CHEMICAL BIOLOGY
Peptide Nucleic Acids
Catalina Achim, Bruce A. Armitage, Danith H. Ly and James W. Schneider, Carnegie Mellon University, Pittsburgh, Pennsylvania
doi: 10.1002/9780470048672.wecb435
Peptide nucleic acid (PNA) is a chimeric molecule that consists of hydrogen-bonding purine and pyrimidine heterocycles attached to a pseudopeptide backbone. The bases allow the recognition of specific DNA or RNA sequences, which results in hybrid multihelical structures. The lack of a negative charge on the PNA backbone eliminates Coulombic repulsion from target DNA and RNA strands, which results in high affinity hybridization. These properties have led to a diverse set of applications for PNA, which includes antisense/antigene inhibition of gene expression, various DNA/RNA detection assays, nucleic acid labeling and purification technologies, and programmed assembly of nanoscale materials. The modular design of PNA has led to the synthesis of several ''next-generation'' analogs that promise to improve PNA's performance in many existing applications as well as to open doors to new applications. Although most prior research on PNA has focused on its nucleic acid-like character, recent developments of the peptide-like aspects of PNA promise to stimulate additionally the work on this fascinating DNA mimic.
The DNA double helix is the first molecular icon, a structure that is readily associated with its function, even by people with little or no science education. Over the past half century, the structure has inspired chemists along three lines that are familiar to those who have worked at the interface between chemistry and biology. The first group of chemists synthesized in the laboratory what nature produces enzymatically, and they were motivated by both fundamental and practical needs. The second group of chemists understood the relationship between structure and function, where synthetic chemistry allows atomic-scale changes in the DNA structure. The third group of chemists identified DNA as a potential target for molecular recognition, with the ability to regulate gene expression using synthetic compounds as the ultimate goal. These lines of inquiry are not necessarily parallel, but rather they become intertwined at various points, most notably in the area of synthetic oligonucleotides. A variety of DNA analogs have been synthesized and characterized, which feature modifications to the deoxyribose ring, the phosphodiester linkage, and the heterocyclic nucleobases. Many of these modifications have been combined within the same structure to optimize the oligonucleotide for a specific application.
Most synthetic DNA analogs represent logical departures from the natural structure. However, one of the most radical structural modifications of DNA to appear in the literature is peptide nucleic acid (PNA, Fig. 1), in which the sugar-phosphate backbone is abolished entirely in favor of a pseudopeptide. PNA was first reported by Nielsen et al. in 1991 from the University of Copenhagen (1). The structure is reminiscent of both proteins and nucleic acids, hence the name, and has led to considerable interest in its possible role in prebiotic chemistry.
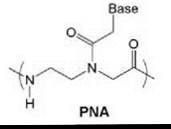
Figure 1. Chemical structure of PNA.
An intriguing molecular structure and origins-of-life scenarios hardly explain the intense research effort dedicated to PNA, whereas the original 1991 Science article has been cited over 1100 times as of September 2006. Rather, it is what PNA does in the presence of DNA and of RNA that has captured the imagination of chemists and of biologists. Although PNA was conceived originally as a triplex-forming oligomer that would bind in the major groove of double-stranded (ds) DNA, the earliest reports demonstrated that homopyrimidine PNA binds to ds DNA by a strand invasion mechanism. First, one PNA strand displaces the homologous homopyrimidine DNA strand locally and forms Watson-Crick base pairs with the complementary homopurine DNA target (Fig. 2) (1, 2). A second PNA strand then binds to the major groove of the hybrid to form a very stable PNA2-DNA triplex. This discovery was very exciting because targeting ds DNA opens the door to the regulation of gene expression at the transcriptional level. Several variations on this “strand invasion” recognition mechanism that ease the sequence requirements have been reported in the ensuing 15 years, and research is ongoing (see Further Reading).
PNA is also capable of binding to complementary single-stranded (ss) DNA and to RNA mixed-sequence (i.e., purine-containing and pyrimidine-containing) targets by Watson-Crick base pairing to form hybrid duplex structures (Fig. 2) (3). As with the triplex structures mentioned above, these hybrids exhibit high thermodynamic stability partly because of the lack of electrostatic repulsion between the PNA and DNA/RNA. High affinity and excellent mismatch discrimination led to many PNA applications that range from the inhibition of mRNA processing and translation to sensing, diagnostics, and imaging. The original Nature article (3) that describes Watson-Crick PNA-DNA and PNA-RNA duplex formation has been cited over 700 times to date.
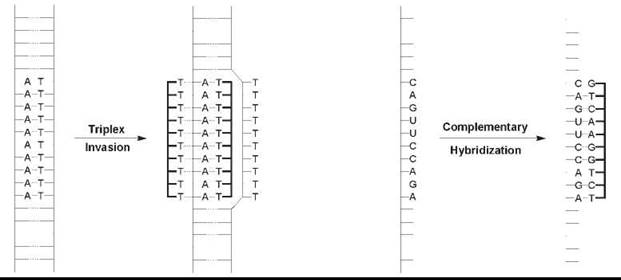
Figure 2. Most common DNA/RNA binding modes. Left: Strand invasion of homopyrimidine PNA into duplex DNA yields a PNA2-DNA triplex. Right: Hybridization of mixed sequence PNA with complementary DNA or RNA produces Watson-Crick base-paired duplex structures.
PNA recognition of DNA and RNA has been extended recently to a new binding mode, named “homologous hybridization” (4, 5). In this format, guanine-rich PNA probes recognize homologous DNA or RNA targets by forming hybrid guanine quadruplexes. Great interest exists in G-quadruplexes for their suspected roles in the regulation of gene expression at the transcriptional and translational levels, so this added binding mode could expand the range of biologic targets for PNA.
This article focuses on the interplay between chemical properties and biologic applications of PNA. Besides strategies to regulate gene expression, PNA-based biotechnologies will be presented. Although the vast majority of these studies have used the original PNA structure, the next-generation PNAs that feature backbone and nucleobase modifications are likely to be used in future applications. Finally, to illustrate the versatility of this fascinating molecule, the use of PNA in materials science will be summarized. The growing footprint of nanotechnology in biologic research warrants careful consideration of the opportunities presented by PNA’s unique properties. As space limitations prevent an exhaustive description of PNA research, readers are referred to several recent reviews for additional information (see Further Reading).
PNA as Antisense and Antigene Agents
Compounds that bind sequence selectively to single-stranded RNA or double-stranded DNA have the potential to regulate gene expression by antisense or antigene mechanisms, respectively. The high affinity and sequence selectivity exhibited by PNA sparked much interest in the possible use of DNA in these applications. Furthermore, the biochemical stability of PNA, as it is not susceptible to cleavage by nuclease nor by protease enzymes, raised hopes that PNAs could be effective in vivo as well as in vitro. Experiments in which the PNAs were targeted either to RNA or to DNA are described in this section.
Antisense applications
In the classic antisense approach, the binding of an oligonucleotide to a complementary region of a mRNA prevents translation via three possible mechanisms: 1) binding of the antisense agent blocks ribosome assembly on the mRNA, 2) an actively translating ribosome cannot proceed past the steric block presented by an antisense agent bound within the coding region of the mRNA, or 3) the hybrid duplex formed by the antisense agent and the mRNA recruits RNase H, which degrades the mRNA strand. PNA-RNA hybrids are not substrates for RNase H, which leaves the first two mechanisms available for PNA antisense agents. Scattered reports indicate that PNAs targeted to the coding region of an mRNA can inhibit translation, presumably by mechanism 2). However, two separate groups have reported that PNAs targeted to either the translation start codon or the 5'-untranslated region exhibited antisense effects, whereas PNAs targeted to the coding region did not (6, 7). The failure of the latter PNAs to block translation could be caused by the weak binding of the PNA to its target sequence, which could occur if the target sequence is part of a stable, folded region of the mRNA. Alternatively, the PNA could bind but be displaced by the ribosome as it translates through the PNA binding site. Regardless of the explanation, these studies indicate that PNAs that interfere with ribosome assembly are most likely to be effective antisense agents.
Three other examples of RNA-targeted PNAs that block gene expression should be mentioned. In the first case, the PNA is not targeted to the mRNA but to the unspliced pre-mRNA present in the nucleus immediately after transcription. Splicing eliminates introns from the pre-mRNA and results in the ligation of exons to produce the mature mRNA. Alternative splicing, in which the same pre-mRNA produces different mRNAs based on the use of different splice sites, increases the complexity of the genome. This process has been implicated in certain genetic diseases, where the use of one splice site leads to a normal protein, whereas the use of a different splice site can lead to a defective protein (and the absence of the normal protein). Cell culture experiments with complementary PNAs targeted to a splice site used to express the membrane isoform of IL-5Ra shifted expression in favor of the soluble isoform, as predicted (8). Meanwhile, antisplicing PNAs targeted to an aberrant splice site in the pre-mRNA for an EGFP reporter gene were shown to shift the expression pattern in favor of the normal splice site, both in cell cultures and in mice (9).
The second example of nonmRNA targeting involved PNAs directed against ribosomal RNA (rRNA), which were investigated as potential antibiotics because bacterial and human rRNA do not have the same sequence. The appeal of targeting rRNA is that in principle the PNA can shut down all protein synthesis, rather than translation of a single mRNA. The strongest effects observed in both cell-free translation and cell growth assays were exhibited by homopyrimidine PNAs with the capacity to form triplex structures with homopurine targets in the peptidyl transferase center and α-sarcin loop (10).
Many different RNA-protein complexes exist in the cell besides the ribosome, and any of these complexes are potential targets for PNA hybridization. Several researchers focused on telomerase, which is responsible for elongating the single-stranded region of the telomere during replication. Telomerase is absent or inactive from most healthy somatic cells, but it is upregulated in many cancer cells. Because the maintenance of telomere length seems to be correlated with a lack of apoptosis, telomerase is an attractive anticancer drug target. Telomerase uses its RNA component as a template to extend the DNA strand, whereas its protein component is a reverse transcriptase enzyme. PNAs targeted to the RNA template, as well as to peripheral sites in the RNA, block telomerase activity at low nanomolar concentrations both in vitro and in cell culture (11).
Antigene applications
PNA exhibits the rare ability to bind sequence selectively to double-stranded DNA. As described, homopyrimidine PNAs can bind to homopurine targets by a triplex invasion mechanism. In addition, PNA pairs can invade specific complementary targets in ds DNA if the affinity of the two PNAs for one another is reduced relative to the PNA-DNA hybrids. This task can be accomplished by incorporating pseudocomplementary bases in place of adenine and thymine (12). (See the “Nucleobase Modifications” section.) A significant challenge to using homopyrimidine or pseudocomplementary PNAs in vivo is the very slow kinetics of strand invasion at physiologic ionic strength and temperature (13). Although the attachment of cationic peptides (14) or intercalating dyes (15) to the PNA seem to accelerate strand invasion into plasmid DNA in vitro, no reports exist on the kinetics or thermodynamics of PNA hybridization to nucleosomes in vitro, let alone in cells. It remains to be seen whether these classes of PNAs will become important components in the chemical biologist’s toolbox.
A recent report showed how to exploit DNA when it is most vulnerable, i.e., when the double helix is unwound transiently and locally (16). This occurs in the vicinity of the transcription start site prior to the transcription of any gene, and the “open complex” is sufficiently long-lived so that a complementary PNA can bind and prevent transcription. PNAs targeted to the promoter for expression of the human progesterone receptor (hPR) exhibited potent antigene effects in a human breast cancer cell line, whereas control PNAs that were not complementary to the hPR promoter did not reduce gene expression. This strategy for inhibiting transcription could be applied to other genes.
In summary, PNAs have shown promising antisense and antigene effects in vitro, in cell culture, and in vivo. Although challenges remain to achieve optimal biodistribution and pharmacokinetics, the ability to modify the PNA backbone directly or to attach peptide transporter groups easily should enhance cell uptake and selectivity in delivering PNAs to targeted cells, which will extend the appeal of PNAs beyond simply having higher affinity than most synthetic oligonucleotides.
Probes, Labels, and Sensors
Hybridization between complementary oligonucleotide strands is the basic process that underlies several biotechnologic techniques that constitute a multibillion-dollar-per-year industry. DNA/RNA microarrays, polymerase chain reactions (PCRs), single nucleotide polymorphism (SNP) assays, and fluorescence in situ hybridization (FISH) are only some techniques that rely on Watson-Crick base pairing. Although each of these methods works when using a synthetic DNA oligonucleotide as a capture or detection probe, the higher affinity and nuclease stability of PNA offers obvious advantages (except in PCR, where a pure PNA oligomer cannot serve as a primer). Besides allowing lower concentrations of probe to be used, which helps to minimize hybridization to unintended targets that have a similar sequence, PNA can invade stable secondary or tertiary structural elements to gain access to its target. These folded structures impose thermodynamic and kinetic penalties to any hybridization probe; the higher affinity of PNA combined with its lack of electrostatic repulsion from DNA and RNA leads to significant advantages of PNA over most oligonucleotides based on polyanionic backbone chemistries. This section illustrates a few applications of PNA in biotechnology.
Fluorescent PNA probes
PNA probes have been used for FISH experiments, and commercial kits are available for microbial targets (17). Typically, the probes present in these kits hybridize to the abundant ribo- somal RNA from bacteria. A recent paper illustrated the value of PNA as a hybridization probe to detect Legionella pneumophila (18). Discrimination between this and other species of Legionella bacteria required a probe that could bind to sites that typically are classified as “low affinity” because of the poor hybridization of DNA probes. PNA probes showed absolute specificity, which identified correctly 47 different strains of bacteria and distinguished between pneumophila and other Legionella species.
PNA FISH probes also have been used for telomere analysis (19). Telomeres consist of hundreds of repeats of a short sequence (e.g., 5'-TTAGGG-3' in humans). Therefore, many copies of a complementary nucleic acid probe can hybridize to each telomere in a cell, which leads to a bright fluorescence signal. The high affinity of PNA allows the use of probes shorter than DNA probes, which means that more copies of the probe can hybridize to the telomeres.
A third application for fluorescent PNA probes is to introduce fluorescent labels into the RNA site specifically (20). For example, PNA probes have been hybridized at exonic sites that flank consecutive splice sites in a pre-mRNA from yeast. Forster resonance energy transfer (FRET) donor and acceptor dyes were attached covalently to the PNAs, and low FRET efficiencies were observed when the PNAs were hybridized to the pre-mRNA. However, when hybridized to the mRNA produced by splicing, large increases in FRET have been observed both in bulk solution and on a glass slide where single-molecule measurements could be made. As in the FISH applications, the ability to use short PNA probes to deliver the fluorescent dye to a desired location decreased the likelihood that the PNA will disrupt the structure and the function of the RNA that is under investigation.
PNA can also be functionalized with “fluorogenic” dyes, that is, dyes that exhibit enhanced fluorescence in response to a change in the environment. Unsymmetrical cyanine dyes developed originally as DNA stains can be attached covalently to PNAs to create fluorogenic probes, and chemistries have been developed that allow attachment of the dye at internal (21) as well as at terminal positions (22).
Finally, the intrinsic properties of PNA led to an interesting variation on the molecular beacon concept. As devised originally, a DNA probe that bears a 5'-terminal and 3'-terminal fluorophore (F) and quencher (Q) groups, respectively, can be designed to fold into a hairpin in which the fluorescence is quenched because of the close proximity of F and Q (23). Subsequent hybridization with a complementary DNA opens the hairpin and the increase in distance between F and Q restores the fluorescence. This design necessitates that the terminal DNA sequences are complementary to stabilize the hairpin conformation by Watson-Crick base pairing. If F and Q groups are attached to a PNA probe, the terminal sequences are not required (24), which is most likely caused by the tendency of PNA to adopt a compact rather than an extended structure in the absence of a complementary strand. These “stemless” PNA beacons ease the sequence constraints that complicate the DNA molecular beacon design.
Probes for nonfluorescent DNA/RNA detection
PNA has been tested in a wide variety of DNA detection modes where unmodified DNA can also be used. However, besides the standard advantages noted above for PNA (high affinity, good mismatch discrimination), many of these assays exploit the ability of PNA to hybridize at low ionic strength, conditions that can denature competing secondary/tertiary structure in the target nucleic acid. Moreover, use of PNA with its neutral backbone as a probe for hybridization at surfaces eliminates the substantial charge repulsion between the DNA/RNA target and immobilized DNA probes.
An interesting new approach to DNA detection involves probes attached to polymer microspheres loaded with a scintillant (25). The hybridization of a complementary, radiolabeled target DNA leads to strong signal amplification because of the close proximity of the scintillant and the radiolabel. Another appealing aspect of this method is that the PNA probe can be synthesized directly on the scintillant-loaded microspheres.
PNA probes have also been used for in vivo imaging of mRNAs (26). In this application, the PNA is functionalized with a cell-penetrating peptide and a magnetic resonance imaging (MRI) contrast agent. Cell uptake and hybridization to the mRNA target allowed MRI imaging of gene expression both in cell culture and in live rats.
Another method for nonfluorescent detection of DNA involves the noncovalent binding of an inexpensive cyanine dye to PNA-DNA duplexes (27). The dye assembles into a helical aggregate in the presence of PNA-DNA, which results in a vivid blue-purple color change. This phenomenon has been used in SNP detection assays.
PCR clamping
An early application for PNA is in a method known as “PCR clamping,” which improves the detection of single nucleotide polymorphisms (SNPs) in samples where mutant DNA of interest is present in a very low amount relative to wild type (28). This situation can occur, for example, when only a few mutant cancer cells are present along with a large excess of healthy cells in a tissue biopsy. The challenge is to detect the low abundance of mutant sequence against the high background of wild-type sequence. One approach to improve the signal-to-noise ratio in such situations is to amplify selectively the mutant DNA by PCR. This is where PNA is particularly helpful. PCR primers are designed to overlap the SNP site present in the cancer cells and then a PNA oligomer that is fully complementary to the wild-type DNA (and therefore has a single mismatch to the mutant DNA) is synthesized. The PNA preferentially hybridizes to the wild-type DNA in competition with the PCR primer that is complementary to the same site, which reduces the amplification of the wild type relative to the mutant DNA. This method allows SNP detection when the wild-type DNA is present in up to 20,000-fold excess relative to the mutant.
Bioseparations
A growing need exists to develop sequence-specific means to purify nucleic acids, both for genomic analysis and for the larger scale purification of plasmid DNA for gene therapy. The sequence-specific binding properties of PNA can be used in affinity separations of DNA as an alternative to standard DNA purification methods such as anion-exchange chromatography and density-gradient techniques. This section describes several such applications of PNA.
Analytical applications
The hybridization of PNA to a complementary DNA or RNA leads to changes in the properties of the nucleic acid. One of these changes occurs in the electrophoretic mobility, where the perturbation in the charge:size ratio because of the PNA hybridization causes significant shifts in the rate at which a given DNA or RNA migrates in an electric field. Adjusting conditions such as temperature allows preferential hybridization of the PNA to fully complementary targets versus those bearing even single mismatches, and it results in large separation during electrophoresis.
Purification applications
In addition to the analytical-scale applications described above, large-scale purification of nucleic acids using PNA probes has also been explored. Applications range from the purification of plasmid DNA to be used in gene therapy to the isolation of RNA and RNA-protein complexes from complex biologic samples. One approach to using PNA in this way is to link it to a solid support such as a polymer bead and then use the PNA-functionalized support to capture complementary DNA from solution. However, both biosensor and surface plasmon resonance data point to a reduction in the specificity and the binding kinetics to PNA probes that are attached directly to solid surfaces. The most effective “purification by hybridization” strategies circumvent this by two-step methods, in which PNA probes with some attachment functionality (e.g., hexahistidine peptide or biotin) bind targets in solution and the resulting PNA-DNA(RNA) hybrids are captured on beads or surfaces by either Ni-histidine (29) or streptavidin-biotin (30) binding.
A simple, more flexible attachment method is to append n-alkanes to PNAs for hybridization in solution (31, 32). The target DNA thereby is modified with a nonpolar tag that may adsorb selectively to nonpolar media, including alkyl-modified Sepharose and surfactant micelles (Fig. 3). In general, the covalent attachment of n-alkanes and other lipophilic materials impacts neither the duplex stability nor the sequence selectivity of PNA-DNA duplexes. The attachment of a 12-carbon alkane to PNA provides adequate resolution to separate 60-mer DNA targets from both noncomplementary oligomers and calf thymus DNA in hydrophobic interaction chromatography (HIC), an aqueous-based method. The resolution is improved greatly when 18-carbon alkanes are attached to PNA probes, even when the PNA is targeted to internal sequences of the target. The strand-invasion ability of PNAs can also be leveraged to purify dsDNA oligomers in HIC. Far greater resolution can be achieved when targeting dsDNA because DNA in duplex form interacts very weakly with the HIC media unless the alkylated PNA is attached.
Finally, using PNA as an affinity capture reagent recently was extended to probing RNA-protein complexes (RNPs) in cells (33). In this application, the PNA is functionalized with a peptide that allows uptake into cells and is complementary to an RNA component of an RNP. The PNA also bears two affinity tags, the first of which is a benzophenone-modified phenylalanine residue that can photocross-link the PNA to a protein present in the RNP. The second tag is a biotin group, which allows the purification of the cross-linked PNA-protein. Subsequent analysis by mass spectrometry identifies both the protein and its cross-linking site. As is the case for PNA used to deliver a fluorophore to a specific site in an RNA, this method requires that the PNA not disrupt the structure being probed.

Figure 3. PNA amphiphiles (representative structure shown) can form mixed micelles with surfactants such as SDS. Hybridization to complementary DNA or RNA allows separation from complex mixtures using open channel capillary electrophoresis.
PNA-encoded libraries
Another application of PNA that has emerged recently is a tag to encode and to screen combinatorial libraries of small molecules (34). For example, fluorescent PNA tags have been attached to individual members of a library of potential protease inhibitors. After incubating the library with a target protein, bound versus free compounds were separated by size-exclusion chromatography and the PNAs obtained from the bound fraction were hybridized to a DNA microarray. Any DNA to which fluorescent PNA was bound allowed decoding of first the PNA and then the compound from the library tagged by the respective PNA. The authors suggested alternative screening, profiling, and assay formats for this promising technology.
Next-Generation PNAs
PNA and its structural analogs are synthesized conveniently by standard solid-phase methods (35). The backbone chemistry of PNA allows the attachment of a diverse array of fluorescent dyes, metal-binding ligands, cell-penetrating peptides, and many other moieties that increase the functional capacity of the PNA. In addition to PNAs that simply are modified by attaching functional groups to the N-terminus or C-terminus, a large number of PNA analogs that feature integral modifications, i.e., alterations in the backbone or nucleobase components, have been reported (36). Most of these analogs were made in attempts to better understand the structure-function relationship of PNA so that the hybridization, solubility, and/or cellular uptake properties of PNA could be improved. In this section, we introduce several second-generation PNAs, classified as either having backbone or nucleobase modifications, and discuss their potential impact on the applications described above.
Backbone modifications
Most backbone-modified PNA analogs fall into one of four classes (Fig. 4). The first class was made by inserting a methylene group into the PNA backbone or the linker that connects the nucleobase to the backbone (Fig. 4, I). These modifications were made to assess the effect of chain length, in the backbone as well as in the linker, on the hybridization properties of PNA. Both structural changes induced significant destabilization of the PNA-DNA and PNA-RNA hybrid duplex. A single backbone modification lowered the Tm of a PNA-DNA duplex by 8-20° C and a PNA-RNA by 6-16° C, depending on where the modification was made. These results suggest that the original PNA design, N-(2-aminoethyl) glycine backbone and carboxymethyl linker, which has the same number of atoms in the backbone and in the linker as the DNA, is optimal for hybridization to DNA and RNA.
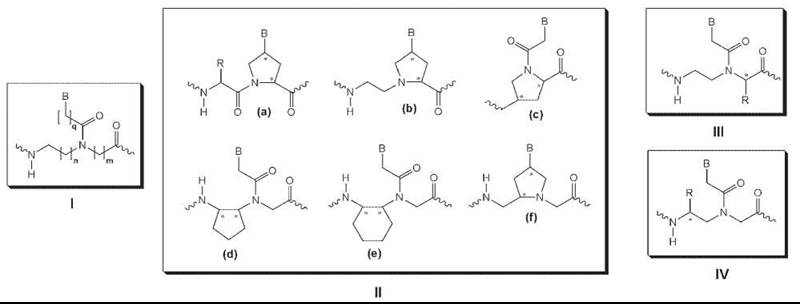
Figure 4. Examples of backbone-modified PNAs.
The second group of backbone modifications was made by introducing methylene bridges that connect the various functional groups in the backbone and in the linker (Fig. 4, II). These modifications were made in attempts to introduce structural preorganization into an otherwise randomly folded PNA backbone structure (37) and led to improvements in the binding affinity (38) and RNA versus DNA selectivity (39). Although this result supports the hypothesis that preorganization of the strands reduces the entropy penalty for duplex formation, additional structural analysis will be required to determine whether these backbone modifications do preorganize PNA into helical structures and whether these helical structures bear close resemblance to that of the targeted molecules, DNA and RNA.
The third group of backbone modifications was made by installing amino acid side chains with R or S configuration at the α-position of the N-(2-aminoethyl) glycine unit (Fig. 4, III) (40). Generally, these chiral PNAs were found to form slightly less stable PNA-DNA complexes than the original unsubstituted PNA for backbones that contain amino acid side chains with bulky, apolar groups. PNAs that contain negatively charged amino acid side chains, such as aspartic or glutamic acid, were shown to induce even greater destabilization, presumably because of the electrostatic and/or steric repulsion. On the other hand, incorporation of positively charged amino acid side chains, such as lysine or arginine, led to the stabilization of the PNA-DNA duplex, with R having a larger effect than S amino acids. Furthermore, the arginine-modified PNAs, also referred to as “GPNAs” because of the presence of guanidinium groups on the PNA backbone, exhibited substantially improved cell uptake relative to unmodified PNAs (41). This is likely because of the similarity between GPNA and various natural and synthetic compounds that feature multiple guanidinium groups and readily enter mammalian cells.
The fourth and perhaps most intriguing class of backbone modifications feature amino acid side chains installed at the γ-position of the N-(2-aminoethyl) glycine unit (Fig. 4, IV). Unlike the unmodified PNA or other PNA derivatives that have been developed so far, generally that do not fold into well-defined conformations, γ-modified chiral PNAs assume helical conformation (42). These chiral PNAs preorganize into either a right-handed or left-handed helix, which depends on the backbone configuration of the amino acids from which the PNA analogs were derived. Those PNAs that are derived from naturally occurring (and less costly) L-amino acids preorganize into a right-handed helix and bind to DNA and RNA with high affinity and sequence selectivity, whereas those derived from unnatural D-amino acids preorganize into a left-handed helix and do not bind effectively to either DNA or RNA.
Nucleobase modifications
Several modified nucleobases have been incorporated into PNA—many of which have been covered in a recent review (36). In general, any modified nucleobases can be incorporated into PNA as long as they can withstand the conditions used commonly in Boc or Fmoc solid-phase peptide synthesis. One of the first modified nucleobases to be incorporated into PNA was pseudo-isocytosine (J, Fig. 5). J is a synthetic analog of N-3 protonated cytosine and forms Hoogsteen base-pairs with guanine in triplexes at neutral pH. This enhances the stability of PNA2-DNA triplex invasion complexes significantly under physiologic conditions, although the kinetics of strand invasion at physiologic ionic strength is relatively slow.
Another important nucleobase to be incorporated into PNA was 2,6-diaminopurine (D, Fig. 5). D is a synthetic analog of adenine (A) that contains an extra exocyclic amine. When base-paired with the complementary thymine nucleobase, D forms three H-bonds as opposed to two H-bonds in base pairs of T with A. The extra H-bond provides additional stability to the hybrid duplex, with a gain in Tm of 3-5°C/bp for PNA-DNA. In addition to providing duplex stability, D has been exploited, in combination with another modified nucleobase, 6-thiouracil (sU), in the recognition of mixed-sequence, double helical DNA in a binding mode called “double-duplex invasion” (12). In this binding mode, both strands of the ds DNA are targeted simultaneously by two PNA strands. Replacement of the naturally occurring T and A nucleobases with D and sU, which form a less stable base pair than D-T and sU-A because of the steric interactions, prevents the two complementary PNA strands from hybridizing to one another to form a PNA-PNA complex. This strategy has been used successfully in the sequence-specific recognition and cleavage of mixed-sequence, ds DNA (43).
More recently, another modified nucleobase termed “guanidino G-clamp” (X, Fig. 5) has been incorporated into PNA (44). Guanidino G-clamp is an analog of cytosine, but unlike cytosine, which can form only three H-bonds with guanine, it can form five H-bonds—three through Watson-Crick and two through Hoogsteen base pairing. Besides the extra H-bonds, the guanidino G-clamp can stack effectively with the adjacent nucleobases because it contains a tricylic phenoxazine ring, and thus, it provides additional stability to the bound complex. No binding studies have been performed on guanidino G-clamp, but the introduction of a similar nucleobase analog that contains an amino instead of a guanidino group enhanced the stability of PNA-DNA and PNA-RNA duplexes by 23° C and 18° C per modification, respectively (45). Guanidino G-clamp is expected to confer even greater stabilization because of its ability to form five H-bonds as opposed to four in the case of amino G-clamp.
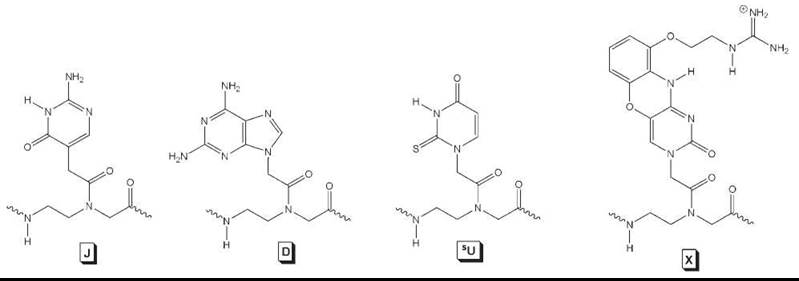
Figure 5. Modified nucleobases incorporated into PNA for improved DNA/RNA binding.
Nanotechnology and Materials Science
The information storage ability conferred to PNA by the nucleobases together with the chemical and biologic robustness of this synthetic nucleic acid have made PNA appealing particularly for use in the organization of chemical objects ranging in size from molecular-scale to nanoscale. The outcome of the interaction between PNA and these chemical entities are materials whose chemical composition and three-dimensional structure are encoded in the nucleobases and/or in the secondary structure of the PNA. The information stored in the PNA is “expressed” into the new structures by relatively weak chemical and physical forces, such as hydrogen or coordination bonding and hydrophobic interactions. The synthesis and the study of the properties of these pre-programmed materials provides information relevant for the understanding of fundamental biologic processes, such as molecular recognition and self-assembly, and of the rules based on which artificial enzymes and molecular electronic devices can be built. The PNA-based materials have the potential of being useful in biology, diagnostics, and therapeutics.
Toward the molecular end of the scale range, PNA has been shown to act as scaffold for transition metal ions situated at the core of PNA duplexes (46) (Fig. 6). The metal ion incorporation was realized by the chemical substitution of nucleobases with ligands. This process is site specific because the ligands have higher affinity for metal ions than the nucleobases. The existence of the metal ions within the PNA duplexes opens the possibility of directional electron transfer mediated by the metal ions, in a manner similar to that in which electron transfer metallo-proteins work in Nature. Also, some of the metal ions enhance electrochemical, optical, or magnetic resonance detection of the PNA strands or duplexes.
At the nanoscale end, the predictable molecular recognition process based on hybridization of ssPNA attached covalently to carbon nanotubes (47) or shell cross-linked nanoparticles (48) has been used to achieve controlled mechanical manipulation of nanosize objects and to organize them in large-scale architectures or on surfaces. The strong interaction between PNA and DNA has also been exploited in the synthesis of DNA-based nanostructures. For example, bisPNAs have been used as tools for rational design of nanosize, DNA-based locked pseudorotaxanes and catenanes. The strand invading ability of homopyrimidine bisPNAs allows them to be used as sequence-specific “openers” of DNA duplexes (49). The resulting single-stranded region of the DNA can in turn bind opposite ends of another single-stranded DNA that is circularized by DNA ligase to produce a catenane-like structure (50).
Biotinylated PNA oligomers have also found use as cross-linking agents to connect avidin with DNA three-way junction nanostructures, which yield hydrogels that grow and shrink in a temperature-dependent manner (51). Avidin-linked enzymes can be integrated into the hydrogel structure, where they can catalyze reactions of small substrates that diffuse into the pores of the gel (52). Cooling below room temperature led to precipitation of the gel and allowed the separation of the product and recovery of the enzyme.

Figure 6. Cartoon representation of a metal-containing, ligand-modified PNA duplex (a) and an example of metal-ligand alternative base pair (b).
Conclusion
The early reports of PNA’s abilities to bind to single-stranded and double-stranded nucleic acids with high affinity and selectivity generated much excitement that led to both in-depth characterization of the binding process as well as an impressive array of biologic, biotechnologic, and nanotechnologic applications. The pace of PNA research as measured by number of publications has grown steadily for the past 15 years and shows no sign of abating. Additional advances are expected to come as next-generation PNAs are developed, particularly those that feature backbone modifications and take advantage of the peptide-like character of PNA. Recent successes in improving antigene/antisense activity and cell uptake bode particularly well for future use of this most versatile of DNA mimics in chemical biology.
References
1. Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991; 254:1498-1500.
2. Cherny DY, Belotserkovskii BP, Frank-Kamenetskii MD, Egholm M, Buchardt O, Berg RH, Nielsen PE. DNA unwinding upon strand-displacement binding of a thymine-substituted polyamide to double-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:1667-1670.
3. Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden B, Nielsen PE. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature 1993; 365:566-568.
4. Datta B, Schmitt C, Armitage BA. Formation of a PNA2-DNA2 Hybrid Quadruplex. J. Am. Chem. Soc. 2003; 125:4111-4118.
5. Marin VL, Armitage BA. RNA guanine quadruplex invasion by complementary and homologous PNA probes. J. Am. Chem. Soc. 2005; 127:8032-8033.
6. Doyle DF, Braasch DA, Simmons CG, Janowski BA, Corey DR. Inhibition of gene expression inside cells by peptide nucleic acids: Effect of mRNA target sequence, mismatched bases, and PNA length. Biochemistry 2001; 40:53-64.
7. Knudsen H, Nielsen PE. Antisense properties of duplex- and triplex-forming PNAs. Nucleic Acids Res. 1996; 24:494-500.
8. Sazani P, Kang SH, Maier MA, Wei C, Dillman J, Summerton J, Manoharan M, Kole R. Nuclear antisense effects of neutral, anionic and cationic oligonucleotide analogs. Nucleic Acids Res. 2001; 29:3965-3974.
9. Sazani P, Gemignani F, Kang S-H, Maier MA, Manoharan M, Persmark M, Bortner D, Kole R. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nature Biotechnol. 2002; 20:1228-1233.
10. Good L, Nielsen PE. Inhibition of translation and bacterial growth by peptide nucleic acid targeted to ribosomal RNA. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:2073-2076.
11. Hamilton SE, Simmons CG, Kathiriya IS, Corey DR. Cellular delivery of peptide nucleic acids and inhibition of human telomerase. Chem. Biol. 1999; 6:343-351.
12. Lohse J, Dahl O, Nielsen PE. Double duplex invasion by peptide nucleic acid: A general principle for sequence-specific targeting of double-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:11804-11808.
13. Demidov VV, Yavnilovich MV, Belotserkovskii BP, Frank-Kamenetskii MD, Nielsen PE. Kinetics and mechanism of polyamide (“Peptide”) nucleic acid binding to duplex DNA. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:2637-2641.
14. Kaihatsu K, Braasch DA, Cansizoglu A, Corey DR. Enhanced strand invasion by peptide nucleic acid-peptide conjugates. Biochemistry 2002; 41:11118-11125.
15. Bentin T, Nielsen PE. Superior duplex DNA strand invasion by acridine conjugated peptide nucleic acids. J. Am. Chem. Soc. 2003; 125:6378-6379.
16. Janowski BA, Kaihatsu K, Huffman KE, Schwartz JC, Ram R, Hardy D, Mendelson CR, Corey DR. Inhibiting transcription of chromosomal DNA with antigene peptide nucleic acids. Nature Chem. Biol. 2005; 1:210-215.
17. Stender H, Fiandaca M, Hyldig-Nielsen JJ, Coull J. PNA for rapid microbiology. J. Microbiol. Methods 2002; 48:1-17.
18. Wilks SA, Keevil CW. Targeting species-specific low-affinity 16S rRNA binding sites by using peptide nucleic acids for detection of Legionellae in biofilms. Appl. Env. Microbiol. 2006; 72:5453-5462.
19. Rufer N, Dragowska W, Thornbury G, Roosnek E, Lansdorp PM. Telomere length dynamics in human lymphocyte subpopulations measured by flow cytometry. Nature Biotechnol. 1998; 16:743-747.
20. Robertson KL, Yu L, Armitage BA, Lopez AJ, Peteanu LA. Fluorescent PNA probes as hybridization labels for biological RNA. Biochemistry 2006; 45:6066-6074.
21. Kohler O, Jarikote DV, Seitz O. Forced Intercalation Probes (FIT Probes): Thiazole orange as a fluorescent base in peptide nucleic acids for homogeneous single-nucleotide-polymorphism detection. ChemBioChem. 2005; 6:69-77.
22. Svanvik N, Westman G, Wang D, Kubista M. Light-up probes: Thiazole orange-conjugated peptide nucleic acid for detection of target nucleic acid in homogeneous solution. Anal. Biochem. 2000; 281:26-35.
23. Tyagi S, Kramer FR. Molecular beacons: Probes that fluoresce upon hybridization. Nature Biotechnol. 1996; 14:303-308.
24. Kuhn H, Demidov VV, Coull JM, Fiandaca MJ, Gildea BD, Frank-Kamenetskii MD. Hybridization of DNA and PNA molecular beacons to single-stranded and double-stranded DNA targets. J. Am. Chem. Soc. 2002; 124:1097-1103.
25. McCairn MC, Hughes MD, Hine AV, Sutherland AJ. A platform for both solid-phase peptide nucleic acid oligomer synthesis and subsequent in situ detection and quantification of nucleic acid sequences. J. Comb. Chem. 2006; 8:639-642.
26. Heckl S, Pipkorn R, Waldeck W, Spring H, Jenne J, von der Leith C-W, Corban-Wilhelm H, Debus J, Braun K. Intracellular visualization of prostate cancer using magnetic resonance imaging. Cancer Res. 2003; 63:4766-4772.
27. Smith JO, Olson DA, Armitage BA. Molecular recognition of PNA-containing hybrids: Spontaneous assembly of helical cyanine dye aggregates on PNA templates. J. Am. Chem. Soc. 1999; 121:2686-5695.
28. Orum H, Nielsen PE, Egholm M, Berg RH, Buchardt O, Stanley C. Single base pair mutation analysis by PNA directed PCR clamping. Nucleic Acids Res. 1993; 21:5332-5336.
29. 0rum H, Nielsen PE, J0rgensen M, Larsson C, Stanley C, Koch T. Sequence specific purification of nucleic acids by PNA-controlled hybrid selection. BioTechniques 1995; 19:472-480.
30. Seeger C, Batz H-G, 0rum H. PNA-mediated purification of PCR amplifiable human genomic DNA from whole blood. BioTechniques 1997; 23:512-517.
31. Lau C, Bitton R, Bianco-Peled H, Schultz DG, Cookson DJ, Grosser ST, Schneider JW. Morphological characterization of self-assembled peptide nucleic acid amphiphiles. J. Phys. Chem. B. 2006; 110:9027-9033.
32. Vernille JP, Kovell LC, Schneider JW. Peptide nucleic acid (PNA) amphiphiles: Synthesis, self-assembly, and duplex stability. Bio- conjug. Chem. 2004; 15:1314-1321.
33. Zielenski J, Kilk K, Peritz T, Kannanayakal T, Miyashiro KY, Eiriksdottir E, Jochems J, Langel U, Eberwine J. In vivo identification of ribonucleoprotein-RNA interactions. Proc. Natl. Acad. Sci. U.S.A. 2006;103:1557-1562.
34. Harris JL, Winssinger N. PNA encoding (PNA = peptide nucleic acid): From solution-based libraries to organized microarrays. Chem. Eur. J. 2005;11:6792-6801.
35. Christensen L, Fitzpatrick R, Gildea B, Petersen KH, Hansen HF, Koch T, Egholm M, Buchardt O, Nielsen PE, Coull J, Berg RH. Solid-phase synthesis of peptide nucleic acids. J. Peptide Sci. 1995; 3:175-183.
36. Beck F, Nielsen PE. Peptide Nucleic Acid (PNA): A DNA mimic with a pseudopeptide backbone. In: Artificial DNA: Methods and Applications. Khudyakov YE and Fields HA, eds. 2003. CRC Press, Boca Raton, FL. pp. 91-114.
37. Kumar VA, Ganesh KN. Conformationally constrained PNA analogues: Structural evolution toward DNA/RNA binding selectivity. Acc. Chem. Res. 2005; 38:404-412.
38. Pokorski JK, Witschi MA, Purnell BL, Appella DH. (S,S)-trans-Cyclopentane-constrained peptide nucleic acids. A general backbone modification that improves binding affinity and sequence specificity. J. Am. Chem. Soc. 2004; 126:15067-15073.
39. Govindaraju T, Kumar VA, Ganesh KN. (SR/RS)-Cyclohexanyl PNAs: conformationally preorganized PNA analogues with unprecedented preference for duplex formation with RNA. J. Am. Chem. Soc. 2005; 127:4144-4145.
40. Haaima G, Lohse A, Buchardt O, Nielsen PE. Peptide nucleic acids (PNAs) containing thymine monomers derived from chiral amino acids: Hybridization and solubility properties of D-lysine PNA. Angew. Chem. Int. Ed. 1996; 35:1939-1942.
41. Zhou P, Wang M, Du L, Fisher GW, Waggoner A, Ly DH. Novel binding and efficient cellular uptake of guanidine-based peptide nucleic acids (GPNA). J. Am. Chem. Soc. 2003; 125:6878-6879.
42. Dragulescu-Andrasi A, Rapireddy S, Frezza BM, Gayathri C, Gil RR, Ly DH. A simple y-backbone modification preorganizes peptide nucleic acid into a helical structure. J. Am. Chem. Soc. 2006; 128:10258-10267.
43. Yamamoto Y, Uehara A, Watanabe A, Aburatani H, Komiyama M. Chemical-reaction-based site-selective DNA cutter for PCR-free gene manipulation. ChemBioChem. 2006; 7:673-677.
44. Ausin C, Ortega JA, Robles J, Grandas A, Pedroso E. Synthesis of amino- and guanidino-G-clamp PNA monomers. Org. Lett. 2002; 4:4073-4075.
45. Rajeev KG, Maier MA, Lesnik EA, Manoharan M. High-affinity peptide nucleic acid oligomers containing tricyclic cytosine analogues. Org. Lett. 2002; 4:4395-4398.
46. Popescu D-L, Parolin TJ, Achim C. Metal incorporation in modified PNA duplexes. J. Am. Chem. Soc. 2003; 125:6354-6355.
47. Williams KA, Veenhuizen PTM, de la Torre BG, Eritja R, Dekker C. Carbon nanotubes with DNA recognition. Nature 2002; 420:761.
48. Turner JL, Becker ML, Li X, Taylor J-SA, Wooley KL. PNA-directed solution- and surface-assembly of shell crosslinked (SCK) nanoparticle conjugates. Soft Matter 2005; 1:69-78.
49. Demidov VV. PNA openers for duplex DNA: Basic facts, fine tuning and emerging applications. Peptide Nucleic Acids: Protocols and Applications, 2nd ed. Nielsen PE, ed. 2004. Horizon Bioscience, Wymondham, UK. pp. 207-226.
50. Demidov VV, Kuhn H, Lavrentieva-Smolina IV, Frank-Kamenetskii MD. Peptide nucleic acid-assisted topological labeling of duplex DNA. Methods 2001; 23:123-131.
51. Cao R, Gu Z, Patterson GD, Armitage BA. Synthesis and characterization of thermoreversible biopolymer microgels based on hydrogen bonded nucleobase pairing. J. Am. Chem. Soc. 2003; 125:10250-10256.
52. Cao R, Gu Z, Patterson GD, Armitage BA. A recoverable enzymatic microgel based on biomolecular recognition. J. Am. Chem. Soc. 2004; 126:726-727.
Further Reading
Armitage BA. The impact of nucleic acid secondary structure on PNA hybridization. Drug Disc. Today 2003; 8:222-228.
Koppelhus U, Nielsen PE. Cellular delivery of peptide nucleic acid (PNA). Adv. Drug Del. Rev. 2003; 55:267-280.
Nielsen PE. The many faces of PNA. Lett. Pept. Sci. 2004; 10:135-147.
Nielsen PE, Egholm M, eds. Peptide Nucleic Acids: Protocols and Applications, 2nd Ed. 2004. Horizon Bioscience, Wymondham, UK.
See Also
Nucleic Acids, Chemistry of
Nucleic Acids, Design and Engineering of
Nucleic Acid Recognition by Peptides and Drugs
Synthetic Chemistry: Building Molecules to Modulate Biological Systems
Synthetic Nucleic Acids to Study Biologic Function