 |
8.05 |
16.2 |
46.7 |
aYields from the literature are for determinations by GC-MS for DNA isolated from chromatin and naked double-stranded DNA. Nil, not detected; b, not detected above levels present in control sample; n.d., lesion measurement not determined. (Reproduced from Reference 1 with permission.)

Figure 5. Some of the many products of oxidation of the bases in DNA.
Oxidative damage to the nitrogenous bases in DNA is readily generated during the handling of this biologic macromolecule under standard laboratory conditions. Despite the extensive use of antioxidants during the isolation and handling of DNA, this problem has seriously complicated accurate measurements of basal steady-state levels of oxidative damage in cells, especially with the advent of sensitive techniques for its measurement, and early estimates reported in the literature should not be relied on (1).
Treatment of DNA with hydrogen peroxide or other free radical-generating systems (such as xanthine/xanthine oxidase) can also result in the formation of an imidazole ring-opened derivative of guanine, designated 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FaPy) (26). This lesion is prominent among the forms of base damage induced by H2O2. Both this reaction and that for the analogous lesion in A proceed through a carbinolamine-type intermediate.
Close to 100 products of DNA base damage caused by ROS have been identified (27), usually by treating bases, nucleosides, or oligonucleotides in vitro with strong oxidants or ionizing radiation (27). It has been estimated that endogenous ROS may result in as many as 200,000 base lesions/day in the genome of the average mammalian cell (1).
Hydrogen abstraction typically affects the deoxyribose sugar units, leading to fragmentation of the sugars, base loss, and strand breaks with a terminal sugar residue fragment (28). Such breaks most often affect single polynucleotide chains of the DNA duplex. In contrast, ionizing radiation, which also generates ROS (1) typically produces clusters of hydroxyl radicals that can cause double-strand breaks. There is an extensive literature on ionizing radiation damage to DNA. This topic is not included in this review because absent unshielded travel in space, organisms are not typically exposed to ionizing radiation. Furthermore, as just noted, many types of DNA damage caused by ionizing radiation are identical to those generated during oxidative stress.
Anaerobic, or hypoxic, cells are resistant to the deleterious effects of ionizing radiation. Ionizing radiation, such as that used in radiotherapy, kills cells by producing DNA damage, particularly DNA double-strand breaks. This damage results from ionizations in the DNA or in water molecules very close to the DNA that produce a radical on the DNA (DNA•).
This radical then enters into a competition for oxidization primarily by oxygen (which fixes, or makes permanent, the damage), or reduction primarily by -SH-containing compounds that can restore the DNA to its original form. Thus, DNA damage, including single- and double-strand breaks, is less in anaerobic cells. This effect of oxygen in sensitizing cells to radiation is quantitated as the ratio of dose in the absence of oxygen to dose in the presence of oxygen needed to obtain the same surviving fraction of cells. For mammalian cells, this ratio is usually 2.5-3.0. The oxygen partial pressure that produces sensitivity midway between the oxic and the anaerobic responses is approximately 3 mm Hg. Clinical trials, particularly with head and neck cancers, have demonstrated that the more hypoxic tumors are more radioresistant than the less hypoxic tumors.
How cells cope with ROS
As mentioned, cells can survive the large amounts of ROS generated during normal cellular metabolism both by directly inactivating ROS by a variety of reactions, and by repairing oxidative damage to DNA. With regard to the former category of cellular defenses, the following have been most extensively studied.
1. Superoxide dismutase (SOD) enzymes that eliminate superoxide by the reaction:
![]()
The several SODs in mammalian cells constitute a major form of protection against ROS. Since superoxide dismutase generates H2O2, its detoxifying effect primarily results from preventing the accumulation of free Fe2+ and the production of peroxynitrite.
2. Removal of hydrogen peroxide by catalase in the reaction:
![]()
In mammalian cells, catalase is largely contained in peroxisomes and does not seem to be nearly as biologically important as SOD (17).
3. The reduction of hydrogen peroxide by organic reductants (RH) such as glutathione, ascorbate, and cytochrome c:
![]()
4. Yet another mode of defense against oxidant stress in mammalian cells is by transcriptional induction of heme oxygenase, which acts to generate heme metabolites with anti-oxidant properties (29).
In addition to these primary antioxidant defenses, various cellular responses (besides multiple DNA repair reactions) are triggered by ROS, which promote the inhibition of cell-cycle progression and the initiation of apoptosis (programmed cell death) (1). Additionally, various regulatory pathways have been identified that are specifically activated when the genome is threatened by oxidative damage. For example, the soxRS and oxyR gene products are redox-sensitive transcriptional activators that respond to ∙O2 and H2O2, respectively (30). Enhanced ∙O2- levels promote conformational changes of SoxR protein, which in turn activates transcription of the soxS gene. Among other genes, soxS enhances expression of superoxide dismutase (sodA) and the DNA repair enzyme endonuclease IV (nfo). The oxyR regulon responds to H2O2 and controls the expression of catalase (katG) and the peroxiredoxin ahpFC (1).
Other Types of Spontaneous Damage to DNA
Spontaneous alkylation of DNA
Alkylating agents are electrophilic compounds that react efficiently with nucleophilic centers in organic macromolecules, including DNA (1). Alkylating agents can be highly toxic to cells and are widely used in the treatment of cancer primarily for this reason. Alkylating agents can be either monofunctional or bifunctional. The former have a single reactive group that reacts covalently with one of the many nucleophilic centers in DNA. Bifunctional agents have two reactive groups, so each molecule can potentially react with two sites in DNA, generating crosslinks. Numerous potential reaction sites for alkylation have been identified in all four nitrogenous bases, but not all of them have equal reactivity. The sites of reaction in DNA for many monofunctional alkylating agents include the following: in A, N1, N3, N6, and N7; in G, N1, N2, N3, N7, and O6; in C, N3, N4, and O2; and in T, N3, O2, and O4. The ring nitrogens of the bases are in general more nucleophilic than the oxygens, with the N7 position of guanine and the N3 position of adenine being the most reactive (1). Alkylation of oxygen in phosphodiester linkages results in the formation of phosphotriesters (1).
Cells are endowed with several distinct enzyme-catalyzed mechanisms that specifically effect the repair of various alkylated bases (1). The alkylating agents used experimentally to generate the multiple substrates attacked by these enzymes are, in the main, not naturally occurring. However, it is intuitively compelling that these enzymes must have evolved to cope with spontaneous forms of alkylation damage to DNA (Table 1). Support for the notion of spontaneous alkylation damage to DNA comes from several studies.
1. Some microorganisms are endowed with a particular methyltransferase activity that catalyzes the methylation of halide ions (31), thereby naturally generating methyl chloride (MeCl), a mutagenic alkylating agent that can react with various atoms in the nitrogenous bases. Large amounts of methyl chloride are also naturally generated during the burning of biomass (32), and reactive methyl radicals can arise by the irradiation or oxidation of methyl compounds such as methylhydrazine (33).
2. Some antibiotics are alkylating agents. A prominent example is streptozotocin, an antibiotic produced by the soil bacterium Streptomyces achromogenes. This compound is a 2-deoxy-D-glucose derivative of the potent alkylating agent methylnitrosourea (MNU).
3. In higher organisms (including mammalian cells), DNA may suffer alkylation damage from inappropriate alkylation by S-adenosylmethionine (SAM) (1, 4). This methyl donor is normally required for the enzymatic methylation of C to yield 5-methylC, and it has been estimated that in proliferating mammalian cells as many as 600 3-methyladenine residues/cell generation may result from inappropriate alkylation, translating to levels of DNA alkylation expected from exposure to 20-nM methyl methanesulfonate (MMS) (34), a potent synthetic alkylating agent. Reactive alkyl radicals and nonradical products generated by lipid peroxidation chain reactions are another potential source of alkylation damage to DNA.
4. Methylating agents can be generated by chemical nitrosation of endogenous metabolites. For example, methylamine produced by the decomposition of organic matter can condense with carbamyl phosphate, a precursor of pyrimidines, to form methylurea, which in turn can be nitrosated to yield methylnitrosourea (MNU). Such nitrosation reactions can be catalyzed by bacterial enzymes (35).
5. Reactive alkyl radicals and nonradical products generated by lipid peroxidation chain reactions are potential alkylating agents. Reactive methyl radicals can also arise by the irradiation or oxidation of methyl compounds such as methylhydrazine (33).
Spontaneous DNA-DNA and DNA-Protein Crosslinks
The covalent interaction of a chemical with bases on both strands of the DNA duplex can result in the formation of interstrand DNA crosslinks. Such lesions can interfere with the separation of the two polynucleotide chains that is required during various DNA metabolic transactions, especially replication and transcription. The biologic consequences of DNA crosslinking have been mainly investigated using synthetic bifunctional chemicals such as bifunctional alkylating agents (nitrogen and sulfur mustards are notable examples). However, several possible endogenous sources of interstrand crosslinks have been identified. For example, dietary nitrites can be converted to nitrous acid under the acidic acid conditions in the stomach. Nitrous acid generates interstrand crosslinks preferentially between the exocyclic N2 amino groups of guanine at CG sequences (36), and it has been estimated that a DNA interstrand crosslink may result for every four deaminations generated by nitrous acid (1). Aldehydes constitute another potential source of interstrand crosslinks in cells. Acetaldehyde, a product of normal cellular glycolysis, can cause DNA interstrand crosslinks (1). Malondialdehyde (another aldehyde) can be generated as products of lipid peroxidation.
DNA-protein crosslinks can constitute another source of spontaneous DNA damage, although one that has not been as extensively studied as the types of DNA damage already discussed. Nonetheless, aldehydes have been shown to cross-link histones and other DNA-bound proteins to polynucleotides. Acrolein and related aldehydes can also be endogenously generated by oxidative degradation of unsaturated lipids. Guanine adducts of acrolein, crotonaldehyde, and trans-4-hydroxynonenal can form crosslinks with peptides (37). This reaction is mediated by a Schiff base linkage between a ring-opened aldehyde moiety and the N-terminal a-amine of the peptide (37).
Relatively unreactive chemicals can be metabolically activated to forms that are highly reactive with DNA
A variety of relatively nonpolar compounds can undergo metabolic activation that is effected by specific metabolizing enzymes (38). The biologic function of these enzymes is to convert potentially toxic, lipid-soluble nonpolar chemicals to more reactive water-soluble and thus excretable forms, frequently esters (38). However, many of these more reactive water-soluble compounds can interact with nucleophilic centers in DNA (38), generating various types of base adducts. A series of membrane-bound proteins endowed with monooxygenase activities have been extensively characterized as components of these detoxification systems. These heme-containing proteins strongly absorb light at ~450 nm and in combination with one or more membrane-bound flavoprotein reductases are often referred to as the cytochrome P-450 system (1).
Among naturally occurring chemical in this category, steroids are becoming increasingly recognized as a source of spontaneous DNA damage. Steroids are derived from cholesterol, which is converted to 17-hydroxyprogesterone by a series of specific oxidation-reduction reactions. Additional such reactions generate the adrenal cortical steroids, as well as the sex hormones estrogen, progesterone, and testosterone. Estrogens, including the natural hormones estradiol and estrone, can be hydroxylated at several positions by cytochrome P-450 enzymes. Hydroxylation at the 4 position to generate catechol estrogens is particularly important with regard to DNA damage. In cells, 4-hydroxylated catechols are oxidized to semiquinones and then to DNA-reactive quinones. These quinone intermediates react at the 3- and 7-positions of the purine bases in DNA, creating unstable bulky adducts that are readily depurinated. The catechol estrogen metabolites may also generate ROS by metabolic redox cycling to the semiquinone form, or by other mechanisms. These estrogen-induced oxidants have the potential to produce the full spectrum of oxidative DNA damage discussed above.
Although not widespread, aflatoxins, among the most potent liver carcinogens known, represent another example of DNA-damaging agents that have their origin as products of natural metabolism. Aflatoxins are toxins produced by the fungi Aspergillus flavus and Aspergillus parasiticus. Contamination of cereal grains such as rice, peanuts, and corn by these fungi can result in human and animal exposure (39). Chemically, the aflatoxins consist of a difurofuran ring system fused to a substituted coumarin moiety, with a methoxy group on the benzene ring (39). Among the aflatoxins of fungal origin, aflatoxin B1 is the most potent.
Aflatoxin B1 can enter cells by passive diffusion and is metabolically converted by mixed function oxygenases of the cytochrome P-450 system, generating aflatoxin B1-8,9-epoxide as a major product (39). G residues in double-stranded DNA are preferentially attacked by this reactive electrophilic epoxide (1). The major product is an N7 adduct on G (8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1) (40). The positively charged imidazole ring weakens the glycosyl bond, promoting depurination and subsequent sites of base loss. Mildly alkaline conditions can result in the formation of formamidopyrimidine derivatives containing an opened imidazole ring, which is a reaction that has also been identified in vivo (39).
DNA Damage Caused by Natural Environmental Agents
Ultraviolet radiation from the Sun
UV radiation from the Sun is a ubiquitous and important source of DNA damage that contributes to the high incidence of skin cancer in exposed individuals. The UV radiation spectrum has been arbitrarily divided into the UV-A component (wavelengths 320-400 nm), the UV-B component (wavelengths 295-320 nm), and the UV-C component (wavelengths 100-295 nm). Penetration of the atmospheric ozone layer is weak for wavelengths below 300 nm. Thus, solar UV radiation primarily comprises the UV-A and UV-B components. However, experimental studies typically involve the use of UV-C radiation from germicidal lamps with a sharp emission maximum at 254 nm. Since this wavelength is very close to the absorption maximum of DNA (260 nm) and is inefficiently absorbed by proteins, UV-C radiation-induced damage is relatively specific for DNA (1).
Major photoproducts in DNA
When cells are exposed to UV-C radiation, the quantitatively most abundant form of DNA base damage involves saturation of the pyrimidine 5,6 double bonds, resulting in covalent linkages between adjacent pyrimidines through a four-membered ring structure (1) (Fig. 6). This lesion is called the cyclobutane pyrimidine dimer (CPD). CPD can exist in cis-syn, cis-anti, trans-syn, or trans-anti configurations, but in double-stranded B-form DNA, they exist predominantly in the cis-syn form (1). However, trans-syn dimers are generated to some extent, mainly in denatured DNA (1), and can be detected in single-stranded regions and in duplex DNA with special conformations, such as the junctions between B-DNA and Z-DNA (1).

Figure 6. Diagrammatic representation of a cyclobutane pyrimidine dimer (CPD) (top) and a (6-4)photoproduct (bottom).
The thermodynamic and spectroscopic properties of short segments of duplex DNA containing a single CPD indicate that cis-syn CPD can be accommodated in the double helical structure of B-DNA such that hydrogen bonding with the opposite A residues is possible (41). The crystal structure of a DNA decamer containing a cis-syn CPD (42) shows that the overall helical axis is bent about 30° toward the major groove and is unwound by about 9°. This structure is consistent with molecular modeling and electron microscopy studies and with the anomalous electrophoretic behavior of CPD-containing oligonucleotides (1). Although Watson-Crick base pairing can occur at the 3' T in a CPD, base pairing of the 5' T with A is severely weakened because one hydrogen bond cannot be made.
CPD are extraordinarily stable to extremes of pH and temperature and to total acid hydrolysis of DNA. They can thus be readily resolved from thymine in such hydrolysates by a variety of chromatographic techniques, and quantitated (1).
The formation of CPD during irradiation of DNA is a reversible process that can be represented by the following reaction:
![]()
This reaction reaches photochemical equilibrium when the thymine-containing CPD content (ToT, ToC and CoT dimers) reaches ~7% of the total thymine content of DNA (1). This steady state reflects a dynamic equilibrium in which the rate of CPD formation (which is pseudo-zero order, to good approximation) and that of CPD reversal (which is first order in dimer content) are equal (1). The yield of ToT CPD is highest, whereas that of C<>C CPD is lowest (1). CPD are not randomly distributed in DNA. Numerous studies at the DNA sequence level have shown that their yields depend on DNA sequence context. In general, the equilibrium level of CPD is higher for TT sites flanked on both sides by A compared with such sites flanked on the 5' side by A and on the 3' side by G or C (1).
Another major form of base damage generated in DNA by UV light is one in which the C6 position of a 5' pyrimidine is covalently linked to the C4 position of the 3' adjacent pyrimidine (Fig. 6). These lesions [which are readily detected by their lability in alkaline conditions at 80-100° C (43)], are called pyrimidine-pyrimidone [6-4] adducts, or simply [6-4] photoproducts ([6-4]PP). The pyrimidine planes in ([6-4]PP) are almost perpendicular. Hence, they result in prominent distortions of the double-helical structure of DNA. ([6-4]PP) can involve adjacent TC, CC, or (less often) TT sequences. Their formation at CT sequences is infrequent. Cytosine methylation at the C5 position inhibits the formation of [6-4]PP (44). The yield of [6-4]PP is proportional to the incident UV fluence in the range 100-500 J/m2 and continues to increase after exposure to several thousand J/m2 (1). In UV-C irradiated DNA, the ratio of CPD:[6-4]PP is ~3:1 (1).
Minor photoproducts in DNA
Exposure of DNA to UV radiation also generates oxidative base damage. In particular, thymine glycol (5,6-dihydroxydihydro-thymine) can be generated by saturation of the 5,6-double bond of some pyrimidines. Under anhydrous conditions, the so-called “spore photoproduct” or 5,6-dihydro-5-(a-thyminyl)-thymine, another type of dimeric pyrimidine photoproduct, can be generated by adduction of the methyl group of a thymine to the C5 position of a neighboring thymine. The formation of this major photoproduct in UV-irradiated bacterial spores of B. subtilis (1) is dependent on the conformation of the DNA, being generated more readily in DNA in the compacted A-form (1). Consequently, the spore photoproduct is readily formed in dry DNA films and in DNA solutions containing high concentrations of ethanol (1).
Another type of photochemical reaction involving a pyrimidine base is the addition of a molecule of water across the 5,6 double bond of C to yield a 5,6-dihydro-6-hydroxy derivative called the cytosine hydrate. The quantum yield for the formation of cytosine hydrates in UV-irradiated DNA is greater in single-stranded than in duplex-DNA (45). Hydrates of cytosine, deoxycytidine, CMP, or dCMP are unstable, readily reverting to the parent form by rehydration (45). However, their half-life is dramatically increased in DNA, and cytosine hydrate may be the major nondimer C photoproduct. Cytosine hydrate can undergo deamination and dehydration to yield uracil (1). The hydrate of 5-methylcytosine may undergo deamination to yield 5-thymine hydrate, which can convert to thymine upon dehydration (1).
A photoproduct called 8,8-adenine dehydrodimer is observed after irradiation of poly(dA). This lesion involves a single 8,8 bond linking the imidazole rings (46). Another uncharacterized lesion involving adenine has been reported after UV irradiation of simian virus 40 DNA (1). The lesion is alkali-labile and occurs at ACA sequences. Other photoinduced lesions involving purines have been identified (47). The fact that they are recognized by DNA repair enzymes suggests that they are physiologically significant. In general, the generation of purine photoproducts lesions is enhanced if they are flanked by two or more contiguous pyrimidines on their 5' side (1).
DNA crosslinks and strand breaks
UV radiation can result in the cross-linking of DNA to proteins (1). Crosslinks between different duplex DNA molecules (DNA-DNA crosslinks) have also been observed, mainly when DNA is irradiated in the dry state, in an extremely densely packed condition (such as in the heads of salmon sperm), or in other special conformations (1). The spore photoproduct, formed in UV-irradiated dry DNA as described above, can arise not only between adjacent pyrimidines in the same chain but as a reaction between pyrimidines in different DNA chains (1).
Irradiation of DNA with very high doses of UV-C light can also result in breakage of the polynucleotide chain (1). However, the amount of UV radiation required to reduce the molecular weight of Streptococcus pneumoniae DNA by 50% is about 100 times that required to reduce the transforming activity of the streptomycin resistance marker of that organism to the same extent (1). No chain breaks are detected in phage T7 DNA exposed to doses of UV radiation that inactivate almost 100% of the phage population (1). Thus, no conclusive evidence shows that the direct formation of DNA strand breaks by UV-C radiation is of biologic consequence. The frequency of strand breaks and DNA-protein crosslinks is dramatically increased by irradiation at longer wavelengths (1). In cells, most strand breaks observed after UV-irradiation are caused by biologic processing, either from interruption or breakdown of stalled DNA replication forks, or as intermediates in the repair of photoproducts.
Conclusions
As discussed, much is known regarding the chemical reactivity of DNA and how these transformations lead to mutation. However, important challenges remain. For example, most of the estimates regarding the rates of DNA damaging processes are derived from simple model studies using oligonucleotides. Do these rates reflect the reality of what occurs in the context of chromatin? These questions are challenging and new technologies will be required to address them. Although not covered in detail in this review, there is much to be done to increase our understanding of the enzymatic mechanisms employed by repair proteins to undo the damage brought about by everyday life. Thus, we anticipate that the field of DNA damage and repair will continue to fascinate both chemists and biologists for some time to come.
References
1. Friedberg EC, Walker GC, Siede W, Wood RW, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis, 2nd ed. 2006. ASM Press, Washington, DC.
2. Watson JD. Molecular Biology of the Gene. 1976. WA Benjamin, Inc., Menlo Park, CA.
3. Watson, JD. The Double Helix. 1968. Atheneum Press, New York.
4. Lindahl T. instability and decay of the primary structure of DNA. Nature 1993; 362:709-715.
5. Duncan BJ, Miller J. Mutagenic deamination of cytosine residues in DNA. Nature 1980; 287:560-561.
6. Shapiro R. Damage to DNA caused by hydrolysis. In: Chromosome Damage and Repair. Seeberg E, Kleppe K., eds. 1981. Plenum Publishing Corp., New York.
7. Shapiro R, Klein RS. The deamination of cytidine and cytosine by acidic buffer solutions. Mutagenic implications. Biochemistry 1966; 5:2358-2362.
8. Lindahl T, Nyberg B. Heat-induced deamination of cytosine residues in DNA. Biochemistry 1974; 13:3405-3410.
9. Lindahl T. DNA glycosylases, endonucleases for apurinic/apyrimidinic sites and base excision repair. Prog. Nucleic Acids. Mol. Biol. Res. 1979; 22:135-192.
10. Ehrlich M, Zhang X-Y, Imander NM. Spontaneous deamination of cytosine and 5-methylcytosine residues in DNA and replacement of 5-methylcytosine residues with cytosine residues. Mutation Res. 1990; 238:277-286.
11. Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry 1972; 11:3610-3618.
12. Lindahl T, Nyberg B. Heat-induced depyrimidination of DNA. Biochemistry 1973; 12:5151-5154.
13. Jones AS, Mian AM, Walker RT. The alkaline degradation of deoxyribonucleic acid derivatives. J. Chem. Soc. Section C. 1968; 2042.
14. Lindahl T, Andersson A. Rate of chain breakage of apurinic sites in double-stranded DNA. Biochemistry 1972; 11:3618-3623.
15. Davies KJ. The broad spectrum of responses to oxidants in proliferating cells; a new paradigm for oxidative stress. IUBMB Life 1999; 48:41-47.
16. Davies KJ. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000; 50:279-289.
17. Henle ES, Linn S. Formation, prevention, and removal of DNA damage by iron/hydrogen peroxide. J. Biol. Chem. 1997; 272:19095-19098.
18. Koppenol WH. The Haber-Weiss cycle-70 years later. Redox Rep. 2001; 6:229-234.
19. Breen AP, Murphy JA. Reactions of oxy radicals with DNA. Free Radic. Biol. Med. 1995; 18:1033-1077.
20. Imlay JA, Linn S. DNA damage and oxygen radical toxicity. Science 1998; 240:1302-1309.
21. Saran M, Bors W. Radical reactions in vivo-an overview. Radiat. Environ. Biophys. 1990; 29:249-262.
22. Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis 2000; 21:361-370.
23. Cadet J, Delatour T, Douki T, Gasparutto D, Pouget JP, Ravanat JL, et al. Hydroxyl radicals and DNA base damage. Mutation Res. 1999; 424:9-21.
24. Kasai H, Nishimura S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984; 12:2137-2145.
25. Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative damage, causes G → T and A → C substitutions. J. Biol. Chem. 1992; 267:166-171.
26. Dizdaroglu M, Rao G, Halliwell B, Gajewski E. Damage to the DNA bases in mammalian chromatin by hydrogen peroxide in the presence of ferric and cupric ions. Arch. Biochem. Biophys. 1991; 285;317-324.
27. Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutation Res. 2003; 531:37-80.
28. Mello-Filho AC, Meneghini R. In vivo formation of single-strand breaks in DNA by hydrogen peroxide is mediated by the Haber- Weiss reaction. Biochim. Biophys. Acta. 1984; 781:56-63.
29. Keyse SM, Tyrrell RM. Heme oxygenase is the major 32 kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:99-103.
30. Demple B, Amabile-Cuevas CF. Redox redux: the control of the oxidative stress response. Cell 1991; 67:837-839.
31. Wuosmaa AM, Hager LP. Methyl chloride transferase: a carbocation route for biosynthesis of halometabolites. Science 1990; 249:160-162.
32. Crutzen PJ, Andreae MO. Biomass burning in the tropics: impact on atmospheric chemistry and biochemical cycles. Science 1990; 250:1669-1678.
33. Sedgwick B. Oxidation of methylhydrazines to mutagenic methylating derivatives and inducers of the adaptive response of Escherichia coli to alkylation damage. Cancer Res. 1992; 52:3693-3697.
34. Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold. Spring Harbor Symp. Quant. Biol. 2000; 65:127-133.
35. Taverna P, Sedgwick B. Generation of an endogenous DNA- methylating agent by nitrosation in Escherichia coli. J. Bacteriol. 1996; 178:5105-5111.
36. Edfeldt NB, Harwood EA, Sigurdsson ST, Hopkins PB, Reid BR. Solution structure of a nitrous acid-induced DNA interstrand DNA cross-link. Nucleic Acids Res. 2204; 32:2785-2794.
37. Kurtz AJ, Lloyd RS. 1,N2-dooxyguanosine adducts of acrolein, crotonaldehyde, and trans-4-hydroxynonenal cross-link to peptides via Schiffbase linkage. J. Biol. Chem. 2003; 278:5970-5976.
38. Anders MW, Dekant W. Conjugation-dependent carcinogenicity and toxicity of foreign compounds. Adv. Pharmacol. 1994; 27:1-519.
39. Smela ME, Currier SS, Bailey EA, Essigman JM. The chemistry and biology of aflatoxin Bi: from mutational spectrum to carcinogenesis. Carcinogenesis 2001; 22:535-545.
40. Essigman JM, Croy RG, Nadzam AM, Busby WF Jr, Reinhold VM, Buchi G, et al. Structural identification of the major DNA adduct formed by aflatoxin B1 in vitro. Proc. Natl. Acad. Sci. U.S.A. 1977; 74:1870-1874.
41. Taylor JS, Garrett DS, Brockie IR, Svoboda DL, Telser J. 1H NMR assignment and melting temperature study of cis-syn and trans-synthymine dimer containing duplexes of d(CGTATTATGC). d(GCATAATACG). Biochemistry 1990; 29:8858-8866.
42. Wang CI, Taylor JS. Site-specific effect of thymine dimer formation on dAn dTn tract bending and its biological implications. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:9072-9076.
43. Lippke JA, Gordon LK, Brash DE, Haseltine WA. Distribution of UV light-induced damage in a defined sequence of human DNA: detection of alkaline-sensitive lesions at pyrimidine nucleoside-cytidine sequences. Proc. Natl. Acad. Sci. U.S.A. 1981; 78:3388-3392.
44. Pfeifer GP, Drouin R, Riggs AD, Holmquist GP. In vivo mapping of a DNA adduct at nucleotide resolution: detection of pyrimidine (6-4) pyrimidone photoproducts by ligation-mediated polymerase chain reaction. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:1374-1378.
45. Fisher GJ, Johns HE. Pyrimidine hydrates. In: Photochemistry and Photobiology of Nucleic Acids. Wang SY, ed. 1976. Academic Press, New York. pp 169-294.
46. Gasparro FP, Fresco JR. Ultraviolet-induced 8,8 adenine dehydrodimers in oligo- and poly-nucleotides. Nucleic Acids Res. 1986; 14:4239-4251.
47. Duker NJ, Gallagher PE. Purine photoproducts. Photochem. Photobiol. 1988; 48:35-39.
Further Reading
Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells.Annu.Rev. Genet. 2004; 38:445-476.
Collins CR. Oxidative DNA damage, antioxidants, and cancer. Bioessays 1999; 21:238-246.
Davies KJ. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000; 50:279-289.
Lindahl T. DNA lesions generated in vivo by reactive oxygen species, their accumulation and repair. In: Advances in DNA Damage and Repair: Oxygen Radical Effects, Cellular Protection and Biological Consequences. Dizdaroglu M, Karakaya A, eds. 1999. Plenum Publishers, New York. pp 251-257.
Pryor WA. Oxy-radicals and related species: their formation, lifetimes, 5 and reactions. Annu. Rev. Physiol. 1986; 48:657-667.
Saran M, Bors W. Radical reactions in vivo-an overview. Radiat. Environ. Biophys. 1990; 29:249-262.
Singer B, Kusmierek JT. Chemical mutagenesis. Annu. Rev. Biochem. 1982; 51:655-693.
See Also
Chemical Views of Biology
Biomolecules Within the Cell
Chemistry of Biological Processes and Systems
Applications of Chemical Biology