CHEMICAL BIOLOGY
Polyketide Biosynthesis, Fungi
Thomas J. Simpson and Russell J. Cox, School of Chemistry, University of Bristol, Bristol, United Kingdom
doi: 10.1002/9780470048672.wecb458
Fungi produce a wide variety of biologically active compounds. Among these compounds, the polyketides form a large and structurally diverse group. These compounds are synthesized by highly programmed, large iterative multifunctional proteins, which are called the polyketide synthases. This review describes the structure and biosynthesis of polyketide fungal metabolites and highlights recent work on the links between gene sequence, protein architecture, and biosynthetic programming for fungal polyketide synthases.
Polyketides have long been recognized as one of the most important classes of secondary metabolites (1). They occur in plants, bacteria, and marine organisms as well as in fungi. Fungal polyketides vary from the simplest monocyclic aromatic compounds, for example, orsellinic 1 and 6-methylsalicylic (6-MSA) 2 acids to polycyclic aromatics such as citrinin 3, alternariol 4, islandicin 5, deoxyherqueinone 6, and norsolorinic acid 7. Although initially associated with the formation of aromatic compounds, many polyketides are nonaromatic (e.g., the macrolide decarestrictine D 8, long-chain polyfunctional molecules exemplified by T-toxin 9 and the decalins, lovastatin 10, and compactin 11). Many other metabolites consist of an aromatic ring attached to a more highly reduced moiety (e.g., zeralanenone 12, dehydrocurvularin 13, and monocerin 14). Additional diversity results from extensive oxidative metabolism of preformed polyketide structures (e.g., penicillic acid 15 and patulin 16), which are formed from cleavage and rearrangement of 6-MSA and orsellinic acid, respectively, and indeed ring cleavage is a very common feature with the potent hepato- toxin aflatoxin B1 17 being derived by extensive ring cleavages and rearrangements of norsolorinic acid 7. Other metabolites contain a polyketide-derived moiety as part of a larger molecule whose biosynthesis is other than polyketide. A classic example is mycophenolic acid 18, in which the branched carboxylic side chain is derived via a cleaved farnesyl moiety. Other compounds of mixed terpenoid-polyketide origin include the mycotoxin viridicatumtoxin 19. Again the origins of the polyketide-derived moiety may be disguised as a result of extensive metabolism as observed in the meroterpenoid metabolites austin 20 and para- herquonin 21, which are all derived via 3,5-dimethylorsellinic acid (2). The xenovulenes 22-24 are an interesting group where it has been shown that the cyclopentananone, benzenoid, and tropolone moieties all have a common biosynthetic origin via ring expansion and ring contaction of 3-methylorcinaldehyde. Other groups contain Krebs’ cycle intermediates, for example, the tetronic acid, carlosic acid 25, and the squalestatins (see 29 below) or amino-acid derived moieties (e.g., fusarin C 26).
Biological Properties
Another important feature of fungal polyketides is their vast range of biological activities both beneficial and harmful. Thus, griseofulvin 27 was one of the first effective antifungal agents. Penicillic acid 15, which was discovered shortly after the penicillins, is also a powerful antibiotic, but unfortunately it proved too toxic for clinical use. Mycophenolic acid 18 has been “rediscovered” as an immunosuppressive agent. The strobilurins (e.g., 28), although not themselves used in the field formed the basis for the development of the widely used methoxyacrylate group of antifungal agents and, of course the statins, as represented by lovastatin 10, are among the most widely prescribed drugs for control of cholesterol levels and associated heart disease. As a cursory inspection of their structures would suggest, the squalestatins (e.g., 29), are effective inhibitors of squalene synthase though their early promise as cholesterol-lowering clinical candidates declined because of inherent toxicity. They contain two separate polyketide chains linked to oxaloacetate. Overall, fungal metabolites and their pharmaceutical and agrochemical derivatives have total sales of many tens of billions of pounds annually. In addition to these beneficial effects, the large group of mycotoxins represented first and foremost by aflatoxin B1 17 and others such as the fuminosins 30, zearaleneone 12, citrinin 3, ochratoxins (e.g., 31), are the cause of many problems in both animal and human health, and spoilage of both growing crops and stored foodstuffs through contamination by mycotoxin producing fungi is a cause of major economic losses worldwide.
Biosynthesis
Although diverse in structure, the class is defined by the common biosynthetic origin of the carbon atoms: These atoms are derived from the CoA thiolesters of small carboxylic acids, such as acetate and malonate. As long ago as 1953, Birch realized that polyketide biosynthesis is related to fatty acid biosynthesis and some of the earliest applications of radioisotopes to natural product biosynthesis were to fungal polyketide metabolites, where the ease of fermentation and isolation of metabolites in pure form, and relatively efficient uptake of simple labeled precursors facilitated the work. In more recent years, fungal metabolites in general, and polyketides in particular, were the focus of the rapidly expanding applications of stable isotope labeling in the 1970s and 1980s beginning with incorporations of singly 13C-labeled precursors with analysis of regiospecifity of labeling being greatly facilitated by the contemporaneous development of Fourier Transform methods and their application to 13C NMR. The application of doubly 13C-labeled precursors led to the concept of bond labeling, which allowed inter alia the mode of cyclization of linear polyketide precursors into polycyclic molecules, bond fragmentation, and rearrangements processes to be detected for the first time through analysis of the resulting 13C-13C coupling patterns. This method was then rapidly followed by applications of isotope induced shifts in 13C NMR which allowed indirect detection of 2H and 18O labels and the use of direct 2H NMR. These new methods (2) allowed stereochemistry and regiochemistry of labeling to be detected and in particular permissible levels of oxidation and reduction in otherwise undetectable biosynthetic intermediates to be determined. Along with similar work with bacterial polyketides, this method laid the basis for the ideas of the processive mode of polyketide biosynthesis to be established. It was a major step because it changed fundamentally the idea that polyketide chains were assembled in their entirety and then subjected to necessary reductive modifications to the simple, in retrospect, idea that these changes occur concomitant with chain elongation rather than post-elongation. Although it is still not uncommon for these classic “Birch” fully oxygenated polyketide intermediates to be invoked it is now evident that in most polyketide metabolites, these have no reality. The concept of processive polyketide assembly and modification brought polyketide biosynthesis even closer to the process of fatty acid biosynthesis in which full reductive processing in each cycle of chain condensation and elongation is the norm. The rapid developments in understanding of the molecular genetics of polyketide biosynthesis particularly in bacteria in the 1990s were fully consistent with the processive mode. Understanding of the genetics of fungal polyketide biosynthesis still lags behind that of bacterial polyketides and the remainder of this article will provide a brief overview of current understanding.
Polyketide assembly
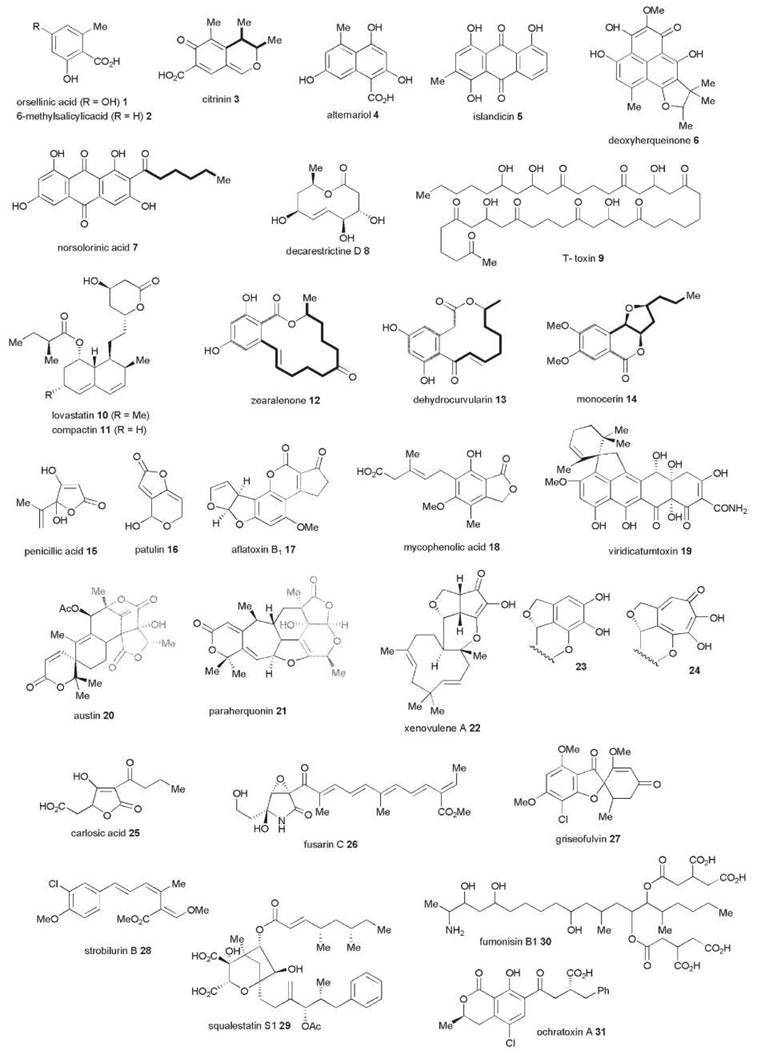
The basic assembly cycle for both polyketide and fatty acid biosynthesis is shown in Fig. 1 in which a starter unit, normally acetate is transferred to the ketosynthase (KS) or condensing enzyme which catalyzes a decarboxylative condensation with malonate, bound after after malonyl tranferase (MT) catalyzed malonylation to the acyl carrier protein (ACP). During fatty acid biosynthesis, the resulting β-ketothiolester 32 is subjected to additional chemical processing while attached to the terminal thiol of the ACP: first: it is reduced by a β-ketoacyl reductase (KR) to a secondary alcohol 34, which then undergoes a dehydratase (DH)-catalyzed dehydration to form an αβ-unsaturated thiolester 35, and final enoyl reduction (ER) yields a fully saturated thiolester 36. Fungal PKS deploy all these reactions, but additionally the chain can be methylated, using a methyl group from S-adenosylmethionine (SAM). This reaction probably occurs after KS, which gives an α-methyl-β-ketothiolester 33. During the biosynthesis of palmitic acid (C16), there are seven cycles of these reactions. The final reaction of FAS is hydrolysis of the thiolester by a dedicated thiolesterase (TE). Apart from the capacity for C-methylation, fungal PKSs in common with other PKSs have the ability to use the condensation and reductive cycle in a highly controlled manner to produce polyketide intermediates in which no reductive modification has occurred to give a a classic poly-β-ketothiolester, or more generally “reduced” or “processive” polyketide intermediates in which a complete spectrum of reduction and/or C-methylation has occurred in each condensation cycle as indicated in Fig. 1 for the squalesatin tetraketide (37) synthase (see below). Another variation is that a range of alternate starter units can be used [e.g., hexanoate in the case of norsolorinic acid 7 or benzoate in the case of the strobilurins or squalestatins (28 and 29)].
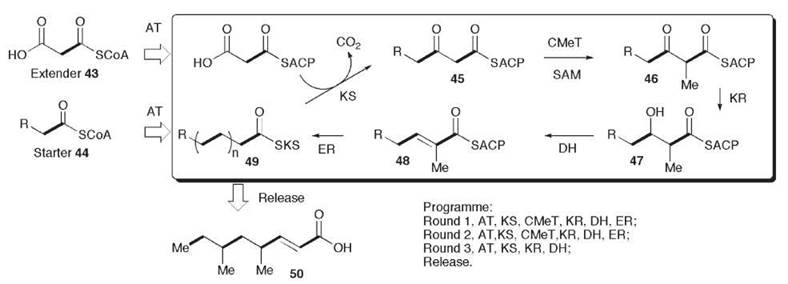
Figure 1. Generic polyketide assembly pathway reactions catalyzed by iterative fungal polyketide synthases. The assembly sequence for the squalesatin tetraketide intermediate 37 is shown for illustration.
Enzymology
The understanding of the relationship between FAS and PKS proteins, the application of molecular genetics, and more latterly genomics, has greatly facilitated the discovery and understanding of polyketide synthases from diverse sources. The homology in catalytic function between FAS and PKS enzymes is preserved in their respective gene sequences. It is now clear that fungal PKSs belong to the class of Type 1 iterative synthases represented by mammalian FASs. Type 1 FAS proteins are large multifunctional proteins in which single (or occasionally two) peptides contain the sequences for KS, ACP, AT, KR, DH, ER, and TE activities—these catalytic functions are carried out by particular functional domains. Similarly, the genes for Type 1 FAS proteins are correspondingly large single open reading frames, and Type 1 PKSs consist of very large multifunctional proteins with individual functional domains. Thus, PKSs use much the same array of chemical reactions as FAS—but the key differences is that of programming: FASs have to control chain length (i.e., the number of extensions), but PKS can additionally control starter unit selection and the extent of reduction during each condensation cycle. Fungal PKS can program the extent of chain methylation and the off-loading mechanism. The issue of programming is key to understanding and exploiting PKSs. In the case of the bacterial modular polyketide synthases, each condensation cycle is catalyzed by a discreet module containing all the catalytic domains required. In this case, the program is explicit in the order and composition of the modules. However, for the iterative Type 1 fungal polyketide synthases, the programme is cryptic—encoded in the PKS itself.
Fungi make some of the simplest and some of the most complex polyketides known (3). It is useful to consider a heirarchy of complexity when considering the structures of fungal polyke- tides, because the complexity in chemical structure is generated by enzymes and ultimately genes. The simplest structures are those such as orsellinic acid 1. Addition of an extra acetate gives a pentaketide such as 1,3,6,8-tetrahydroxynaphthalene, which is a compound widely distributed in fungi and which is involved in melanization—a key component of apressorium formation and invasion of plant cells by plant pathogen such as Magnaporthe grisea, Colletotrichum lagenarium, and others. Additional complexity is represented by compounds such as 6-MSA acid 2, in which a single, programmed reduction reaction occurs during biosynthesis. More reduction is then observed in compounds such as T-toxin 19 produced by Cochliobolus heterostrophus and lovastatin 10 produced by Aspergillus terreus. In these compounds, many more carbon atoms are used and many more reduction and dehydration reactions occur. Additionally in many cases (e.g. fusarin C 26), pendant methyl groups have been added from the S -methyl of methionine, catalyzed by a C-methyl transferase domain.
Linking PKS genes and compounds in fungi
The huge structural variety of fungal polyketides is caused by differences in programming of their PKS proteins—apparent increases in structural complexity are caused by increasing use and control of reductive, dehydrative, and methylating steps by the PKS. This inrease must be beacuse of differences in PKS protein sequence and structure. This fact has been exploited in the development of rapid methods for the cloning of fungal PKS genes associated with the biosynthesis of particular fungal polyketide types.
Bingle et al. (3) realized that these subtle protein sequence differences should be reflected in DNA sequence, and polymerase chain reaction (PCR) primers could be designed to amplify fragments of fungal PKS genes selectively from fungal genomic DNA (or cDNA). In early work in this area, they hypothesized that fungal polyketides could be grouped into two classes: nonreduced (NR) compounds such as orsellinic acid 1, norsolorinic acid 7, and 1,3,6,8-tetrahydroxynaphthalene, and partially reduced (PR) compounds, such as 6-MSA 2. At the time, very few fungal PKS genes were known, and based on very limited sets of sequences, they designed degenerate PCR primers that were complimentary to conserved DNA sequences in the KS domains in fungal PKS responsible for the biosynthesis of NR and PR compounds. Later, the same analysis was extended to the KS domains of highly reduced (HR) compounds, such as lovastatin 10, when DNA sequence data became available for the lovastatin nonaketide and diketide synthases (LNKS and LDKS, respectively) (4). The availability of these sequences also allowed the development of selective PCR primers for CMeT domains.
This sequence analysis has been significantly extended as genomic approaches have been applied to fungi recently (5). Full genome sequences have now been obtained for more than a dozen fungi. In each organism, many PKS genes have been discovered. For example, Aspergillus niger contains 34 PKS genes, so several hundred fungal PKS genes are known. Sequence comparison of all these new PKS genes, however, shows that the three classes of fungal PKS genes predicted by Bingle et al. (3) are the same three classes observed in the most recent sequence comparisons (5). Despite the fact that so many fungal PKS genes have been discovered, however, relatively few genes have been definitively linked to the biosynthesis of specific metabolites. Because the NR, PR, and HR nomenclature is useful for describing both the chemical products and their cognate genes, the state of knowledge of fungal PKS is reviewed in this way.
Fungal NR-PKS
The tetraketide orsellinic acid 1 is the simplest tetraketide, which requires no reductions during its biosynthesis. One of the first discovered fungal PKS, orsellinic acid synthase (OSAS) was isolated from Penicillium madriti in 1968. Despite the early work with the protein, however, the OSAS-encoding gene has not yet been discovered, and nothing is known of the catalytic domains or their organization. However, genes involved in the biosynthesis of several other nonreduced polyketides are now known, and a general pattern of domain organization has emerged. In all known cases, these genes encode Type 1 iterative PKS proteins. At the N-terminus, a domain is present that seems to mediate the loading of a starter unit (Fig. 2a). It seems that the starter unit can derive from either a dedicated FAS, another PKS, or an acyl CoA. The starter unit loading domain (SAT) is followed by typical KS and AT domains responsible for chain extension and malonate loading. Beyond the AT is a conserved domain known as the product template (PT) domain. This domain is followed by an ACP. Some NR-PKS seem to terminate after the ACP, but many feature a diverse range of different domains that include cyclases, methyl transferases, and reductases. Thus, it seems that these synthases are arranged with an N-terminal loading component; a central chain extension component that consists of KS, AT, PT and ACP domains; and a C-terminal processing component.
NR-PKS loading component
Feeding experiments with isotopically labeled precursors have shown that many NR fungal polyketides are formed by the use of “advanced” starter units. In the classic case of norsolorinic acid 7 biosynthesis, it has long been known that hexanoate forms the starter unit. Differential specific incorporation of acetate into the early and late positions in compounds such as citrinin 3 have been used to argue that these compounds may have been formed by more than one PKS so that one PKS makes an advanced starter unit, which is passed to a second PKS for additional extension.
Evidence for this suggestion is growing. Townsend has defined the molecular basis for the ability of NR-PKS to use starter units derived from other FAS or PKS systems. Two genes in the aflatoxin biosynthetic gene cluster of Aspergillus parasiticus (stcJ and stcK) encode the α and β components of a typical fungal FAS (HexA and HexB). Clustering of these FAS genes with the NSAS PKS suggested that HexA and HexB probably produced hexanoate for use as the norsolorinic acid starter unit. The protein complex formed between NSAS, HexA, and HexB, which is known as NorS, was isolated and characterized (6). This 1.4-MDa protein complex synthesizes norsolorinic acid 7 from malonyl CoA, acetyl CoA, and NADPH. Townsend showed that hexanoyl CoA is not a free intermediate produced by NorS, which suggests that hexanoate produced as an ACP derivative by the HexA/HexB FAS must be passed directly to NSAS. In the absence of NADPH, hexanoate cannot be formed by the FAS components; thus, no norsolorinic acid is formed.
The N-terminal domain of NSAS posesses canonical acyl transferase sequence motifs so that this domain could be a candidate for the required starter unit transferase. This domain was cloned and expressed along with the ACP and was shown to catalyze the transfer of hexanoate from CoA onto the the ACP. Site-directed mutagenesis experiments to remove the proposed catalytic cysteine of the transferase resulted in loss of catalytic activity. The N-terminal transferase showed significant selectivity for the transfer of hexanoate over longer or shorter acyl chains (7). Thus, the N-terminal domain of NSAS acts as a starter unit:ACP transacylase (SAT) component.
Sequence comparison with other known NR PKS suggests that such SAT domains are common. In the few cases where the PKS sequence has been correlated with product structure the presence of SAT domains now explains prior results from feeding experiments, which suggested the use of advanced starter units. For example it is now known (8) that two PKS genes are involved in the biosynthesis of zearalenone 12—one of these is a HR-PKS (see below) and probably provides a highly reduced hexaketide as a starter unit. The second zearalenone PKS is a NR-PKS possessing an N-terminal SAT domain, which likely loads the hexaketide ready for three further extensions. Most NR PKS seem to possess potential SAT domains whether they require an acetate starter unit or not. For example, polyketide synthases involved in the biosynthesis of YWA1 39 (WAS) and tetrahydroxynaphthalene (THNS). In the case of THNS from C. lagenarium, in vitro experiments have implied that the purified protein uses malonyl CoA as the starter unit. The THNS SAT domain may therefore be involved with loading and decarboxylation of malonate to use as a starter unit, much as the bacterial Type 2 KSβ component does.
NR PKS chain extension component
The extension components of NR polyketide synthases consist of KS, AT, PT, and ACP domains. Sequence analysis of PT domains (450-550 residues) from a range of NR PKS genes in which the chemical products are known suggests that it is conceiveable that the PT domain is involved in chain-length determination. Comparison of the PT domains from the Acremonium strictum PKS1, citrinin PKS, zearalenone PKS-B, NSAS, sterigmatocystin PKS, dothistromin PKS, THNS from C. lagenarium and Wangiella dermatitidis, and WAS suggests that these domains group into clades that correspond with chain length (9). Thus the citrinin, ASPKS1, and zearalenone-B groups correspond to tetraketide synthases; the NSAS, sterigmatocystin, and dothistromin PKS are all octaketide synthases; the THNS are hexaketide synthases; and the wA synthase (WAS) forms an outgroup, which is a heptaketide.
The domains found after the chain extension components at the C-termini of fungal NR-PKS are highly varied. These components include putative Claisen-cyclase/thiolesterases (CLC/ TE), C-methyl transferases (C-MeT), reductases (R), and additional ACP domains. Recent work indicates that these processing components act after chain assembly to modify either a poly-keto or a cyclized intermediate.
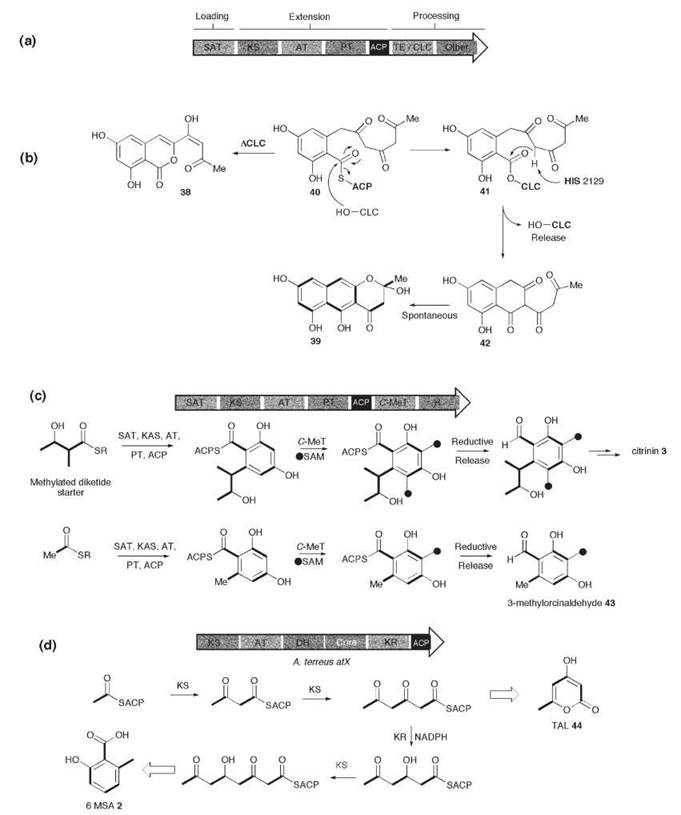
Figure 2. (a) General architecture of NR PKS genes; proposed mechanism of, (b) the WAS CLC domain, (c) the Monascus purpureus citrinin PksCT and A. strictum MOS synthases; and (d) domain architecture of A. terreus and P. patulum MSAS, and proposed mechanism of 6-MSA 2 and TAL 44.
CLC/TE domains
The first fungal NR PKS gene to be cloned was Aspergillus nidulans wA (encoding WAS). Limited domain analysis was carried out to determine the presence of KAS, AT, and ACP domains. Later, it was shown that wA also possess a TE domain, which form one of the most common processing components of NR PKS. The TE domain can either operate as a standard thiolesterase or be involved in a cyclization-release mechanism. Fujii et al. (10) expressed A. nidulanswA (encoding WAS) in Aspergillus oryzae. Initially, the expression strain produced the isocoumarin 38, which indicated that WAS is a heptaketide synthase (Fig. 2b). It was then realized that the expression construct used in this experiment had a deletion that resulted in the expressed WAS missing the final 67 amino acids of the C-terminal TE domain. When the complete wA gene was expressed, however, the heptaketide naphthopyrone YWA1 39 was produced (10). A series of experiments that involve step-wise shortening of the C-terminus of WAS showed that deletion of as few as 32 amino acids resulted in production of the isocoumarin 38. Site-directed mutagenesis of a conserved serine and histidine in the C-terminal domain also resulted in a switch from naphthopyrone production to citreoisocoumarin production.
Isotopic feeding experiments using 13C-labeled acetate indicated the folding pattern shown in Fig. 2b for the naphthopyrone 39. Thus, both the isocoumarin 38 and the naphthopyrone 39 must result from the cyclization of the common intermediate 40. This finding suggests that the WAS chain extension component produces a heptaketide and catalyzes the cyclization and aromatization of the first ring. The C-terminal domain must therefore catalyze a second (Claisen) cyclization reaction to form 42. The involvement of conserved serine and histidine residues suggests involvement of the CLC-bound intermediate 41 shown in Fig. 2b. Thus, the TE domain has been renamed as Claisen Cyclase (CLC). These domains also occur in the known NSAS and THNS proteins in which the same chemistry must occur to provide the observed products.
C-MeT domains
Few NR PKS are known to possess C-methylation domains, although many known fungal nonreduced polyketides are C-methylated, such as 3,5-dimethylorsellinic acid. A small group of NR PKS have been identified in genome sequences (5), which feature a C-MeT domain located after the ACP (Fig. 2c). The first correlation between a gene sequence and a compound came from citrinin 3, in which the PKS involved in citrinin biosynthesis in Monascus ruber was reported (11). Here, the C-MeT domain must be programmed because it acts twice during polyketide biosynthesis when a probable methylated diketide starter unit is extended. It is not yet clear whether the C -MeT domain acts during extension, after chain extension but before aromatization, or after aromatization. 1,3-Dihydroxyaromatics are known to tautomerize easily to keto forms, and it is conceivable that it could act as the nucleophile for the reaction with SAM.
R domains
Reductases are currently rare as part of the processing component of NR PKS. Evidence for the role of these reductase domains has been obtained by the isolation of the PKS responsible for the formation of the tetraketide component found in xenovulene A 22. The PKS gene (MOS) was found to have SAT, KS, AT, C-MeT, and R domains (Fig. 2c), and heterologous expression of the gene in A. oryzae resulted (9) in a high yield of 3-methylorcinaldehyde 43. Although not described in the literature, sequence analysis of the citrinin PKS sequence discussed above shows that it also possesses a C-terminal thiolester reductase domain.
Similar domains are known from NRPS systems in which reductase domains are sometimes used as chain release mechanisms, which release an aldehyde or primary alcohol. In the case of MOS and citrinin biosynthesis the reductive release mechanism makes good sense as this provides the products with C-1 at the correct oxidation state (Fig. 2c).
Fungal PR-PKS
The domain structure of PR PKSs is much closer to mammalian FAS, with an N-terminal KS followed by AT, and DH domains (Fig. 2d). A so-called “core” domain follows the DH, which is followed by a KR and the PKS terminates with an ACP domain. The domain structure differs considerably from the NR PKS—no SAT domain or PT domain exists, and the PKS terminates after the ACP and seems not to require a TE/CLC domain. No obvious catalytic machinery exists for off-loading the product. The DNA sequence of the KS domain is distinguishable from NR-PKS and FAS KAS domains using selective PCR primers and DNA probes (3). Overall, however, catalytic domains closely match those of the mamalian FAS, although the “core” domain is different. Remarkably, the fungal MSAS genes are closely related to recently discovered bacterial genes for the synthesis of the nonreduced tetraketide orsellinic acid 1 (12) apart from lacking a ~450-amino acid region that encompasses the KR domain.
Although many PR PKS genes are known from genome sequencing projects, only three genes have been matched to chemical products—in all cases, the tetraketide 6-MSA 2 MSAS from Penicellium patulum is one of the smallest Type 1 PKS at 188 KDa. It proved relatively easy to isolate, and many of the earliest in vitro studies of the enzymology of any PKS were carried out with this enzyme (13, 14). The PKS is evidently programmed—acetate must be extended three times, and the KR must only act once, after the second extension. In the absence of NADPH, the KR reaction cannot occur, the tetraketide is not produced, and triketide lactone (TAL) 44 is produced instead (Fig. 2d). This reaction reveals that chain extension and reduction are linked, and it indicates that the KR must act during chain extension. Mammalian FAS also produces TAL 44 under the same circumstances.
Chain length determination seems to use a “counting mechanism” as in the case of the Type 2 actinorhodin PKS. Incubation of various acyl CoA starter units with malonyl CoA with MSAS in the absence of NADPH and acetyl CoA resulted in two chain extensions to produce the corresponding substituted TALs.
Moriguchi et al. (15) have carried out an interesting series of expression experiments in the yeast Saccharoveyces cerivisiae in which two copies of the MSAS gene can be expressed simultaneously. This inspection has allowed complementation experiments in which they show that up to 44 amino acids from the N-terminus can be removed and activity retained. However, at the C-terminus, deletion of as few as nine amino acids caused loss of activity because of removal of key ACP residues. Use of combinations of deletion mutants, with the knowledge that MSAS forms homotetramers, provided evidence that the ACP of one peptide chain must interact with the KS of another chain. In addition, a short core domain region was identified (15). This finding was essential for successful complementation and suggested that it acts as a motif required for subunit-subunit recognition similarly to the core region of mammalian FAS, which has been shown to mediate assembly of the synthase.
Fungal HR-PKS
The final class of fungal PKS produces complex, highly reduced compounds such as lovastatin 10, T-toxin 19, fumonisin B1 30, and squalestatin 29. In all cases, these fungal compounds are produced by iterative Type 1 PKS. These PKSs have an N-terminal KS domain, followed by AT and DH domains (Fig. 3). In many cases, the DH is followed by a C-MeT domain. Some HR PKSs possess an ER domain, but those that lack it have a roughly equivalent length of sequence with no known function. This domain is followed by a KR domain, and the PKS often terminates with an ACP. No domain similar to the PT domain of the NR PKS or the core domain of the PR PKS seems to exist, and there is no N-terminal SAT domain as found in the NR PKS. As with the NRs and PR PKSs, many HR PKS genes are known from the numerous fungal genome sequences, but as yet few gene sequences have been linked to the production of known compounds. However, of the few cases in which both gene and chemical product are known, some progress has been made in understanding function and programming.
Figure 3. Expression of lovB in A. nidulans, (a) in the absence and, (b) the presence of lovC; (c) lovD catalyzed acyl transfer of lovB diketide 50 to monacolin J 49; (d) proposed roles of FUSS domains in the biosynthesis of "pre-fusarin" 60.
The lovastatin polyketide synthases
Lovastatin 10 (also known as mevinolin) is produced by A. terreus. The related compound compactin (mevastatin) 11, produced by Penicillium citrinum is identical to lovastatin apart from the C-12 methyl group absent in compactin. Isotopic feeding experiments have shown that two polyketide chains are required: a nonaketide and a methylated diketide. The requirement for two polyketide synthases is evident in the gene clusters associated with biosynthesis of 10 and 11 where two PKS genes are found (4), lovB and lovF, which encode LNKS and LDKS in the case of lovastatin (Fig. 3b).
LNKS formally should synthesize a fully elaborated nonake- tide such as 47, which could undergo a biological Diels-Alder reaction to form dihydromonacolin L 48—which is the observed first PKS-free intermediate. Note that it is possible that the Diels Alder reaction actually occurs at the more activated hexaketide stage. When lovB was expressed in the heterologous fungal host A. nidulans (4) however, the polyunsaturated compounds 45 and 46 were isolated. These pyrones are related to the expected nonaketide (e.g. methylation has occurred at the correct position), but it is evident that reductions, specifically enoyl reductions and later keto reductions, have not occurred correctly and that chain extension has terminated prematurely (Fig. 3a), which is becauase the NADPH binding site of the ER domain of LNKS is impared. A separate ER encoding gene, lovC, occurs downstream from lovB, and Kennedy et al. (4) showed that in coexpression experiments the lovC protein could complement the missing ER domain and the expected dihydromonacolin L 48 was produced (Fig. 3b). It is evident that the lovC protein must interact with LNKS and control programming by ensuring enoyl reduction at the correct positions and allowing complete chain extension. It now seems that lovC type proteins are a common feature of HR PKS systems in fungi.
In the bacterial modular PKS systems, usually a C-terminal TE domain is involved in off-loading the product—either to another PKS or to solution. LNKS seems to possess part of an NRPS condensation (C) domain (see below) immediately downstream of the ACP, and this domain has been proposed to be involved in product release, presumably by either activating water as a nucleophile or the C-5 hydoxyl rather than the nitrogen of an aminothiolester as activated by most NRPS C domains. LDKS is closely related to LNKS, but its ER domain seems to be intact. LDKS is unusual among fungal PKS because it is not iterative—a single round of extension and processing affords the diketide 50 (Fig. 3b). In this respect, LDKS closely resembles a single module of a bacterial modular PKS (1). LDKS again lacks an obvious product release domain, which ends immediately after the ACP. A specialized acyl transferase, encoded by lovD, transfers 50 from LDKS onto the C-10 hydroxyl of monacolin J 49 (16).
The squalestatin SI polyketide synthases
Squalestatin S1 29 is a potent inhibitor of mammalian squalene synthase. It is produced by Phoma species, and like lovastatin, consists of two polyketide chains: a main chain hexaketide and a sidechain tetraketide. Like lovastatin, both chains are methylated, but unusually for a fungal HR polyketide, the main chain is formed from a non-acetate starter unit—benzoate is incorporated at this position.
Cox et al. (17) cloned a HR PKS gene from Phoma sp, PhPKSl. This gene was expressed in the heterologous fungal host A. oryzae, which produced the tetraketide 37. PhPKSl thus encodes the squalestatin tetraketide synthase (SQTKS). SQTKS is highly homologous to LDKS, which lacks the C-terminal NRPS condensation domain of LNKS. Like LDKS, SQTKS posseses a functional ER domain, but SQTKS carries out three extensions. Like LDKS, all modification reactions occur after the first extension. The stereochemistry of the branching methyl group is the same in each case. Two more extensions occur—all modifying reactions occur again after the first of these, but neither ER or C-MeT are used after the final extension (Fig. 1).
HR PKS from Alternaria solani
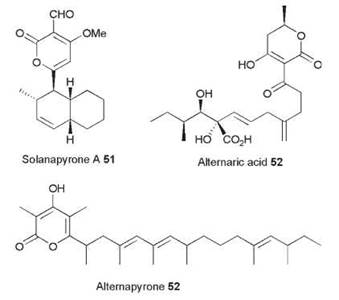
Alternaria solani is a plant pathogen and the causitive agent of early blight in solanum species. It produces numerous polyketides, such as solanopyrone A 51 and alternaric acid 52, and it is thus an ideal target species for speculative PKS gene-fishing expeditions. Fujii et al. (18) have conducted just such investigations, using PCR primers based on conserved PKS sequences as probes with genomic DNA libraries (18). An early investigation yielded two hits—one a HR PKS gene named alt5, and another, a NR PKS named pksA. The alt5 gene encodes a typical HR PKS, which is known as PKSN, with the usual array of catalytic domains. Inspection of the ER sequence suggested that it should be functional like those from LDKS and SQTKS. Expression of alt5 showed this to be correct—a single compound was synthesized in good yield (~15 mgl-1), which proved to be the octamethylated decaketide pyrone 53, named alternapyrone. This compound is the most complex polyketide yet reported to be produced by an iterative PKS, which shows programmed chain length control, keto-reduction, methylation, and enoyl reduction.
HR PKS-NRPS
Fungi produce a wide range of bioactive compounds derived from polyketides fused to amino acids. Examples include fusarin C 26, equisetin 54, and tenellin 55. Fusarin C 26 consists of a tetramethylated heptaketide fused to homoserine and is produced by strains of the plant pathogens Fusarium moniliforme and Fusarium venenatum. Genomic DNA libraries from these organisms were used to isolate a gene cluster centered around a 12-Kb ORF encoding a HR PKS fused to a nonribosomal peptide synthetase (NRPS) module (19). The PKS region is homologous to LNKS: KAS, AT, and DH domains are followed by CMeT, a defective ER, KR, and ACP domains. Like LNKS, the ACP is upstream of an NRPS condensation (C) domain, but in this case the NRPS module is complete, which features downstream adenylation (A), thiolation (T), and C-terminal thiolester reductase (R) domains (Fig. 3c). Directed knockout of the PKS-NRPS gene proved it to be involved in the biosynthesis of 26, and it was thus named fusarin synthetase (FUSS). The disfunctional ER domain and the fact that no lovC homolog seems to exist in the cluster, is consistent with the polyunsaturated nature of the polyketide moiety.
It is probable that FUSS assembles a tetramethylated heptaketide 56 attached to the ACP (Fig. 3c)—the structure of which is similar to the heptaketide pyrone 46 produced by LNKS in the absence of the LovC protein. In parallel, the A domain of the NRPS module seems to select, activate, and attach homoserine 57 to the thiolation domain. The C domain then uses the amide of homoserine to form an amide with the ACP-bound polyketide, which forms a covalently bound intermediate peptide 58. The final reaction catalyzed by FUSS may be reductive release of the thiolester, which forms peptide aldehyde 59. Finally, Knoevenagel cyclization would give the putative prefusarin 60. Other genes in the FUSS cluster are presumably responsible for the required additional transformation of 60 to fusarin C 26 itself: epoxidation; oxidation of a pendant methyl to a carboxylate and esterification; and hydroxylation a to nitrogen.
A highly homologous PKS-NRPS gene has been shown to be involved in the biosynthesis of equisetin 54 in Fusarium heterosporum (20). EQS posseses the same catalytic domains as FUSS, but examination of the structure of 54 indicates that the pyrollidinone carbon derived from the carboxylate of the amino acid (serine in this case) is not reduced, which indicates either a reoxidation mechanism, or the fact that the R domain does not produce an aldehyde intermediate in this case.
Conclusion
Recognition that the fungal PKS can be categorized into three subsets has allowed more detailed consideration of the programming elements. The NR PKSs are arranged into loading, extension, and processing components, and to some extent this hypothesis has been verified by expression and study of individual catalytic domains and by the construction of hybrid NR PKS genes. However, the programming elements of the PR and HR PKS remain obscure. The first experiments to probe programming in HR PKS have involved domain swaps, but few conclusions have yet been drawn. Little information is known about the three-dimensional structure of fungal PKS—whereas sequence and domain organization similarity with mammalian FAS mean that broad descriptions of the architecture of the catalytic domains can be modeled, detailed hypotheses that involve individual domains or peptide motifs cannot yet be linked with programming.
References
1. Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 2001; 18:380-416.
2. Simpson TJ. Applications of multinuclear NMR to structural and biosynthetic studies of polyketide microbial metabolites. Chem. Soc. Rev. 1987; 16:123-160.
3. Bingle LEH, Simpson TJ, Lazarus CM. Ketosynthase domain probes identify two subclasses of fungal polyketide synthase genes. Fung. Genet. Biol. 1999; 26:209-223.
4. Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR. Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science 1999; 284:1368-1372.
5. Kroken S, Glass SNL, Taylor JW, Yoder OC, Turgeon BG. Phylogenomic analysis of type 1 polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:15670-15675
6. Watanabe CMH, Townsend CA. Initial characterisation of a type 1 fatty acid synthase and polyketide synthase multienzyme complex NorS in the biosynthesis of aflatoxin B1. Chem. Biol. 2002; 9:981-988.
7. Crawford JM, Dancy BCR, Hill EA, Udwary D, Townsend CA. Identification of a starer unit-acyl carrier protein transacylase domain in an iterative type 1 polyketide synthase. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:16728-16733.
8. Gaffoor I, Trail F. Characterisation of two polyketide synthase genes involved in zearalenone biosynthesis in Gibberella zeae. App. Env. Microbiol. 2006; 72:1793-1799.
9. Bailey AM, Cox RJ, Harley K, Lazarus CM, Simpson TJ, Skellam E. Characterisation of 3-methylorcinaldehyde synthase (MOS) in Acremonium strictum: first observation of a reductive release mechanism during polyketide biosynthesis. Chem. Commun. 2007; 4053-4055.
10. Fujii I, Watanabe A, Sankawa U, Ebizuka Y. Identification of a claisen cyclase domain in fungal polyketide synthase WA, a naphthpyrone synthase of Aspergillus nidulans. Chem. Biol. 2001; 8:189-197.
11. Shimizu T, Kinoshita H, Ishihara S, Sakai K, Nagai S, Nihira T. App. Env. Microbiol. 2005; 71:3453-3457.
12. Ahlert J, Shepard E, Lomovskaya N, Zazopoulos E, Staffa Bachmann BO, Huang K, Fonstein L, Czisny A, Whitwam RE, Farnet CM, Thorson JS. The calicheamicin gene cluster and its iterative type 1 enediyne PKS. Science 2002; 297:1173-1176.
13. Beck J, Ripka S, Siegner A, Schiltz E, Schweizer. The multifunctional 6-methylsalicylic acid synthase gene of Penicillium patulum. Its gene structure relative to that of other polyketide synthases. Eur. J. Biochem. 1990; 192:487-498.
14. Child CJ, Spencer JB, Bhogal P, Shoolingin-Jordan PM. Structural similarities between 6-methylsalicylic acid synthase from Penicillium patulum and vertebrate type 1 fatty acid synthase: evidence from thiol modification studies. Biochemistry 1996; 35:12267-12274.
15. Moriguchi T, Ebizuka Y, Fujii I. Analysis of subunit interactions in the iterative type 1 polyketide synthase ATX from Aspergillus terreus. ChemBioChem. 2006; 7:1869-1874.
16. Xie X, Watanabe K, Wojcicki WA, Wang CCC, Tang Y. Biosynthesis of lovastatin analogues with a broadly specific acyl transferase. Chem. Biol. 2006; 13:1161-1169.
17. Cox RJ, Glod F, Hurley D, Lazarus CM, Nicholson TP, Rudd BAM, Simpson TJ, Wilkinson B, Zhang Y. Rapid cloning and expression of a fungal polyketide synthase gene involved in squalestatin biosynthesis. Chem. Commun. 2004; 2260-2261.
18. Fujii N, Shimomaki S, Oikawa H, Ebizuka Y. An iterative type 1 polyketide synthase PKSN catalyses synthesis of the decaketide alternapyrone with regiospecific octa-methylation. Chem. Biol. 2005; 12:1301-1309.
19. Song ZS, Cox RJ, Lazarus CM, Simpson TJ. Fusarin C biosynthesis in Fusarium moniliforme and Fusarium venenatum. ChemBioChem. 2004; 5:1196-1203.
20. Fillmore JP, Warner DD, Schmidt EW, Sims JW. Equisetin Biosynthesis in Fusarium heterosporum. Chem. Commun. 2005; 186-188.
Further Reading
For general review of polyketides:
O’Hagan D. The Polyketide Metabolites. Ellis Horwood Ltd. Chichester, UK, 1991.
For comprehensive listings of fungal polyketide molecules:
Turner WB. Fungal Metabolites. Academic Press, London, 1971.
Turner WB, Aldridge DC. Fungal Metabolites Part II. Academic Press, London, 1983.
For a review of mycotoxins: Steyn PS, Vleggar R. Mycotoxins and Phycotoxins, Elsevier, Amsterdam, 1986.
For further discussion of fungal PKS genes: Cox RJ. Polyketides, proteins and genes in Fungi: programmed nano-machines begin to reveal their sectors. Org. Biomol. Chem. 2007; 5:2010-2026
Polyketide Biosynthesis, Enediyne Polyketides
Polyketide Biosynthesis, Aromatic Polyetides
Polyketide Biosynthesis, Modular Polyketide Synthases
Polyketide Biosynthesis, Polyethers
Polyketides as Drugs