CHEMICAL BIOLOGY
The Pentose Phosphate Pathway: An Overview
John F. Williams, Research School of Chemistry, Institute of Advanced Studies, The Australian National University, Canberra, Australia
doi: 10.1002/9780470048672.wecb433
The elementary textbook treatment of the pentose phosphate pathway (PPP) describes its nature and occurrence in the cytoplasm of most cells. The reaction scheme is of two segments, oxidative and nonoxidative. The reactions of the oxidative segment are few and involve the decarboxylation of glucose 6-phosphate (Glc 6-P) to ribulose 5-phosphate (Ru 5-P) and CO2 via 6-phosphogluconolactone and 6-phosphogluconate (6-PG). Concomitant production of two moles of NADPH and H+ for each mole of Glc 6-P converted to Ru 5-P. The nonoxidative reactions are depicted classically with an ordered reaction sequence of reversible steps for the interconversion of other pentose phosphate products that originate from Ru 5-P by epimerase and isomerase enzymes. The reactions of the PPP also link the formation of sugar phosphate intermediates (glycolyl units) containing 3-7 carbon atoms that are generated by freely reversible equilibrium reactions catalyzed in fixed order by Transketolase and Tansaldolase. These reactions permit the following three biosynthetic PPP functions to emerge: cellular energetics, growth, and repair. Specifically via 1) the contribution by the oxidative segment of a high NADPH / NADP+ redox potential that provides electrons for most reductive anabolic processes;2) the formation of ribose 5-phosphate (Rib 5-P) for all nucleotide and nucleic acid biosynthesis, which is a demand that does not usually exceed 2% of all Glc metabolized by PPP activity; and (c) a storage pool of diverse phosphorylated glycolyl units that may be used directly or as signals for biosynthetic and energy-yielding reactions by other pathways. Finally, selected reactions of the nonoxidative segment are also part of the most extensive synthetic and life-sustaining event on the planet, namely the photosynthetic reductive path of CO2 assimilation in all C-3 plants.
The history of the unravelling of many of the above events reveals instances of profound weakness in evidence used to justify the nonoxidative pathway description. These instances are identified and discussed in this overview and show that textbook depictions of PPP are so incomplete and misleading that the scheme for the pathway is deemed erroneous.
A figurative narration describing some of the elementary biochemistry of the pentose phosphate pathway (PPP) of glucose metabolism (Figs. 1 and 2) (1) has appeared, with ever diminishing scope of treatment, in all textbooks on general biochemistry since 1954. It is a scheme of reactions exclusively confined in the cytoplasm of most cells. The few reactions of the oxidative segment (Fig. 1) are established thoroughly and are beyond contention. No such certainty can be claimed for the assignment of enzyme composition, correct identification of all reactants, nor the reaction order for the nonoxidative segment (Fig. 2). Otto Warburg, a German biochemist and Nobel Laureate, was the dominant figure in the unravelling of the chemistry of the Fig. 1 reactions. Bernard L. Horecker, an American enzymologist, can justifiably claim much of that distinction for the Fig. 2 scheme (2).
The Reactions and Significance of the Oxidative Segment
The first step in the discovery of the PPP commenced in the autumn of 1929 when Otto Warburg arrived in Baltimore to deliver the Herter Lecture at John’s Hopkins Hospital. As part of the visit, he was invited to the laboratory of E.S.G. Barron to witness a demonstration of some remarkable and puzzling results that Barron and Harrop had published recently (3). The demonstration involved a simple system, namely, the reaction of glucose with non-nucleated erythrocytes in the presence and absence of methylene blue (MeB). Methylene blue is one of a large number of organic dyes that form electromotively active oxidation-reduction systems. MeB had been used successfully by Torsten Thunberg, during the previous decade, for the identification of many dehydrogenase reactions. However, considerable diplomacy was needed for a peaceful presentation of the Barron reaction system because Warburg had been publicly scathing in condemnation of biochemical reactions that included MeB. Notwithstanding this prejudice, when Warburg observed the outcome of the Barron experiments, he was truly astonished. In 1929, it was accepted that all glucose metabolism in erythrocytes was by glycolysis with the formation of two equivalents of lactate, which were diffused by the cells. Contrary to this expectation, the Barron data with MeB as a reactant showed a 50% suppression of the extent of glycolysis and lactate formation while the rate and extent of glucose utilisation was maintained and accompanied by a rapid oxygen uptake. Florkin (4) wrote, “In Baltimore Warburg clearly saw, for the first time in his experience, a dehydrogenase at work even though methylene blue was involved.”
Warburg returned to Berlin where, with his colleague W. Christian, he repeated and extended the Barron work and over the next 18 months showed that the phenomenon, which was first witnessed in Baltimore, comprised the following sequence of reactions [reactions (1)-(5)].


Reactions (1)-(5) encompass the disclosure of 1) the reactions of the oxidative segment of the PPP, 2) the discovery of a new pyridine nucleotide-NADP+, and 3) the detection of a flavin mononucleotide (FMN) flavoprotein (initially called “Old yellow enzyme”) and the recognition of its role as a catalytic carrier of reducing equivalents from reduced pyridine nucleotide to molecular oxygen via MeB. No contradiction can exist that the unravelling of the reactions of reactions (1)-(5) marked a historic moment of high achievement in biochemical research. The achievment was to lead Warburg and Christian to discover and characterize glucose 6-phosphate dehydrogenase (Zwischenferment) (Glc 6-PDH) (EC 1.1.1.49) and NADP+ (Wasserstoffubertragendes).
In 1931, Warburg and Christian (5) reported the first discoveries on Glc 6-PDH and the conversion of glucose 6-phosphate (Glc 6-P) to 6-phosphoglucono 8-lactone, Keq = 6 x 107 (Fig. 1). Warburg thereby established the existence of new reactions for the oxidative metabolism of glucose that differed from those of the Emden-Meyerhof glycolytic pathway. During the next decade, Warburg, Dickens, Lipmann, Diche, and others showed, among other things, that the above lactone, a potential acid, was hydrolyzed by lactonase (EC 3.1.1.17) to form 6-phosphogluconic acid (6-PG). The thermodynamic constants for the combined actions of Glc 6-PDH and 6-phosphogluconate-δ-lactonase are ∆G = — 6.35 Kcal mol-1 and Keq 1.7 x 104 at pH 7 (6). Decarboxylation of 6-PG by an NADP+-dependent 6-phosphogluconate dehydrogenase (6-PGDH) (EC 1.1.1.44) formed a pentose 5-phosphate, which was not identified correctly until 1951 (7). This latter step is essentially irreversible, and the oxidative energy released is irredeemably lost because the pathway does not include the primitive, albeit effective, mechanism for ATP generation and capture by substrate-level synthesis of acyl-phosphoanhydride or acyl-thioester.

Figure 1. Reactions of the oxidative segment of the pentose phosphate pathway. The conversion of [2-14C] glucose to CO2 and [1-14C] ribulose 5-phosphate is shown. Detail of the reaction steps are discussed in the text. Reprinted from International Journal of Biochemistry, 19. Williams John. F., Arora Krishan K. and Longnecker John P. The pentose pathway: A random harvest, 69 Pages, 1987, with permission from Elsevier, http://www.sciencedirect.com/science/journal/13572725
The sum reaction for the oxidative segment of the PPP is shown by reaction (1). Using insightful planning and remarkable chemistry, Warburg et al. (8) had also isolated and characterized (from 100 L of horse erythrocytes) the new pyridine nucleotide coenzyme NADP+ for the above reactions. He recognized and chemically established that the new coenzyme was functionally different than the NAD+of the glycolytic fermentation pathway that produced ethanol or lactate. However, this brilliant discovery also led to the first of many contentious episodes that have marked the unravelling of the PPP. With prophetic force and confidence, Warburg et al. (8) defined the biologic role of NADPH as the source of reducing equivalents for respiration and thereby put forth an erroneous view that persisted for the next 16 years. This misjudgement was also supported by Engelhardt and Barkash (9) who had proposed the title “Hex- osemonophosphate Shunt” for the oxidation of Glc 6-P by Fig. 1 reactions. The currency of the error only ceased when Lehninger (10) showed that NADH of mitochondria is the redox source of reducing equivalents for the electron-to-oxygen pathway of respiration and ATP production.
Only gradual recognition occurred (Reference 11, pp. 679683) that the principal function of the dehydrogenase enzymes of Fig. 1 was the provision and maintenance of a very high NADPH/NADP+ratio (approximately 80/1, compared with 8.6 x 10—4 for the cytoplasmic NADH/NAD+ ratio) as the source of cytoplasmic reducing power (electrons) for biosynthetic and other reactions that require reducing equivalents in aerobic organisms. (A reducing equivalent is equal to one electron or one hydrogen atom.) Although the PPP is the most usual source of biosynthetic reducing power in cytoplasm, a singular outstanding exception to this generality was found in adipose tissue by Flatt and Ball (12) who showed that 50% of the very large demand for NADPH was supplied by the action of L-Malate:NADP+ oxidoreductase [oxaloacetate decarboxylating (EC 1.1.1.40.)], and the remaining half was supplied by the PPP.
Glc 6-P DH exists in multiple molecular forms and is subject to wide genetic, endocrine, and nutritional variations. Its deficiency is the most common enzymopathy in humans. It is also the most sensitively regulated enzyme in the whole array of reactions comprising the oxidative and nonoxidative segments of the PPP (Figs. 1 and 2). More than a dozen pathways of biosynthesis use NADPH as a substrate (e.g., synthesis of fatty acids, steroids, some photosynthetic products, isoprenoids, sphingosines, phenylalanine and tyrosine, seminal fructose, 3-hydroxykynurenine, and NAD+). A high level of unbound NADPH is mandatory for the reductions of oxidized glutathione, dihydrofolate, D-glucuronate, ribonucleotides, cytochrome P450, nitrate and nitrite, glucose production from pyruvate via malate, and the hydroxylation of some fatty acids. The cellular oxidant/antioxidant balance is also dependent on NADPH production principally derived from PPP activity. The regulation of the following reactive oxygen system (ROS) enzymes: glutathione reductase, glutathione peroxidase, catalase, superoxide dismutase, NADPH oxidase, and nitric oxide synthetase depend on the high value of the NADPH/NADP+ redox couple. Thus, Glc 6-PDH has a pivotal role in the management of oxidative stress and its consequences in normal physiology and pathophysiologic events such as carcinogenesis and aging. Consult Reference 13 for a review of Glucose 6-phosphate dehydrogenase and Reference 14 for a brief review of all other enzymes shown in Figs. 1 and 2.

Figure 2. Reactions of the non-oxidative segment of the pentose phosphate pathway. The reaction sequence for the F-type (Classic) PPP is based on data in references (40 and 41). Conventionally the flow of reactants is from [1-14C] ribulose 5-P (the product of Fig. 1) to labelled hexose and triose phosphates. The 14C distributions into labelled intermediates is also shown. Reaction details are discussed in the text. Reprinted from International Journal of Biochemistry, 19. Williams John. F., Arora Krishan K. and Longnecker John P. The pentose pathway: A random harvest, 69 Pages, 1987, with permission from Elsevier,http://www.sciencedirect.com/science/journal/13572725
The Reactions of the Nonoxidative Segment
A synoptic survey of the unravelling of the nonoxidative segment is presented in the latest comprehensive review of the PPP (1). The treatment covers the chemistry of reactions, enzymes, methods, tissue distributions, and the mathematical theory for the quantitation of metabolism by a theoretical connection of the pathways of Figs. 1 and 2 into a pentose phosphate cycle (PC).
Early Discoveries
The initial investigations that lead to chemical knowledge of the pathway of Fig. 2 were indirect and not planned to that end as the following narration will show. Warburg’s proposition linking NADPH and respiration [reactions (2)—(4)] was shared by Erwin Haas who was a member of Warburg’s Berlin-Dahlem laboratory. Haas departed Germany in 1938 and proceeded to the laboratory of Professor T. R. Hogness (University of Chicago), where he met Bernard Horecker, who had just completed Ph.D. training in enzymology. Haas was a meticulous scientist who possessed much of Warburg’s data and methods (of which he was something of a master) and a plan of research to test the proposed role of NADPH in respiration. Haas and Horecker set out to isolate a putative NADPH cytchrome-c reductase [reaction (6)] in order to demonstrate the existence and nature of an enzyme that was hypothesized to be the missing link in a respiratory pathway between reduced pyridine nucleotide and oxygen via the cytochrome system [reactions (7) and (8)].
NADPH-Cytochrome C reductase (EC 1.6.2.4)
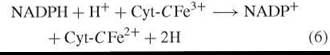
Cytochrome Oxidase (EC 1.9.3.1)
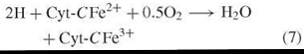
Sum Reaction
![]()
The search for an enzyme activity was successful, and a reasonably pure flavoprotein, NADPH-cytochrome c reductase (EC 1.6.2.4), was isolated from yeast (15). It was then 1940, and basic research was interrupted and not taken up again until the end of the Second World War. During that interval, Horecker worked on war-related programs, was appointed to the staff of the National Institutes of Health, and in 1945 returned to basic research in enzymology. He proceeded to isolate NADPH-cytochrome c reductase from acetone-dried extracts of pig liver (16), at a time that was almost coincident with the report of Lehninger (10) that NADH was the substrate of respiration. As listed above, the true acceptor for NADPH-cytochrome c is cytochrome P450. This natural disappointment drew attention to a much larger problem, namely an inquiry into the chemistry, enzymology, and metabolic fate of the pentose phosphate product of Fig. 1. The British biochemist, Frank Dickens (Courtauld Institute, London U.K.), had been making pioneering investigations on this topic since 1936. However, a solution of the problem was going to be made in the United States, where metabolic biochemistry and enzymology flourished in the post-war vigour of the1950s. It is helpful to note the intense interest at the time in the nature and significance of the new pathway of Glc 6-P oxidation. Important findings were going to be made in the laboratories of Bernard Horecker, Ephraim Racker, Seymour Cohen, Bernard Axelrod, and Gilbert Ashwell. Moreover, a most promotive stimulus was the outstanding investigation then being made by Melvin Calvin and colleagues at UC Berkeley. Their work was to lead to a Nobel Prize and to our current chemical appreciation of the path of reductive carbon fixation by photosynthesis (PS). Calvin’s great progress, during 1950-1955, was heavily dependent on success by the above biochemists to solve the chemical and enzymo- logical problems posed by the reactions of Fig. 2. A notable number of the intermediates and enzymes of Fig. 2 were identified as reactants in the path of carbon in PS (17). Also, Horecker made early and successful attempts to compete with Calvin in the quest to map the enzymology of the carbon reduction cycle.
By 1950 all investigators possessed strong clues that were to serve as signposts for an ultimate elucidation of a PPP reaction scheme. These signposts were 1) clear evidence that an alternative path of Glc 6-P oxidation existed in yeast, some bacteria, red cells, liver, and other animal tissues. 2) Dickens (18) had confirmed that 6-PG was decarboxylated oxidatively at carbon one to yield ribose 5-phosphate (Rib 5-P) and other sugar phosphate products (all unresolved), including a putative tetrose-P. He also demonstrated that Rib 5-P was oxidized at five times the rates of arabinose 5-phosphate (Ara 5-P) and xylose 5-P, both of which are theoretical products of 6-PG decarboxylation. 3) Finally Dische (19) reported that inosine and inorganic phosphate (Pi) were converted by red cell lysates to triose and hexose phosphates. This last important finding of possible end products of Rib 5-P dissimilation was confirmed by Waldvogel and Schlenk (20) who showed Glc 6-P formation from Rib 5-P using rat liver extracts.
Post-1950 Discovery of Reactions for the Nonoxidative Segment of the PPP
With the above background, between 1950 and 1955, a breathtaking series followed of discoveries of enzyme and substrate reactivities that were destined to be incorporated into a reaction scheme (mechanism) for the classic nonoxidative PPP (Fig. 2). The Fig. 2 diagram has also been given the prefix F-type (for fat-cell) PPP Williams et al. (1), because its ordered reaction sequence was later shown to measure uniquely a large contribution (50%) to metabolism when Glc was converted to fatty acids and triglyceride by insulin-stimulated adipocytes (12, 21, 22). An assembly of reaction steps for the scheme of Fig. 2 may be theorized using the conjunction of results from the following temporal list of events. In 1951, Cohen’s group (23) showed that Rib 5-P and Ara 5-P were formed from 6-PG oxidation. Rib 5-P formation was confirmed by Horecker et al. (7) who also proved unequivocally that Ru 5-P was the first pentose -P formed when 6-PG was decarboxylated. A new enzyme, Ribose 5-phosphate isomerase (R 5-P I) (EC 5.3.1.6), that catalyzed the interconversion of the above two pentose phosphates was also proposed (7) (Fig. 2). A very active preparation of R 5-P I from Alfalfa (24) was used to demonstrate its reaction and equilibrium. Horecker and Smyrniotis (25) used a liver enzyme preparation with Rib
5-P to note the formation of an initially high concentration of sedoheptulose 7-phosphate (Seh 7-P). This seven-carbon ketulose ester had been found originally and was characterized by Andy Benson et al. (26) in Calvin’s laboratory, and it was shown to be an early product of PS carbon fixation. Seh 7-P was formed by the action of Transketolase TK (EC 2.2.1.1) (Fig. 2). TK was discovered by Racker and his collaborators (28-29) who demonstrated that it catalyzed the transfer of a two-carbon fragment (an active glycolaldehyde group) from appropriately structured ketulose-sugar donors to a wide selection of aldo-sugar acceptors (Reference 1, p. 754). Two of its donor transfer actions, using different aldo-acceptors, are shown as blue rectangular panels in Fig. 2. TK requires Mg2+ and thiamine pyrophosphate (TPP) as a coenzyme, and a two carbon group is transferred to its acceptor via an hydroxyethyl-TPP intermediate. A list of 15 of its glycolaldehyde acceptor substrates is tabulated (Reference 1, p. 754). Horecker’s group (30) discovered a second broad specificity group transferring enzyme, namely Transaldolase (TA) (EC 2.2.1.2), which catalyzed the reversible transfer of a three-carbon dihydroxyacetone-enzyme-bound moiety (shown as red panels in Fig. 2) from Seh 7-P to glyceraldehyde 3-phosphate (Gra 3-P). This reaction formed Fru 6-P and a presumed tetrose phosphate that owed its assigned identity more to arithmetic than to biochemistry because it was neither isolated nor identified. The availability of synthetic erythrose 4-phosphate (Ery 4-P) enabled Kornberg and Racker (31) to demonstrate the reversal of the TA reaction [reaction (9)]; thus, an authentic reason for its inclusion as an intermediate in the reaction scheme of Fig. 2 was provided. Ery 4-P probably only exists in exceedingly low concentration in any tissue, and to date, no evidence exists that it has ever been measured correctly in, nor isolated from, any preparation carrying out PPP or PS metabolism in vivo or in vitro (32, 33).
![]()
A third ketopentulose ester, xlulose 5-phosphate (Xlu 5-P) Fig. 2, was isolated as a product of Rib 5-P metabolism by Ashwell and Hickman (34). It was also shown by Srere et al. (35) that Xlu 5-P, rather than the earlier assigned Ru 5-P, was a definitive substrate of TK. Ribulose 5-phosphate-3'-epimerase (Fig. 2) catalyzed the formation of Xlu 5-P and imparted the transconfiguration to the hydroxyls at carbons 3 and 4, which is a necessary stereochemical condition for substrate reactivity with TK. The 3'-epimerase was purified from a bacterial source by Stumpf and Horecker (36) and from muscle by Dickens and Williamson (37). In summary, the above research seemed to uncover an array of substrates and enzymes that satisfied a minimum requirement for inclusion in a new pathway that connected the product of 6-PG decarboxylation with the formation of hexose and triose phosphates. A prescient Racker (38) was the first to present Fig. 2 as a theoretical cyclic metabolic pathway with stoichiometry for an ensemble of the above reactants and enzymes.
Search for an Order of Reactions in the Nonoxidative PPP
It is possible to draw various theoretical schemes that oblige the arithmetic conjunction of five-carbon sugars with a summary outcome of sugar products that contain six-carbon and three-carbon atoms, respectively (39). That variety is greatly enhanced if reactions catalyzed by aldolase (Ald) (E C 4.1.2.13) are included. Aldolase occupies the same cellular compartment as all other enzymes of the PPP; it is a dihydroxyacetone 3-phosphate (DHAP) group transferring enzyme, with a catalytic capacity that is usually much greater than TK or TA (a notable exception is adipose tissue where Ald activity is low (21, 22) and only approximates the activity of TK and TA). Ald also has a broad substrate array of aldo-sugar phosphate acceptors (Reference 1, p.754), most of which are the same substrates as those involved in TK and TA reactions. It has never been clear why the pioneering investigators of the nonoxidative PPP assigned aldolase a role of catalytic silence.
The results of the only foundational experiments that explored the identity of a reaction sequence (mechanism) for the PPP were published by the Horecker group (40, 41). Detailed treatment of the conduct and conclusions of these important, but rarely discussed, experiments is given in References 1 and 42. A prediction labeling technique that used [1-14C]- and [2,3-14C]-Rib 5-P as substrates was adopted. These substrates were reacted with fractionated enzyme extracts of acetone powder preparations from rat liver, pea leaf, and pea root tissues and formed, among other intermediates, C14-labeled Glc-6-P (Fig. 3) and glyceraldehyde 3-P (Gra 3-P) (41). It was anticipated that both the position and the amount of 14C-label imparted to the Glc 6-P formed by the above substrates would reveal the nature and order of the reactions involved in its formation.
Because the enzyme extracts were made from acetone-dried powders, they were free of all nucleotides (40, 42). This strategy confined the dissimilation of labeled Rib 5-P to a hexose 6-P end point. Glc 6-P could not recycle, and the labeled prediction pattern was not scrambled. Mg2+ was also omitted from the reaction mixture to inhibit the activity of Fructose bisphosphatase (EC 3.1.3.11) and production of a contaminating Fru 6-P from Fru 1,6-P2 formed by aldolase and the triose-P products of the TK reactions. It is curious that Fru 1,6-P2, which is an Ald product, was formed (Fig. 3) by the liver and pea tissue reaction mixtures but was omitted from the final construction of the scheme for Fig. 2.
The experiments with liver enzyme preparation were of a 17-hour duration (Fig. 3). Ribose 5-P was used rapidly during the initial 3 hours, and Seh 7-P also accumulated during this early period. Only with the slow decline in Seh 7-P from 6 hours was there increased linear production of Glc 6-P, that was harvested after 17 hours and degraded carbon atom by carbon atom (42) to produce the C14distribution pattern of the whole molecule. The degradation data using [1-C14]-Rib 5-P as substrate showed that the Glc 6-P product was labeled with C14 isotope at carbons 1 and 3 with a C-1/ C-3 isotope ratio of 3 (74% of the C14 isotope in C-1 and 24% in C-3) (40). With a sense of caution, Horecker (40) tentatively proposed an ordered set of equations for the reactions of the TK and TA steps (Fig. 2), where the C-1 of the combined two species of Fru 6-P ended up with twice the level of label as that at C-3. Fair agreement was claimed (40) between the experimental values (ratio 3) and the theoretical prediction of Fig. 2 (ratio 2). Clearly no agreement was reached, fair or otherwise: The value 3 can never be 2.

Figure 3. Conversion of ribose 5-phosphate to sugar phosphate products by rat liver enzyme preparation (RLEP) (40). Details of the incubation, protein content and methods used are described in (42). For measurement of the max. catalytic capacity of RLEP see (43). Reprinted from International Journal of Biochemistry, 19. Williams John. F., Arora Krishan K. and Longnecker John P. The pentose pathway: A random harvest, 69 Pages, 1987, with permission from Elsevier, http://www.sciencedirect.com/science/journal/13572725
The above difference is serious, and the experimental isotope distributions cannot be reconciled with the Fig. 2 reaction scheme, nor can the difference be excused by pleading inaccuracies in the procedures of analysis and degradation, because the percentage error in the determination of C-1 is only 2%; C-2, 2.7%; and C-3, 1%, with a maximum cumulative percentage error of 12% for the estimate of all carbons of the molecule (42). The companion experiment (41) used [2,3-14C]-Rib 5-P as the prediction substrate and 4-hour incubations with enzyme extracts of acetone powders of liver, pea root, and pea leaf tissues. The results (1, 41) deviated so radically from the path predicted by Fig. 2 that they cannot be deemed to offer any support for the proposed PPP reaction sequence. It is emphasized that in the publications of these two studies, an ordered series of chemical equations for the nonoxidative PP was only tentatively proposed and a metabolic map was not drawn. However, in 1955, two important and substantial reviews of carbohydrate metabolism were published by Horecker and Mehler (44, 45), and each presented Fig. 2 as the metabolic map for the PPP (still with the tentative caveat). That illustration is still the contemporary chart of the PPP or pentose cycle (PC). It is astonishing that such substandard and unacceptable disagreement between practice and theory was so uncritically ignored by the general community of biochemists. Harland Wood (46) was an exception; he reported a tolerant suspicion of the [1-14C] Rib 5-P data and total rejection of any claim for predictive value by the [2,3-14C] Rib 5-P results. It was suggested (46) that an as yet unidentified pathway was operated by the above enzyme fractions. Notwithstanding the rejection by Wood (46), without anymore revision, a placid acceptance followed as well as an equally prompt inclusion of F-type PPP into the canon of metabolic biochemistry. Two independent investigators did test the mechanism of the PPP; the first by Katz et al. (47) used [1-14C]-Rib metabolism in liver slices, and the other by Hiatt (48) used the same labeled substrate in mouse liver in vivo. These investigators did not find 14C distributions in the glucose product with twice as much isotope in C-1 as C-3, but instead they found these carbons were equally labeled. These independent failures to confirm the predictions of Fig. 2 were also ignored. Finally it is important to note that inclusion of several PPP enzymes and reactants in the Path of Carbon in PS gratuitously added an aura of confidence and prestige to the status of the tentative PPP. Thus, 1955 marked an end of the second contentious period in the unraveling of the pathway.
Other than immensely valuable research by Patricia McLean and her collaborators at the Courtauld Institute, U.K., and investigations by Karl Brand at the Max Planck Inst, Dortmund, Germany, fundamental research on the mechanism of the PPP essentially ceased by 1957 and was not resumed for another decade. Instead the era of the quantitative measurement of pathways of carbohydrate metabolism had dawned and PPP measurements featured hugely. This emphasis on quantitation is best summarized in the following quotation from H. G. Wood (46): “The determination of the relative role of different pathways in normal living cells is without doubt of the greatest fundamental importance to our understanding of life processes and will in the future require more attention in all fields of metabolism.” Later Wood and Katz collaborated (49, 50) and during the next eight years developed theory and methods for the measurement of an entity denoted by Wood (46) and later stringently defined by Wood and Katz (49) as PC. The PC definition directed the following set of ordered events: (I) the entry of three moles of Glc 6-P into metabolism by the oxidative segment (Fig. 1) and a characteristic product formation of CO2, Gra 3-P, NADPH, and H+ [reaction (13)]. (II) All Fru 6-P formed in the nonoxidative segment of the cycle is converted to Glc 6-P [reaction (12)]. Compliance with directives I and II determined the reaction sequences of the partial reactions of reactions (10) and (11).
Oxidative Segment PPP
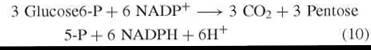
Nonoxidative PPP
![]()
Glucosephosphate isomerase (EC 5.3.1.9)
![]()
Pentose Cycle
![]()
In a review (51) of theory for estimating the PC, the following boundary conditions were defined: 1) three moles of CO2; six moles each of NADPH and H+ are formed per mole of Glc 6-P oxidized by the cycle. 2) All C14-labeled glucose metabolized by the cycle is in isotopic and chemical equilibrium with Fru 6-P [reaction (8)]. 3) The reaction steps shown at reactions (6)-(9) are unidirectional. 4) All C14 -labeled substrate, intermediary, and product stages of the reactions of reactions (6)-(9) are deemed to be in metabolic (chemical) and isotopic equilibrium, and finally 5) the original position of the label in the substrate can only be randomized by the mechanism of the PC [reaction (9)].
Measurement of the Pentose Cycle: Theory and Practice
A detailed treatment of this topic, including the derivation of some important mathematical expressions is given in Reference 1 (pp.753-762), and only a summary of the essentials will be presented here. Between 1958 and 1979, a dozen theoretical papers were published that provided the mathematical basis and formulas for measuring the F-type PC by the application of 14C-specifically labeled substrates. The method of measurement depended on an exact solution of the problem posed by the recycling and, thus ,changes to 14C isotope distributions in Glc 6-P, which emerge from the metabolism of the substrates [2-14C]-glucose or [3-14C]-glucose in PC. Calculating the different distributions of labeled carbon to infinite cycles, for all percentage contributions of PC, is a difficult mathematical problem that was solved entirely by Joseph Katz. Katz is not only a gifted biochemist but also an equally talented mathematician and innovative metabolic theorist. The acceptance of a PC definition imposed agreement that all 14C-labeled Fru 6-P formed by the Fig. 2 reaction sequence is converted to Glc 6-P and recycled again through the oxidative segment reactions. The quintessence of all measurement methods has involved the development of mathematical expressions that describe the rhythmical and ordered redistributions of either carbons 2 or 3 from labeled substrate glucose, into positions 1, 2, and 3 of the hexose 6-P products for any percentage contribution of PC. Such a theoretical distribution is a unique property of the PC. Experimental data for the C-1/C-2 and C-3/C-2 ratios, which have definite limits and values imposed by Katz and Wood theory (49, 50), are used in specially derived equations that calculate the PC contribution relative to the total metabolism of glucose. The above statements cannot be qualified. They derive from the fixed order of the reactions of Fig. 2, which is the mechanistic basis for all the formulas of all the measurement papers (Reference 1, p. 753). The amounts of 14C in C-1, C-2, and C-3 of Glc 6-P or its derivatives and the precise ratios of their isotopic labeling are the identity badge of the classically defined PC reaction sequence. They are also the foundation of its theories of existence and quantitation. It was therefore intriguing to note that, notwithstanding the initial ambiguities in the reaction order of Fig. 2 using liver enzymes plus the failed efforts of Katz (47) and Hiatt (48) to support it, that no less than ten independent measurements over nearly 30 years failed to find any significant level of PC in liver (Reference 1, p. 766). Liver is a very rich source of the enzymes of the PPP (6, 14, 42, 52), and it provides, among other things, a ready display of the reactions of Figs. 1 and 2. Thus, the failure by all measurement investigations to find an F-type PC in liver was mystifying. The mystery deepened when it was noted that the formulation of their elegant measurement theory required Katz and Wood to abandon, indeed fail to mention, their earlier reservation and criticism of the substandard evidence for the reaction sequence of PPP (46) and the failure (by JK) to confirm its presence in liver (47).
Search for an Alternative Reaction Scheme for the Pentose Pathway
A resolution of the mystery was sought in the author’s laboratory, with investigations that commenced with the propositions that the scheme of Fig. 2 may be an erroneous prediction of the PPP mechanism and the coupling of reactions of Figs. 1 and 2 into a metabolic cycle (PC) [reaction (13)] had no firm experimental support. For example, the McLean group (Reference 14, p. 110) frequently reported, during a decade of investigations, that the enzymes of Figs. 1 and 2 did not behave as “constant proportion groups.” Rather their group values showed a well-defined segregation into unlinked oxidative and nonoxidative enzyme clusters with activities that independently varied.
The L-Type Pentose Cycle
The following three sets of findings summarise selected aspects of progress in the unraveling of a new PPP reaction sequence in liver that was found to measure 20-30% of total glucose metabolism. The experimental history of these events is fully recorded by Williams et al. in References 42 and 53 and in Reference 1 (pp. 766-790).
First, the foundational experiment that established the F-type PPP was repeated using [1-14C]-Rib 5-P and the same rat liver enzyme preparation. However, the reaction mixture was sampled for the labeled Glc 6-P product at a series of much shorter time intervals and right up to the 17-hour termination point that was described for the original work. The results (Table 1) showed a patterned assortment of label distributions in Glc 6-P, which drifted from 8 hours to 17 hours toward the isotope composition in C-1 and C-3 that was noted originally by Horecker et al. (40). However, in this study, the C-1/C-3 ratio at 17 hours was the prized value of 2. Moreover in the seven time samples, which commenced at 1 minute, Glc 6-P was heavily labeled in C-2, C-4, and C-6, whereas C-1 and C-3 only began to accumulate 14C-isotope after 3 hours of reaction. Although this was a study in vitro, it is obvious that liver cells in vivo do not take between 3 and 17 hours to elaborate a path of metabolism and that more enlightening events were being revealed by the isotope distributions in the samples analyzed between 1 minute and 30 minutes of reaction. Second, a carbon balance analysis of all compounds in the various reaction mixtures showed that the intermediates of Fig. 2 only accounted for 80% of the carbon in the Rib 5-P substrate (42). The compounds comprising the missing 20% were identified as sugar phosphates, mostly ketuloses (Fig. 4). These new reactants were isolated, shown to be radioactive, and identified as Seh 1,7-P2, D-manno-Heptulose 7-P, D-glycero-D-altro-Octulose 1,8-P2 (D-g -D-a -Oct); D-glycero-D-ido-Octulose 1,8-P2 (D-g -D-i - Oct) and a small amount of Ara 5-P (42). Octulose monophosphates and bisphosphates and Seh 1,7-P2 were measured in fresh liver by Paoletti et al. (54) and in spinach chloroplasts by Flanigan et al. (33). Last, the structures and order of the reactions of these sugar esters in a new and much modified reaction scheme for the PPP in liver are shown in Fig. 4. The new intermediary compounds were isolated easily from all incubations from 30 minutes to 17 hours. The scheme of Fig. 4 shows the new PPP with prediction14C-labeling patterns in the intermediates and products of the reactions. The reaction scheme of Fig. 4 was formulated initially from the distributions of 14C in the labeled Glc 6-P (42) and D-g -D-i –Oct 1,8-P2 (55) formed from [1-14C]-Rib 5-P during the early intervals of the repeat of the foundational experiment (42). The new pathway, called L-(liver) type PPP, is distinguished from depictions of the classic F-type PPP by the inclusions of Seh-1,7-P2 and octulose-(Oct)-monophosphates and bisphosphates together with Ara 5-P as new intermediates. Aldolase, phosphotransferase (PT), and D-Arabinose-5-phosphate ketol isomerase (EC 5.3.1.13) are new enzymes. The effects of mass transfer catalysis by TA were absent, but TA-exchange reactions (TAx) (56), which contributed the [4,6-14C]-labeling pattern to Glc 6-P during the first 8 hours were active (1, 42, 56). The TK reaction forming hexose 6-P in the L-type PPP (Fig. 4) used D-g -D-i -Oct 8-P as substrate. TK and Ald were also very active exchange catalysts (57-59).
Table 1. Percentage distribution of 14C in glucose 6-phosphate formed during the time course of reactions of [1-14C]-ribose 5-phosphate with rat liver enzyme preparation (42)
|
Carbon number |
1 min |
2 min |
3 min |
30 min |
3h |
8h |
17h |
|
1 |
1.40 |
0.70 |
1.30 |
1.20 |
28.50 |
40.50 |
56.50 |
|
2 |
44.10 |
43.80 |
13.00 |
11.80 |
17.70 |
17.60 |
12.40 |
|
3 |
1.10 |
0.10 |
3.30 |
1.10 |
10.50 |
16.50 |
24.40 |
|
4 |
12.20 |
2.60 |
5.10 |
7.10 |
5.50 |
4.00 |
2.00 |
|
5 |
0.50 |
7.10 |
0.40 |
0.60 |
0.00 |
4.60 |
1.60 |
|
6 |
40.70 |
45.70 |
76.90 |
78.20 |
37.80 |
21.80 |
3.10 |
|
Recovery (%) of 14C |
105.1 |
100.8 |
106.2 |
103.4 |
96.9 |
98.7 |
100.5 |
A clear demonstration that aldolase is a mandatory catalyst in liver PPP involved the immunochemical evidence of Bleakely et al. (43) who showed the total cessation of hexose 6-P formation when liver aldolase antibody titrated the removal of aldolase from the system where Rib 5-P was reacted with the same rat liver enzyme preparation that established the Fig. 2 scheme. Irrespective of the other contrary data, this evidence alone showed another reaction mechanism involved Ald in liver PPP. The claim that aldolase is an essential enzyme in the PPP was also supported by data of (60) in which, using an in vitro construction of a PPP preparation for the complete oxidation of Glc, noted the formation of Oct-P and the need to include aldolase and sedoheptulose 1,7-bisphosphatase for the construction system to work.

Figure 4. L-type pentose phosphate pathway reactions. The [1-14C] ribulose 5-P substrate is the product of reactions shown at Fig. 1. The reaction sequences illustrate the distinctive distribution of 14C in hexose 6-P and the following new intermediates: octulose monophosphate, sedoheptulose and octulose bisphosphates. Transaldolase exchange reactions involving ribose and arabinose 5-phosphates are shown. Other detail of the L-type pathway is given in the text. Reprinted from International Journal of Biochemistry, 19. Williams John. F., Arora Krishan K. and Longnecker John P. The pentose pathway: A random harvest, 69 Pages, 1987, with permission from Elsevier, http://www.sciencedirect.com/science/journal/13572725
Exposing the Problem of Assigning a Reaction Scheme to the Nonoxidative PPP
The reactions and enzymes shown in Fig. 4 occur in liver cytoplasm and are expected items in the soluble enzyme compartments of most animals. Adipocytes as well as perhaps the lactating mammary gland and some microorganisms may be exceptions. However, the failure to find incontrovertible evidence for both F-pathways and L-pathways that permitted the placement of C-3 to C-8 glycolyl phosphates in a reaction order and with stoichiometry that satisfied flux demands of a metabolic pathway or cycle is the crux of this 50-year-old enigma. Finding an answer to the problem proved to be quick, obvious, and simple. Uncovering irrefutable evidence and proof for the explanation was a more pressing task that was solved by Flanigan et al. (61). It was the universal use of 14C isotopes in prediction-labeling experiments to both inquire into mechanism and predicate theories for quantifying the F-type and L-type PPP that was the first fundamental error. The second misjudgment was lack of practical attention to the consequences of the glycolyl-group exchange reactions that are actively catalyzed by TKX, TAX, and AldX. The subscript X is used to distinguish exchange catalysis from the mass transfer activity of these enzymes. Despite some anecdotal warnings by the authors of References (49-51) that both exchange and mass transfer activities could be anticipated in PPP and PS investigations, the warnings were unheeded by the PC measurers. A rare exception, in which an exchange-enzyme control accompanied measurements of the L-type PC, is discussed by Williams (1) and Longnecker and Williams (62).
13C-NMR spectroscopy was used by Flanigan et al. (61) for the kinetic investigations of exchange reactions by group- transfer enzymes. Adoption of NMR technology silenced the unfailing criticism of14C labeling patterns that were measured by wet chemical methods. The maximum catalytic capacities for exchange by the three enzymes were all quantified in reaction mixtures at mass-transfer equilibrium. TAx was measured by the exchange rate of the 13C-TA dihydroxyacetone group with unlabeled Seh 7-P. TKx was measured by the rate of incorporation of a (2-13C)-TK glycolaldehyde group to unlabeled Fru 6-P and Aldx by the exchange of an unlabeled DHAP-Ald group from D-g -D-a-Oct 1,8-P2 to [1-13C]-Rib 5-P and measurement of the rate of formation of [4-13C]-D-g-D-a-Oct 1,8-P2. A comparison of the exchange capacities of these enzymes with the maximum nonoxidative PPP flux rates in three liver preparations showed that TKx and Aldx exceeded flux by 9-19 times in liver cytosol and acetone powder enzyme preparations in vitro and by 5 times in hepatocytes. TAx was less effective in exchange, only exceeding the flux rate by 1.6 times and 5 times in liver cytosol and acetone powder preparations, respectively. Values for the ratios of the rates of group exchange and pathway flux in liver are important because of the feature roles of hepatic tissue and of these preparations in the establishment and status of the Fig. 2 and Fig. 4 schemes in biochemistry. The prevalence of exchange activity was also investigated using the dominant TKx rates relative to the maximum PPP flux rates of normal and regenerating liver, Morris hepatoma, mammary carcinoma, melanoma, colonic epithelium, spinach chloroplasts, and epididymal adipocytes. TKx rates in these preparations exceeded PPP flux by 5-600 times (61).
Conclusion
A surfeit of evidence has accumulated slowly during the last 53 years to show that predictions and calculations based on 14C-distributions in PPP and PS products and intermediates are pointless and misleading. The isotope patterns cannot reveal the order of the reaction sequences, which can only be measured by an uncompromised net flow of carbon. Instead the 14C-patterns mainly reflect a composition of disorder, induced by random combinations of group exchanges that scramble, and obliterate, any underlying slow flux influences. The 14C ratios, which are used by measurement theoreticians, in the PC reaction products merely measure the net radioisotopic equilibrium of the combination of reactions catalyzed by TAx, TKx, and Aldx. Thus, the description and metabolic map of F-type (and the L-type PPP) has been an erroneous presentation since its first appearance in biochemical texts. In his monograph (11), Terry Wood correctly concluded that “radioisotopic studies of PPP (mechanism) have only created problems and their use should be eschewed and replaced by investigations that use pure enzymes and rigorous chemical procedures.”
How should we now view the PPP—with or without conceptual structures?
The reactions of Fig. 1 are not in contention. The enzymes and intermediates of Figs. 2 and 4 are probably present in most tissues, particularly where Ald activity is greater than that of TA. No secure evidence exists, however, that the individual reactions of either Figs. 1 or 2 are so linked that they constitute a metabolic pathway, with a flux generating step, ordered reactant, and end-product stoichiometry or fixed direction of operational flux. Nor is there a basis for belief that the PPP can operate as a metabolic cycle. The reaction array is best perceived as a surviving example of the unstructured primordial pools of reactants that preceded the evolution of primary metabolic pathways and cycles currently encountered in biochemistry. The pentose phosphate pool is a reservoir of glycolyl phosphates, with as many accessible inputs and exits as there are intermediates in Figs. 2 and 4. This reservoir includes a link with hexose and triose phosphates of the glycolytic pathway that may react reversibly to form or be formed from pentose phosphates by near-equilibrium mass-action effects and in the process mix with other glycolyl phosphates that are neither constrained by stoichiometric obligation to connecting reaction sequences nor removed freely into or supplied by the actions of other metabolism.
References
1. Williams JF, Arora KK, Longnecker JP. The pentose pathway: a random Harvest. Impediments which oppose acceptance of the classical (F-type) pentose cycle for liver, some neoplasms and photosynthetic tissue. The case for the L-type pentose pathway. Int. J. Biochem. 1987; 19:749-817.
2. Horecker BL. The pentose phosphate pathway. J. Biol. Chem. 2002; 277:47965-47971.
3. Barron ESG, Harrop GA. Studies on blood cell metabolism II. The effect of methylene blue and other dyes upon the glycolysis and lactic acid formation of mammalian and avian erythrocytes. J. Biol. Chem. 1928; 79:65-87.
4. Florkin M. The respiratory chain. In Comprehensive Biochemistry, vol 31. Part III. Florkin M, Stotz EH, eds. 1975. Elsevier/ North Holland Biomedical Press, Amsterdam. p. 370.
5. Warburg O, Christian W. Uber activierung du Robisonschon hexose-mono-phosphorsaure in roten blutzellenerung und die gewinnung aktivierender fermentlosungen. Biochem. Z. 1931; 242: 206-227.
6. Casazza JP, Veech RL. The interdependence of glycolytic and pentose cycle intermediates in ad libitum fed rats. J. Biol. Chem. 1986; 261:690-698.
7. Horecker BL, Smyrniotis PZ, Seegmiller JE. The enzymatic conversion of 6-phosphogluconate to ribulose 5-phosphate and ribose 5-phosphate. J Biol. Chem. 1951; 193:383-396.
8. Warburg O, Christian W, Griese A. Wasserstuffubertrangendes co-ferment seine zusammensetzung und wirkungsweise. Biochem. Z. 1935; 282:157-205.
9. Engelhardt WA, Barkash AP. Oxidative breakdown of phosphogluconic acid. Biokhimiya 1938; 3:500-521.
10. Lehninger AL. Phosphorylation coupled to oxidation of dihydrodiphosphopyridine nucleotide. J. Biol. Chem. 1951; 190:345-359.
11. Klingenberg M, Bucher T. Biological oxidations. Ann. Rev. Biochem. 1960; 29:669-708.
12. Flatt JP, Ball EG. Studies on the metabolism of adipose tissue XV. An evaluation of the major pathways of glucose catabolism as influenced by insulin and epinephrine. J. Biol. Chem. 1964; 239:675-685.
13. Levy HR. Glucose-6-phosphate dehydrogenase. In Advances in Enzymolology. Vol. 431. MeisterA, ed. 1979. John Wiley & Sons, New York. pp. 97-192.
14. Wood T. The Pentose Phosphate Pathway. 1985. Academic Press, New York.
15. Haas E, Horecker BL, Hogness TR. The enzymatic reduction of cytochrome c reductase. J. Biol. Chem. 1940; 136:747-774.
16. Horecker BL. Triphosphopyridine nucleotide-cytochrome c reductase in liver. J. Biol Chem. 1950; 183:593-605.
17. Calvin M. The photosynthetic carbon reduction cycle. J. Chem Soc. 1956; 1895-1915.
18. Dickens F. Oxidation of phosphohexonate and pentose phosphoric acids by yeast enzymes. I. Oxidation of phosphohexonate. II. Oxidation of pentose phosphoric acids. Biochem. J. 1938;32:1626-1644 and Biochem. J. 1938; 32:1645-1653.
19. Dische Z. Phosphorylierung der im adenosin enthaltenden d-ribose und nachfolger zerfall des esters unter triosephosphatbildung im blut. Naturwiss 1938; 26:252-253.
20. Waldvogel MJ, Schlenk F. Enzymatic conversion of ribose into hexose-monophosphate. Arch. Biochem. 1947; 14:484-485.
21. Ball, EG. Regulation of fatty acid synthesis in adipose tissue. Adv. Enzyme Regl. 1967; 4:3-18.
22. Blackmore PF, Williams JF, Schofield PJ, Power PA. Mechanism and contribution of the pentose phosphate cycle to glucose metabolism in epididymal fat tisue. Int. J. Biochem. 1982; 14: 171-186.
23. Scott DBM, Cohen SS. Enzymatic formation of pentose phosphate from 6-phosphogluconate. J. Biol. Chem. 1951; 188:509-530.
24. Axelrod B, Jang R. Purification and properties of phosphoriboisomerase from alfalfa. J. Biol. Chem. 1954; 209:847-855.
25. Horecker BL, Smyrniotis PZ. The enzymatic formation of sedoheptulose phosphate from pentose phosphate. J. Am. Chem. Soc. 1952; 74:2123.
26. Benson AA, Bassham JA, Calvin M. Sedoheptulose in photosynthesis by plants. J. Am. Chem. Soc. 1951; 73:2970.
27. Benson AA, et al. The path of carbon in photosynthesis XV. Ribulose and sedoheptulose. J. Biol. Chem. 1952; 196:703-715.
28. Racker E. Enzymatic formation and breakdown of pentose phosphate. Fed. Proc. 1948;7:180.
29. Racker E, de la Haba G, Leder IG. Thiamin pyrophosphate, a coenzyme of transketolase. J. Am. Chem. Soc. 1953; 75:1010-1011.
30. Horecker BL, Smyrniotis PZ. Transaldolase: the formation of fructose-6-phosphate from sedoheptulose-7-phosphate. J. Am. Chem. Soc. 1953; 75:2021-2022.
31. Kornberg HL, Racker E. Enzymic reactions of erythrose 4-phosphate. Biochem. J. 1955;61:iii-iv.
32. Williams JF, Blackmore PF, Duke CC, MacLeod JK. Fact, uncertainty and speculation concerning the biochemistry of D-erythrose 4-phosphate and its metabolic roles. Int. J. Biochem. 1980; 12:339-344.
33. Flanigan IL, MacLeod JK, Williams JF. A re-investigation of the path of carbon in photosynthesis utilyzing GC/MS methodology. Unequivocal verification of the participation of octulose phosphates in the pathway. Photosynth. Res. 2006; 90:149-159.
34. Ashwell G, Hickman J. Formation of xylulose 5-phosphate from ribose 5-phsphate in spleen extracts. J. Am. Chem. Soc. 1954; 76:5889.
35. Srere PZ, Cooper JR, Klybas V, Racker E. Xylulose 5-phosphate, a new intermediate in the pentose phosphate pathway. Archiv. Biochem. Biophys. 1955; 59:535-538.
36. Stumpf PK, Horecker BL. The role of xylulose 5-phosphate in xylose metabolism by Lactobacillus pentosus. J. Biol. Chem. 1956; 218:753-768.
37. Dickens F, Williamson DH. Pentose phosphate isomerase and epimerase from animal tissues. Biochem. J. 1956; 64:567-578.
38. Racker E. Alternate pathways of glucose and fructose metabolism. Adv. Enzymol. 1954; 15:141-181.
39. Mittenthal JE, Yuan AO, Clark B, Scheeline A. Designing metabolism: Alternative connectivities for the pentose phosphate pathway. Bull. Math. Biol. 1998; 207:393-403.
40. Horecker BL, Gibbs M, Klenow H, Smyrniotis PZ. The mechanism of the conversion of pentose phosphate to hexose monophosphate I. With liver enzyme peparation. J. Biol. Chem. 1954; 207:393-403.
41. Gibbs M, Horecker BL. The mechanism of the conversion of pentose phosphate to hexose monophosphate II. With pea leaf and pea root preparations. J. Biol. Chem. 1954; 208:813-820.
42. Williams JF, Clark MG, Blackmore PF. The fate of 14C in glucose 6-phosphate synthesised from [1-14C. ribose 5-phosphate by enzymes of rat liver. Biochem. J. 1978; 176:241-256.
43. Bleakley PA, Arora KK, Williams JF. Evidence that aldolase and D-arabinose 5-phosphate are components of pentose pathway reactions in liver in vitro. Biochem. Int. 1984; 8:491-500.
44. Horecker BL, Mehler AH. Carbohydrate metabolism. Ann. Rev. Biochem. 1955; 24:207-274.
45. Gunssalus IC, Horecker BL, Wood WA. Pathways of carbohydrate metabolism in microorganisms. Bacteriol. Revs. 1955; 19:79-128.
46. Wood HG. Significance of alternate pathways in the metabolism of glucose. Physiol. Revs. 1955; 35:841-859.
47. Katz J, Abraham S, Hill R, Chaikoff IL. The occurrence and mechanism of the hexose monophosphate shunt in rat liver slices. J. Biol. Chem. 1955; 214:853-868.
48. Hiatt HH. Glycogen formation via the pentose phosphate pathway in mice in vivo. J. Biol. Chem. 1957; 224:851-859.
49. Wood HG, Katz J. The distribution of 14C in the hexose phosphates and the effects of recycling in the pentose cycle. J. Biol. Chem. 1958; 233:1279-1282.
50. Katz J, Wood HG. The use of glucose 14C for the evaluation of the pathways of glucose metabolism. J. Biol. Chem. 1960; 235:2165-2177.
51. Wood HG, Katz J, Landau BR. Estimation of pathways of carbohydrate metabolism. Biochem. Zeit. 1963; 338:809-847.
52. Novello F, Mc Lean P. The pentose phosphate pathway of glucose metabolism: measurement of the non-oxidative reactions of the cycle. Biochem. J. 1968; 107:775-791.
53. Williams JF, Blackmore PF, Clark MG. New reaction sequences for the non-oxidative pentose phosphate pathway. Biochem. J. 1978; 176:257-282.
54. Paoletti F, Williams JF, Horecker BL. Detection and estimation of sedoheptulose and octulose mono- and bisphosphates in extracts of rat liver. Archiv. Biochem. Biophys. 1979; 198:620-626.
55. Williams JF, Clark MG, Arora KK. 14C-labelling of octulose bisphosphates by L-type pentose pathway reactions in liver in situ and in vivo. Biochem. Int. 1985; 11:97-106.
56. Williams JF, Clark MG, Arora KK, Reichstein IC. Glucose 6-phosphate formation by L-pentose pathway reactions of rat liver in vitro: further evidence. Hoppe Seyler’s Zeit. Fur Biochemie. 1984; 365:1425-1434.
57. Clark MG, Williams JF, Blackmore PF. The transketolase exchange reaction in vitro. Biochem. J. 1971; 125:381-384.
58. Clark MG, Williams JF, Blackmore PF. Exchange reactions in metabolism. Catal. Rev. Sci. Eng. 1974; 9:35-77.
59. Bartlett MRE, Collins JG, Flanigan IL, MacLeod JK, Williams JF. CO2 labelling of octulose bisphosphates during photosynthesis. An NMR study using intact spinach leaves. Biochem Int. 1989; 8:491-500.
60. Cori D, Racker E. The oxidative pentose phosphate cycle. V. Complete oxidation of glucose 6-phosphate in a reconstructed system of the oxidative pentose phosphate cycle. Archiv. Biochem. Biophys. 1959; 83:195-205.
61. Flanigan IL, Grant-Collins J, Arora KK, MacLeod JK, Williams JF. Exchange reactions catalysed by group transferring enzymes oppose the quantitation and unravelling of the identity of the pentose pathway. Europ. J. Biochem. 1993; 213:477-485.
62. Longnecker JP, Williams JF. Measurement of oxidative and non-oxidative pentose phosphate cycle. [5-14C] Glucose in isolated hepatocytes. Biochem. J. 1980; 188:859-865.
Further Reading
Bassham JA. Mapping the carbon reduction cycle: a personal perspective. Photosynth. Res. 2002; 76:35-52.
Florkin M. The pentose phosphate cycle. In Comprehensive Biochemistry, vol. 33A. Part V. Florkin M, Stotz EH, eds. 1979. Elsevier/ North Holland Biomedical Press, Amsterdam. pp. 61-79
Horecker BL. Alternative pathways of carbohydrate metabolism in relation to evolutionary development. Proc. 5th Int Congress of Biochemistry. 1961. Moscow, Pergamon Press, Oxford. pp. 86-93.
Katz J. The use of glucose 14C in the study of the pathways of glucose metabolism in mammalian tissues. Radioactive Isotopes in Physiology, Diagnostics and Therapy. Schweigk H, Turba F, eds. 1961. Springer, Berlin. pp. 705-751.
Veech RL. A humble hexose monophosphate pathway metabolite regulates short- and long-term control of lipogenesis. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:5878-5880.
See Also
Glucose Homeostasis, Chemistry of
Lipid Homeostasis, Chemistry of
Metabolic Labelling of Sugars
Metabolism, Cellular Organisation
Nucleic Acid Metabolism, Chemistry of
Redox Regulation and Signalling: Reactive Oxygen Species (R O S)