CHEMICAL BIOLOGY
Polyketide Biosynthesis, Polyethers
Alison M. Hill, School of Biosciences, University of Exeter, Stocker Road, Exeter, United Kingdom,
doi: 10.1002/9780470048672.wecb460
This review covers the biosynthesis of terrestrial and marine polyethers and discusses their biologic properties and the molecular genetics and enzymology of the proteins responsible for their formation. The biosynthesis of monensin, nanchangmycin, nonactin, and the marine polyether ladders are discussed in detail. Novel enzymes found only in type I polyketide polyether gene clusters that are responsible for the epoxidation and cyclization of polyene biosynthetic intermediates are described. The macrotetrolide biosynthetic gene cluster, which is an ACP-less type II polyketide synthase that functions noniteratively is reviewed.
The first polyether antibiotic to be isolated was nigericin 1 in 1951. By 1983, more than 70 terrestrial polyether antibiotics had been reported and Cane et al. (1) had proposed a unified stereochemical model to account for their biosynthesis, including the polyene-polyepoxide model of polyether formation. It took almost 20 years for the first polyether gene cluster to be reported (2). In contrast, the first marine polyethers were reported in 1981, and a model to explain the biosynthesis of marine polyether ladder compounds was proposed in 2006 (3); no genetic information is available currently for the marine polyethers.
Biological Background
Interest in these compounds (Fig. 1) derives from their ability to transport ions across biologic membranes, and some terrestrial polyethers have been used widely in veterinary medicine. Marine polyethers are responsible for numerous cases of human food poisoning and toxic algal tides, which cause massive fish kills.
Terrestrial polyethers
At physiological pH, the ionophores are ionized with the fat- soluble part of the molecule residing in the lipid bilayer of the membrane and the ionized moiety in the aqueous milieu (4). Binding of the metal ion takes place at the membrane surface: As successive ether oxygen atoms from the ionophore bind to the metal, it loses its solvated water molecules, thereby forming a neutral zwitterionic metal-ionophore complex. Transport across the membrane can now take place, and at the opposite surface, the process is reversed to leave the metal cation and the anionic ionophore on the other side of the membrane. The ionophore must bind another cation (usually a proton) to return to its original starting point.
The anionic ionophores are highly selective for particular metal cations, and both kinetic and thermodynamic terms determine which cation is selected. Note that the thermodynamic stability of the metal-ionophore complex does not always determine the transport rate. For example, nigericin forms a much more stable complex with potassium but transports sodium much more quickly. In the presence of both sodium and potassium, however, the extra stability of the nigericin-potassium complex results in the preferential transport of potassium over sodium (4).
Selectivities of ionophores are shown in Table 1 (5, 6). The carboxyl group may (e.g., nigericin) or may not (e.g., monensin 2) be involved in cation liganding via an ionic bond. Monensin has six liganding oxygen atoms but none for ionic bonds, and it is a weaker complexing agent than is nigericin. Lasalocid 4 can form a singly charged complex with doubly charged cations such as Ca2+. The broad specificity of lasalocid results from the metals sitting on top rather than within the oxygen system of the ionophore. For nonactin 5, the ester carbonyl oxygen atoms and the ether oxygen atoms are all liganding and form the apices of a cube. Nonactin adopts a conformation resembling the seam of a tennis ball (5).
Table 1. Ionophore selectivities (5, 6)
|
Ionophore |
MW |
Selectivity sequence |
|
Nigericin 1 |
724 |
K > Rb > Na > Cs >> Li Na >> K > Rb > Li > Cs Cs > Rb ≈ K > Na > Li; Ba > Sr > Ca > Mg NH4 > K ≈ Rb > Cs > Na Ca > Mg > Na |
|
Monensin A 2 |
670 |
|
|
Lasalocid 4 |
590 |
|
|
|
|
|
|
Nonactin 5 |
736 |
|
|
Tetronasin 10 |
602 |
Commercial interest in the polyketide polyethers stems from their antibiotic activity. Many polyethers of the class are potent coccidiostats and can control coccidial infections in poultry when added at approximately 100 ppm to the feed (7). Cocciodosis is caused from an infection in the digestive tract by parasitic protozoa, most commonly in poultry from the genus Eimeria. Monensin was the first polyether antibiotic to be patented and was released onto the market in 1971. It is still widely used today and is marketed under the tradename of Coban® (Elanco, Toronto, Ontario, Canada). After monensin treatment, coccidia have been observed to literally explode at one stage in their lifecycle because of the increase in osmotic pressure generated from the exchange of sodium outside the organism for protons inside (4). Lasalocid was launched in 1976 under the tradename Avatec® (Alpharma, Inc., Bridgewater, NJ) (7), and unlike monensin, it cannot be used for laying hens and must be withheld from poultry for three days before slaughter. The use of these compounds has led to some controversy as small amounts of the antibiotics have been detected in food for human consumption. Lasalocid residues have been detected in eggs, including organic eggs, and in 2004, the Soil Association recommended that maximum residue limits and acceptable daily intake levels should be set in line with Australian limits of 50 μg/kg in eggs and 1 μg/kg, respectively.
Ionophores can also be used in small doses to control bacteria in the rumen of cattle, which alters the ruminal fermentation to increase the amount of propionate (6). It results in an improvement in the feed efficiency and a reduction in methane output. Rumensin® (Elanco) is the tradename for monensin. Nigericin and the divalent antiporter tetronasin 10 also exert the same effect. Changes in ruminal fermentation patterns have been shown not to be cation specific, but the ionophore must have sodium or potassium and proton antiporter activity.
More recently, antimalarial activity has also been reported for monensin, nigericin, and lasalocid (8, 9). Plasmodium falciparum is sensitive to these compounds at all stages, but the schizont stages were most sensitive with 12 hours required for complete parasite clearance (8). In infected mice treated with 10mg/kg of monensin, all mice were cured compared with one third of the mice given nigericin. A combination of the two drugs was shown to be even more effective allowing the dose to be decreased. The biological effect is thought to develop from the depletion of protons from the parasite’s intracellular vesicles that results in acidification of the parasite cytosol, thereby inhibiting hemoglobin digestion, mitosis, and endocytic and exocytic pathways (9).
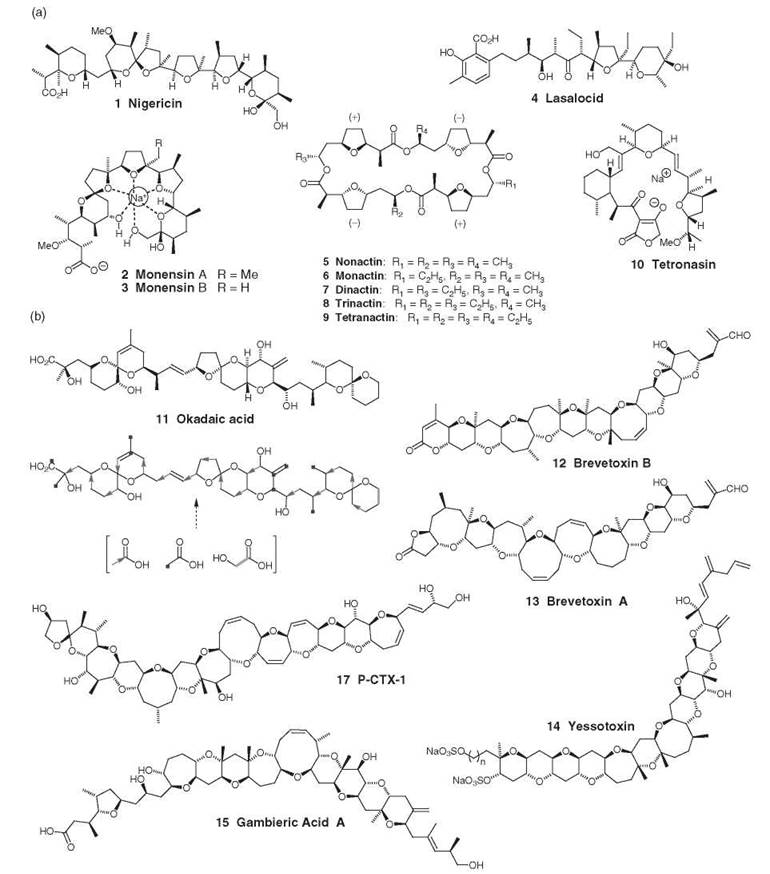
Figure 1. (a) Terrestrial polyethers; (b) marine polyethers.
Marine polyethers
Isolation of marine polyethers (Fig. 1b) is more recent than from terrestrial sources with structures for okadaic acid 11 and brevetoxin B 12 reported in 1981. Since that time, numerous polyether structures have been isolated from marine sources and several have had their biosynthetic pathways investigated through classical feeding experiments [e.g., okadaic acid (10)].
Considerable interest in these compounds exists as many are implicated in more than 60,000 cases of poisoning every year, of which 1.5% of these are fatal (11). These toxins are produced by marine microalgae, predominantly dinoflagellates, and are associated with red tides and lead to massive fish kills and mollusc contamination. Most dinoflagellate toxins are polyketides and include many polyethers with distinctive ring junctions resembling the rungs of a ladder. The dinoflagellate toxins are hazardous if inhaled or ingested. They are odorless, tasteless, and not destroyed by cooking or autoclaving. These toxins are extremely toxic in minute quantities, and no acceptable exposure limits have been established.
Okadaic acid 11 is a potent and specific inhibitor of protein phosphatases produced by the dinoflagellates Prorocentrum lima, Dinophysis fortii, and Dinophysis accuminata. It accumulates in bivalves and is one of the main toxins responsible for diarrhetic shellfish poisoning (DSP) (11). It has a highly unusual biosynthesis that has generated a lot of speculation because of the presence of “isolated” acetate chain methyl carbon atoms (10). Okadaic acid is a potent inhibitor of protein phosphatase A (11).
The brevetoxins (e.g., brevetoxin A 13 and brevetoxin B 12) are a family of lipid soluble neurotoxins produced by Karenia brevis (formally known as Gymnodinium breve) that are structurally similar to yessotoxin. They are thought to exert their biological action through the depolarization of the sodium channels of the excitable membranes (11). Binding of brevetoxin to the voltage-gated sodium channels changes its function by shifting the activation voltage for channel opening to a more negative value and inhibiting the inactivation of opened channels (resulting in persistent activation). If ingested, they cause neurotoxic shellfish poisoning. Brevetoxin derivatives have been patented as a treatment for cystic fibrosis, mucociliary dysfunction, and pulmonary diseases.
Yessotoxins 14 are produced by dinoflagellates of the genera Protoceratium and Gonyaulax. They cause selective disruption of the E-cadherin-catenin system in epithelial cells (11). In common with the other ladder shaped marine polyethers, yessotoxin interacts with transmembrane helix domains.
The gambieric acids 15, which are isolated from Gambierdiscus toxicus, are potent antifungal compounds that are particularly effective against filamentous fungi but inactive against yeasts. They are also cytotoxic, but they do not exhibit the neurotoxicity that is associated with other large marine polyethers such as yessotoxins, brevetoxins, and maitotoxins (11).
With a molecular weight of 3422 Da, maitotoxin 16 (see Fig. 4a later on in this article) is the largest nonproteinaceous natural product isolated (3). It is a marine polyether ladder produced by the dinoflagellate G. toxicus. Maitotoxin is a powerful activator of voltage-insensitive Ca2+ channels and exerts its biological effect through the increase of intracellular Ca2+ concentration (11). It can be used as a tool for studies on cellular events associated with Ca2+ flux. It stimulates synthesis and secretion of the nerve growth factor. The minimum lethal dose of the toxin in mice is 0.17 μg/kg (intraperitoneally).
The ciguatoxins (CTXs) are responsible for the symptoms of ciguatera fish poisoning caused by ingesting certain tropical and semitropical fish from the Indo-Pacific Oceans and Caribbean Sea. Ciguatoxin poisoning is the most frequent food-borne illness related to fin-fish consumption although it is rarely fatal. Symptoms include neurological, cardiovascular, and gastrointestinal disorders, and ciguatoxins can be detected in all body fluids (12). Most people recover slowly over time, which can take from weeks to years. P-CTX1 17 is the most toxic of all ciguatoxins with an LD50 of 0.25 μg/kg intraperitoneally. The toxins derive from the Gambierdiscus spp., which produce less-potent compounds that are biotransformed in the livers of the fin-fish to the more toxic ciguatoxins. The compounds activate the voltage-gated sodium channels that result in an increase of intracellular sodium. In high doses, ciguatoxins block the voltage-gated potassium channels that lead to membrane depolarization and that contribute to a lowering of action potential threshold (12).
Chemistry
Classical feeding experiments with both stable and radioactive isotopic labels (7) enabled the biosynthetic origin of the polyethers to be elucidated and for a general stereochemical model to be proposed (1). More recent work on this class of compounds has focused on a genetic approach, and unusual and interesting genes specific to polyether biosynthesis have been isolated from these clusters.
Monensin
Monensins A 2 and B 3 are polyether ionophores produced by Streptomyces cinnamonensis that differ only in the sidechain at C16 (ethyl/methyl). Monensin acts as a specific ionophore to dissipate ionic gradients across cell membranes and is used widely in veterinary medicine and as a food additive in animal husbandry (7). Antimalarial activity has also been reported (8, 9). Monensin is the best studied of the polyether ionophore antibiotics, and it was the first to have its gene cluster sequenced (2).
Early feeding studies established that monensin A is biosynthesized from a classical polyketide pathway and is derived from five acetate, seven propionate, and one butyrate (for monensin B, an additional propionate unit replaces the butyrate) (7). Four of the nine oxygen atoms are derived from molecular oxygen, with the remaining five deriving from the corresponding carboxylic acid precursors (Fig. 2a). Based on these initial experiments, it was proposed that the monensin PKS produced a linear E,E,E-triene precursor 24 that was oxidized and cyclized to give the final structure (1). Alternative proposals using Z,Z,Z - and E,Z,Z-trienes have been made (2).
Publication of the gene cluster for monensin (Table 2) (2, 13-17) showed that the PKS comprised 12 modules in eight contiguous open reading frames consistent with the production of the linear triene premonensin 25. The loading module contains an N-terminal KSQ domain that functions as a malonylCoA decarboxylase to generate starter units in situ.
Table 2. Monensin gene cluster (2, 13-17)
|
Polypeptide (size aa) |
Proposed function |
|
MonAI (3025) |
|
|
Loading module |
KSQ, AT(A), ACP |
|
Module 1 |
KS, AT(P), DH,* KR, ACP |
|
MonAII (2238) |
|
|
Module 2 |
KS, AT(P), DH, ER, KR, ACP |
|
MonAIII (4132) |
|
|
Module 3 |
KS, AT(A), DH, KR, ACP |
|
Module 4 |
KS, AT(P), DH, ER, KR, ACP |
|
MonAIV (4038) |
|
|
Module 5 |
KS, AT(B),1 DH, KR, ACP |
|
Module 6 |
KS, AT(A), DH, ER, KR, ACP |
|
MonAV (4106) |
|
|
Module 7 |
KS, AT(P), DH, KR, ACP |
|
Module 8 |
KS, AT(A), DH, ER, KR, ACP |
|
MonAVI (1700) |
|
|
Module 9 |
KS, AT(A), KR, ACP |
|
MonAVII (1641) |
|
|
Module 10 |
KS, AT(P), KR, ACP |
|
MonAVIII (3753) |
|
|
Module 11 |
KS, AT(P), KR, ACP |
|
Module 12 |
KS, AT(P), DH,* ER,2 KR, ACP |
|
MonAIX (268) |
PKS, Type II TE |
|
MonAX (267) |
|
|
MonBI (144) |
Epoxide hydrolase/cyclase3 |
|
MonBII (140) |
|
|
MonCI (496) |
Non-haem epoxidase |
|
MonCII (299) |
Chain terminating TE4 |
|
MonD (458) |
Hydroxylase |
|
MonE (276) |
Methyltranferase |
|
MonH, MonRI, MonRII |
Transcriptional regulators |
|
MonT (511) |
Efflux protein, resistance protein |
Reproduced from Reference 17 with permission from the Royal Society of Chemistry.
*“Unnecessary” domain (sequence is indistinguishable from active Mon DH domains).
1The AT of module 5 should incorporate an ethylmalonate extender unit, and it contains a signature sequence very close to the propionate sequence.
2Predicted to be inactive.
3Originally assigned as double-bond isomerase.
4Originally assigned as epoxide cyclase.
The monensin PKS does not contain an integrated C-terminal thioesterase domain at the end of module 12; instead an unusual 121 amino acid extension, rich in glycine, asparagines, and glutamine, was found (13). MonCII, which was originally assigned as an epoxide cyclase, has been shown to hydrolyze monensinyl NAC thioester 21 (Fig. 2c) as well as two other model substrates (14). Deletion of monCII gave a mutant that produced none or only trace amounts of monensin, which is consistent with its role as a chain-terminating TE. Complementation of monCII on a plasmid restored monensin production. Moreover, cell-free extracts from the monCII mutant treated with KOH gave significant amounts of monensin that resulted from the hydrolysis of a monensinyl ester or thioester present in the cell-free extract. Two ORFs, monAIX and monAX, have been shown to function as Type II thioesterases. Deletion of monAIX and/or monAX resulted in a modest drop in the monensin titer, which is consistent with their editing Type II TE role (14).
Genes for late steps such as methylation (monE) and hydroxylation (monD) and several regulatory genes (monH, RI, RII) were found in the cluster (2, 13). MonT is proposed to be involved in monensin export and consequently ensures the producing strain is self-resistant. The monCI gene product is a flavin-dependent epoxidase. Deletion of monCI results in complete loss of monensin production and the accumulation of the linear E, E, E-triene lactones (22, 23, Fig. 2d), which differ from premonensin 25 only by the different cyclization pattern of the polyketide chain: δ-lactone (22, 23) compared with a hemi-ketal (25) (15). Strains in which the adjacent genes monBI and/or monBII are deleted in addition to monCI give the same results. No oxidized derivatives of 22 and 23 were found, which suggests that the epoxidase encoded by monCII is necessary and sufficient for epoxidation of all three double bonds. These experiments are consistent with an E,E,E-configured triene as originally proposed by Cane et al. (1). Heterologous expression of MonCI in Streptomyces coelicolor gave a strain that could convert linalool to linalool oxide with a 10-20-fold greater conversion activity than studies carried out previously with S. cinnamonensis (13).
MonBI and monBII are highly homologous to each other and significantly similar to a ∆3-3-ketosteroid isomerase of Co- momonas testosteroni. Originally, it was proposed that monBI and monBII interconverted E double bonds to Z via an extended enolate ion during polyketide biosynthesis in two modules (resulting in an E,Z,Z-triene intermediate) (2). Their assignment has been revised in light of new experimental evidence that has shown that they are involved in the epoxide ring opening and concomitant polyether ring formation (16). Deletion of monBI and/or monBII gave strains that produced no monensin. Instead, C-3-O-demethylmonensins 18, C-9-epi-monensins 19, and C-26-deoxy-epi-monensins 20 (Fig. 2b) were produced in addition to numerous minor components. The major products from these mutant strains were the C-3-O-demethyl analogs. Hence, in the absence of the C-3-methoxy group, nucleophilic attack by the C-5 hydroxyl group from the si face of the C-9 carbonyl giving the natural epimer of the spiroketal takes place (Fig. 2e). In contrast, the presence of the methoxy group at C-3 leads to the C-5 hydroxy attacking the C-9 carbonyl from the re face leading to the unnatural spiroketal epimer 19. The C-26-deoxy-epi-monensins result from them being poor substrates for MonE. Treatment with acid converted the epi -monensins into the more thermodynamically stable corresponding monensins. Additional minor metabolites were detected in the cell culture but were produced in insufficient quantities to allow for structure determination. LC-MS analysis revealed that their molecular weights were identical to mon- ensins A/B and C-3-O-demethylmonensins A/B, which suggests that they were intermediates with the correct oxidation state but cyclized incompletely. Treatment of these minor metabolites with acid led to their conversion to monensins A/B and C-3-O-demethylmonensins A/B. Taken together, these results show that the MonB enzymes are epoxide hydrolase/cyclase enzymes that accelerate, but do not change, the stereochemical course of polyether ring formation (16). Since the pattern of products in MonBI and MonBII mutants seem to be identical, it seems that their actions are somehow coordinated and could in principle exist as a heterodimer in S. cinnamonensis. Although the arrangement of the proteins is not known, it is clear that ring closure in monensin biosynthesis occurs stepwise in a precise order under enzymatic control from one or both MonB enzymes. The timing of the hydroxylation step at C-26 has also been shown to occur before either methylation or polyether ring formation (16). The biosynthetic pathway for monensin is shown in Fig. 2e.

Figure 2. (a) Classic biosynthesis of monensin A (7); (b) novel monensins (16); (c) monensinyl N-acetylcysteamine thioester (14); (d) triene lactones produced by monCI mutant (15); (e) biosynthesis of monensin: The timing of the methylation and hydroxylation steps and release from the PKS are not known for certain (2,13-16).
Streptomyces nanchangensis produces the polyether antibiotic nanchangmycin 28, the 16-membered macrolide meilingmycin, and at least two other antibiotics of unknown structure (18).
The nanchangmycin gene cluster comprises 30 ORFs in a region of 132kb of DNA (Table 3) (19, 20). The type I PKS, encoding 14 modules in 11 ORFs, is arranged in two distinct groups (nanA1-A6, nanA7-A11) with the sugar biosynthesis genes located in between. A KSQ domain is located in the loading domain in NanA1 and so decarboxylation of malonate provides the acetate starter unit. The type II ACP (NanA10) is proposed to act as an independent ACP for module 13 (nanA9), which lacks an ACP function. Either side of nanA10 three unique polyether genes are located: nanI, nanO, and nanE, which are homologous to monBI/BII, monCI, and monCII. The epoxidase encoded by nanO oxidizes the triene precursor 30, which is then cyclized by NanI (Fig. 3b). Originally, the chain-releasing (CR) domain located in module 14 was thought to carry out the release of the polyether chain from the nanchangmycin PKS (19). Recent work has shown that although the CR domain is required in vivo for the efficient production of nanchangmycin, it does not catalyze the release of the polyketide chain from the PKS (20). NanE, is in fact responsible for the release of the fully processed nanchangmycin. It is homologous to both MonC11 and NigCII (which carries out an analogous role in nigericin biosynthesis). A gene encoding a cytochrome P450 (nanP) is located adjacent to the second PKS cluster and is thought to catalyze the oxidation of the C30 methyl group (19).
It is proposed that the biosynthesis of nanchangmycin (Fig. 3b) (19, 20) begins with loading of malonyl CoA onto NanA1. Subsequent decarboxylation and 13 rounds of extension would result in a tetradecaketide intermediate 30 attached to the type II independent ACP NanA10. Two epoxidations of the triene intermediate by NanO and ring opening of the di-epoxide 31 by NanI take place while the PKS intermediate is still attached to the independent ACP NanA10. The polyether chain 32 is then transferred to the ACP of module 14 for the final round of chain extension. Release from the PKS (NanE) followed by hydroxylation at C-30 by the putative monooxygenase NanP would give the completed aglycone polyether 33. Biosynthesis of the 4-O-methyl-L-rhodinose from D-glucose-1-phosphate (NanG1-G4, NanM) and attachment to the aglycone by NanG5 provides the completed nanchangmycin.
The novel nanchangmycin aglycone 29 (Fig. 3a) containing a keto group at C-19 was produced from a mutant containing an in-frame deletion of the module 6 KR (19).
Table 3. Nanchangmycin gene cluster (17, 19, 20)
|
Polypeptide (size aa) |
Function |
|
NanAl (2902) |
|
|
Loading Module |
KSQ, AT(A), ACP |
|
Module 1 |
KS, AT(P), DH, KR,* ACP |
|
NanA2 (2223) |
|
|
Module 2 |
KS, AT(P), DH, ER, KR, ACP |
|
NanA3 (4032) |
|
|
Module 3 |
KS, AT(A), DH, KR, ACP |
|
Module 4 |
KS, AT(P), DH, ER, KR, ACP |
|
NanA4 (3956) |
|
|
Module 5 |
KS, AT(P), DH, KR,* ACP |
|
Module 6 |
KS, AT(A), DH,* ER, KR, ACP |
|
NanA5 (3979) |
|
|
Module 7 |
KS, AT(P), DH, KR, ACP |
|
Module 8 |
KS, AT(A), DH, ER, KR, ACP |
|
NanA6 (l665) |
|
|
Module 9 |
KS, AT(A), KR,* ACP |
|
NanA7 (l646) |
|
|
Module 10 |
KS, AT(P), KR, ACP |
|
NanA8 (3455) |
|
|
Module 11 |
KS, AT(P), KR, ACP |
|
Module 12 |
KS, AT(P), DH, KR, ACP |
|
NanA9 (802) |
|
|
Module 13 |
KS, AT(P) |
|
NanA10 (104) |
ACP |
|
NanA11 (2187) |
|
|
Module 14 |
KS, AT(P), DH, ER, KR, ACP, CR |
|
NanE (290) |
Type I TE1 |
|
NanG1-G5 |
4-O-methyl-L-rhodinose biosynthesis |
|
NanI (313) |
Epoxide hydrolase/cyclase2 |
|
NanM (305) |
Methyl transferase |
|
NanO (478) |
Epoxidase |
|
NanP (423) |
Cytochrome P450 |
|
NanR1-R4 |
Regulation |
|
NanT1-T5 |
Transporters, chemoreceptor, regulator |
Reproduced from Reference 17 with permission from the Royal Society of Chemistry.
*Predicted to be inactive.
1Originally assigned as an epoxide hydrolase.
2Originally assigned as a ketosteroid isomerase.
Nonactin
The macrotetrolides 5-9 are a family of macrocyclic polyethers produced by S. griseus that exhibit a broad spectrum of biological activities (antibacterial, antitumor, antifungal, and immunosuppressive) (5, 21). They can act as ionophores and are effective against Gram-positive bacteria, mycobacteria, and fungi. The parent compound, nonactin 5, is an achiral molecule assembled from four molecules of enantiomeric non-actic acid 34 (Fig. 3c) in a (+)(-)(+)(-)-ester linkage. The homologues 6-9 are derived from the substitution of ethyl groups for methyl groups on the macrocyclic backbone.
Classical biosynthetic studies with 14C-labeled compounds and later with stable isotopes established that nonactic acid is derived from two acetates (or malonates), succinate and propionate (Fig. 3d) (21). Feeding studies with 36 (Fig. 3e) has shown that the first committed step of macrotetrolide biosynthesis is the coupling of the succinate unit with an acetate (or malonate) to give a-ketoadipate 40 (22). The late steps of non-actate biosynthesis (Fig. 3 g) were shown to involve the cyclization of 42a into (-)-nonactate and 42b into (+)-nonactate, which demonstrates that a pair of enantiocomplementary pathways are operating in nonactate biosynthesis (21). (±)-Non-actic acid is incorporated efficiently into nonactin. Both (+) and (-)-nonactic acids as well as their dimers have been isolated from cultures of S. griseus.
Genetic characterization of the nonactin gene cluster has revealed that an ACP-less Type II PKS is used to assemble the macrotetrolides (21). The non-PKS is highly unusual, catalyzing both C-C and C-O bond-forming reactions, functioning noniteratively and acting on acyl CoA substrates (which is typical of Type III PKS). Twenty-three non genes were identified, which includes five discrete KS proteins, NonJKPQU, and four discrete KR proteins, NonEMNO. Heterologous expression of these 23 nongenes in S. lividans resulted in macrotetrolide production, thereby confirming that only these genes are required for macrotetrolide biosynthesis; sequestration of an ACP from elsewhere on the S. griseus genome does not occur.
The proposed biosynthetic pathway (Fig. 3 g) requires three condensation and four reduction steps that would require at least three KS and four KR genes. It has not been established whether the five KS and four KR proteins function independently, but it has been demonstrated that all nine enzymes are required for macrotetrolide biosynthesis. Shen and Kwon (21) have suggested that the macrotetrolide PKS comprises noniteratively functioning subunits, which is unprecedented in all Type II PKSs known to date. NonJ and NonK have been assigned roles in dimerization (see below), which leaves NonPQU to carry out the three C-C bond-forming reactions. A mutant lacking the NonPQU genes was able to convert non-actic acid into nonactin, whereas deletion of NonJK resulted in no macrotetrolide formation. Hence, NonPQU represents a novel Type II minimal PKS that acts noniteratively, does not have an ACP, and uses acyl CoA substrates directly for polyketide biosynthesis.
NonJ has been shown to catalyze the first dimerization step to give (-)-non-actyl-(+)-non-actyl CoA 44, and NonK is responsible for the stereo-specific cyclodimerization to afford nonactin (21). All C-C bond forming KSs are characterized by a Cys-His-His (Asn) catalytic triad (including Non- PQU), whereas NonKJ are characterized by a mutated catalytic triad: Cys-Gly/Tyr-His. Replacement of the conserved cysteine residue in NonJ or NonK with glycine gave mutants that could not catalyze the transformation of (±)-non-actic acid into non- actin. Taken together, NonJK cannot function as a decarboxylase and instead catalyze the C-O bond-forming steps in nonactin biosynthesis using the same active site cysteine that other KSs use for C-C bond formation. In common with NonPQU, NonJK act noniteratively on CoA substrates.
The product of the minimal PKS, 41, is the branch point of the pathway that diverges into a pair of enantio-specific pathways, each of which involves two KRs and affords (+)- or (-)-42. The nonactate synthase, NonS, catalyzes the intramolecular Michael addition of (-)-nonactic acid 43a from (-)-42a. A nonS mutant supplemented with (±)-non-actic acid could produce nonactin, monactin, and dinactin, but not trinactin and tetran- actin, which require one/two (-)-homononactate moieties, respectively. This process suggests that NonS can cyclize (-)-42a into (-)-nonactate 43a and (-)-homononactate 35 only and that another nonactate synthase is required for the cyclization of (+)-42b to give (+)-nonactate 43b and (+)-homononactate.
Based on its high sequence homology to CoA ligases, NonL was identified as a CoA ligase catalyzing the transformation of (±)-non-actic acid into (±)-nonactyl CoA. The sequence of reactions for the biosynthesis of nonactin and the other macrotetrolides in S. griseus is shown in Fig. 3 g.
S. griseus is likely to be protected from nonactin by two mechanisms. The first mechanism seems to involve pumping excess antibiotic from the cell. Two genes, orf5 and orf6, which are clustered with the rest of the nonactin biosynthesis genes, show homology with ABC transporter genes. The second mechanism is the enzyme catalyzed hydrolysis of nonactin and homologues by NonR, which is homologous to serine protease and esterase enzymes and confers tetranactin resistance to S. lividans TK24. Overexpressed NonR has been shown to stereospecifically catalyze the breakage of the macrotetrolide ring into homochiral nonactate dimers 37 (Fig. 3f) in a two-step process (23). The macrotetrolides are converted initially into their seco-tetramer species and subsequently hydrolyzed to the dimer; in both cases, it is the bond between the alcohol of the (+)-nonactate and the carboxylate of (-)-nonactate that is cleaved. No trimer or monomers were detected.
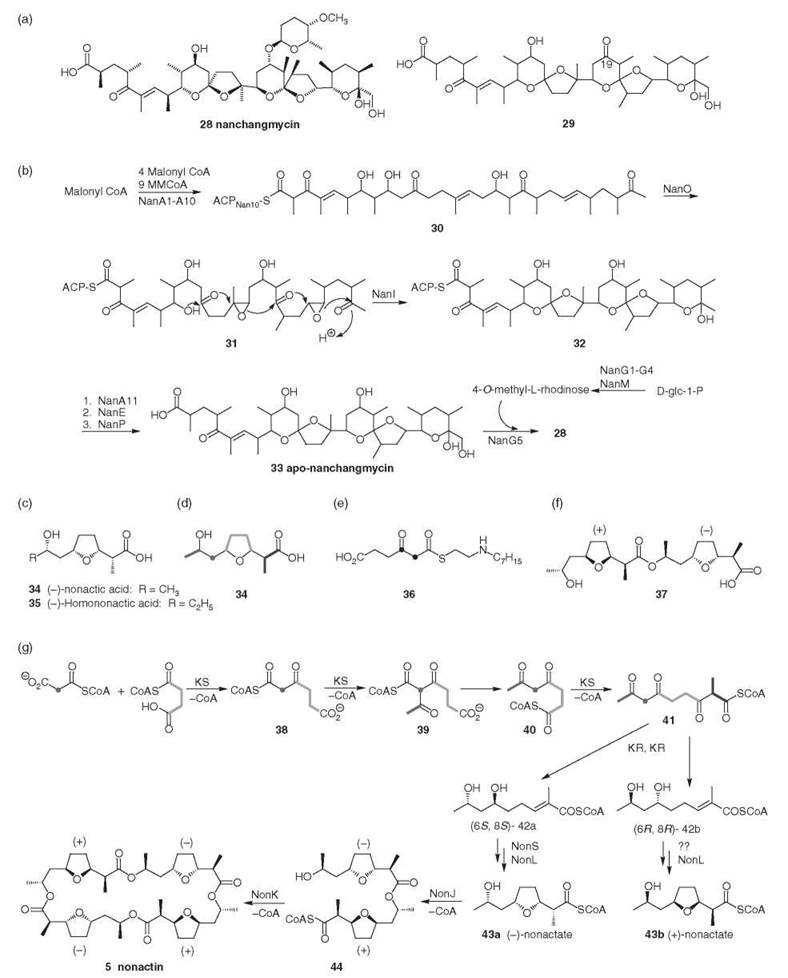
Figure 3. (a) Nanchangmycin and novel nanchangmycin aglycone. (b) Nanchangmycin biosynthesis (19, 20). (Adapted from Reference 17 with permission from the Royal Society of Chemistry.) (c) Structure of building blocks for the macrotetrolides. (d) Biosynthetic origin of carbon skeleton of nonactic acid (21). (e) (2,3-13C2)-3-ketoadipate N-caprylcysteamine thioester (22). (f) Homochiral nonactate dimmer (23). (g) Nonactin biosynthesis (21). (Adapted from Reference 17 with permission from the Royal Society of Chemistry.)
All marine polyether ladders thus far characterized (e.g., maitotoxin 16 (Fig. 4a), the brevetoxins 12 and 13 and hemibrevetoxin B, the yessotoxins 14 (and the truncated adriatoxin), the Pacific and Caribbean ciguatoxins, the gambieric acids 15 and gamberiol (the gymnocins and brevenal) can be grouped into 14 backbone structures (3).
Retrobiosynthetic analysis of these structures has led to the development of a model for the biosynthesis of these complex structures from the cyclization of a polyepoxide precursor (3). The model accounts for the conserved stereochemistry of the numerous ring junctions in these polyethers, which are syn/trans (Fig. 4b). The required configuration of the rings can be derived from stereochemically identical all (R,R)- or (S,S)-trans epoxides, which are derived from the appropriate polyene (which may contain over 20 double bonds). Epoxidation of the polyene precursor takes place from the same face, and consequently, it is possible for a single monooygenase with broad specificity to produce all trans epoxides. Ring closure requires an endo-selective opening of each epoxide.
Independently, Prasad and Shimizu (24) as well as Lee et al. (25) proposed that brevetoxin A is biosynthesized from the cyclization of a polyepoxide precursor in a series of SN2 (R,R)-trans epoxide openings (Fig. 4c). Gallimore and Spencer (3) argue that the nine disfavored endo-tet closures required for this mechanism makes it mechanistically unlikely and point out that an alternative cascade of SN2 epoxide openings in the opposite direction from all (S,S)-trans epoxides yields the same structure.
Although it can be envisaged that a trans-polyene is converted by a monooxygenase to a polyepoxide intermediate, which is processed by an epoxide hydrolase (cf. MonB in monensin biosynthesis) to the polycyclic ether, because of the reactivity of the polyepoxide intermediate (which may contain in excess of 20 reactive epoxide groups), this seems unlikely. An alternative mechanism is for the epoxidation and cyclization steps to be coupled in an iterative process whereby the production of an epoxide is followed by ring closure (Fig. 4 d). Gallimore and Spencer (3) suggest that it may be possible for this process to be affected by the monooxygenase only: After epoxidation, the bound enzyme stabilizes the endo transition state relative to the exo as the hydroxyl nucleophile attacks. Once the ring has been closed, the enzyme would dissociate and move on to the next double bond. In this manner, a single enzyme could be responsible for the conversion of a polyene chain to a polyether ladder. In both cases, no restriction on the number of double bonds can be processed, and hence, the number of contiguous rings can be (in theory) infinite.
Of all the marine polyether ladder metabolites examined, only a single ring junction in the largest natural product, maitotoxin, could not be explained using the model (3). Three of the four ladders in maitotoxin conformed to the stereochemical model; however, ladder C requires an epoxide with the opposite stereochemistry to the other centers, which gives rise to the only exceptional ring junction (“the J-K ring junction”) in any of the known polyether ladders. To explain this anomaly, Gallimore and Spencer (3) refer back to the original stereochemical assignment by Satake et al., which is described as challenging, and they suggest that the assignment be reexamined.
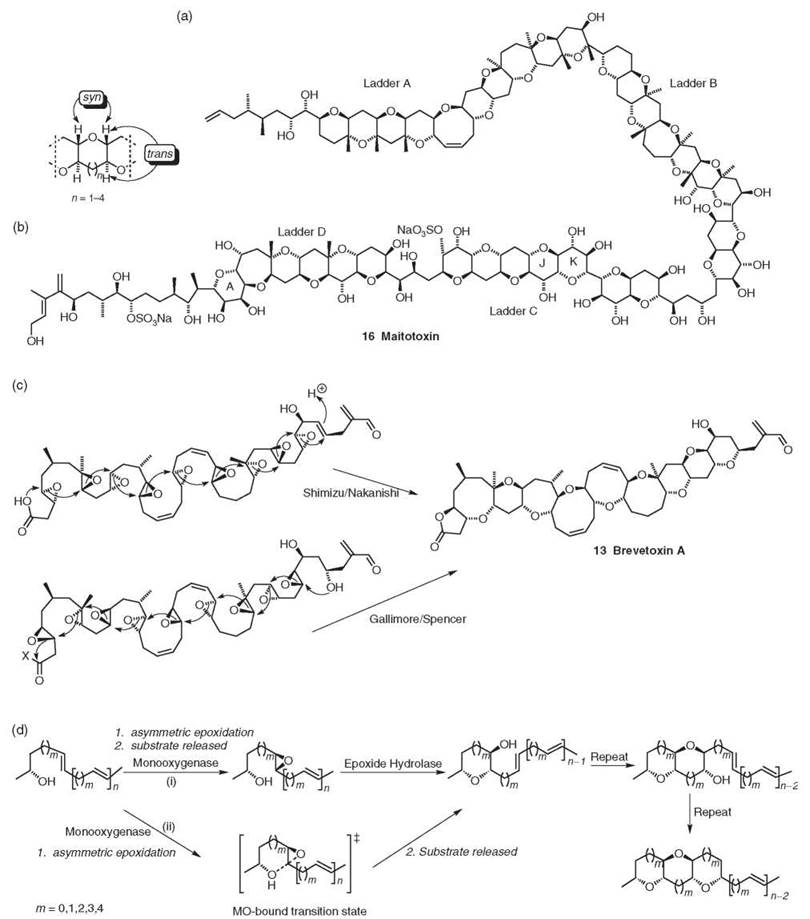
Figure 4. (a) Maitotoxin. (b) Common structure feature of marine polyether ring junctions (3). (c) Brevetoxin A biosynthesis as proposed by 1) Shimizu/Nakanishi (24, 25) and 2) Gallimore and Spencer from all (S,S)-trans epoxides (3). (d) Possible enzymatic routes to a fused polyether using 1) a mono-oxygenase and an epoxide hydrolase or 2) a monooxygenase only (3).
Early work on polyethers relied on the isolation and structure determination of the natural products followed by classical feeding experiments using both radioactive and stable isotopes to elucidate their biosynthesis (7). Today, advances in genetic techniques have allowed the gene sequences of the terrestrial polyethers to be studied although the number of published polyether clusters is small compared with macrolides and mixed NRPS/PKS systems. No marine polyether has had its gene cluster characterized, which reflects the difficulties with working with marine organisms and the lack of molecular tools available for these systems.
An interdisciplinary approach is required to study the polyethers involving chemists, biochemists, and molecular biologists. The first step is the isolation and structure determination of the polyether by natural product chemists. For the marine polyether ladders, this can be a huge undertaking in itself. Maitotoxin 16, for example, has four ladders, 32 rings, and a molecular weight of 3422 Da! Sophisticated NMR experiments were required to assign all stereocenters, and yet it was only detailed retrobiosynthetic analysis by Gallimore and Spencer that raised the possibility that one ring junction may have been assigned incorrectly (see above) (3).
Locating the polyether gene cluster can be done by using probes specific for polyketide genes to screen a cosmid library of total DNA [for both monensin 2 (2, 13) and nanchangmycin 28 (19), probes from the erythromycin PKS were used]. Alternatively, the polyether’s resistance gene can be used to locate the gene cluster [e.g., nonactin 5 (26)]. Sequencing and mapping of the cosmids in the library allows the gene cluster size and composition to be determined. More than one PKS sequence can be revealed, so care must be taken to ensure that the gene cluster of interest is being sequenced. For example, eight clusters were identified from the nanchangmycin producer S. nanchangensis, and the nanchangmycin gene cluster was identified through gene disruption (18). The organization and assignment of function of each ORF is carried out by sequence comparison. Cloning and overexpression of individual enzymes in the gene cluster is an important step in confirming the function and mechanism of a particular ORF. For example, sequence comparisons of both the monensin and the nanchangmycin clusters revealed no TE function required for the release of the polyketide from the PKS. However, cloning, overexpression, and characterization of MonCII (14) and NanE (20) resulted in the reassignment from an epoxide cyclase to Type I TE.
The preparation of mutants is an important tool to investigate the role of individual enzymes, and it is used to ascertain which genes in the cluster are essential for the biosynthesis of the polyether and whether modified/truncated products are formed. The effected gene can then be supplied in trans on a plasmid to see whether activity can be restored. Chemists are required to analyze the fermentation mixtures for minor and truncated metabolites. In this manner, the triene lactones 22 and 23 were isolated from a 7L culture of a mutant strain of S. cinnamonensis lacking MonCI, and their structure was determined by NMR (15).
HPLC-MS is an essential technique for the separation and identification of these trace compounds, but unless sufficient quantities can be isolated for structure determination by NMR, chemists are also required to synthesize standards for comparison. Characterization of the gene cluster requires chemists to synthesize intermediates (often with stable isotopes at specific places) to be used as substrates for purified enzymes or to confirm the structure of products formed from modified genes.
References
1. Cane DE, Celmer WD, Westley JW. Unified stereochemical model of polyether antibiotic structure and biogenesis. J. Am. Chem. Soc. 1983; 105:3594-3600.
2. Leadlay PF, Staunton J, Oliynyk M, Bisang C, Cortes J, Frost E, Hughes-Thomas ZA, Jones MA, Kendrew SG, Lester JB, Long PF, McArthur HAI, McCormick EL, Oliynyk Z, Stark CBW, Wilkinson CJ. Engineering of complex polyketide biosynthesis — insights from sequencing of the monensin biosynthetic gene cluster. J. Indus. Microb. Biotechnol. 2001; 27:360-367.
3. Gallimore AR, Spencer JB. Stereochemical uniformity in marine polyether ladders - implication for the biosynthesis and structure of maitotoxin. Angew. Chem. Int. Ed. 2006; 45:4406-4413.
4. Riddell FG. Ionophoric antibiotics. Chem. Br. 1992; 28:533-537.
5. Pressman BC. Biological applications of Ionophores. Ann. Rev. Biochem. 1976; 45:501-530.
6. Fellner V, Sauer FD, Kramer JKG. Effect of nigericin, monensin, and tetronasin on biohydrogenation in continuous flow-through ruminal fermenters. J. Dairy Sci. 1997; 80:921-928.
7. Westley JW. Polyether antibiotics - biosynthesis. Antibiotics IV. JW Corcoran, ed. 1981. Springer-Verlag, New York. pp. 41-73.
8. Gumila C, Ancelin M-L, Delort A-M, Jeminet G, Vial HJ. Characterisation of the potent in vitro and in vivo antimalarial activities of ionophore compounds. Antimicrob. Agents. Chemother. 1997; 41:523-529.
9. Adovelande J, Schrevel J. Carboxylic ionophores in malaria chemotherapy: the effects of monensin and nigericin on plasmodium falciparium in vitro and plasmodium vinckei petteri in vivo. Life Sci. 1996; 59:309-315.
10. Needham J, Hu T, McLachlan JL, Walter JA, Wright JLC. Biosynthetic studies of the DSP toxin DTX-4 and an okadaic acid diol ester. J. Chem. Soc., Chem. Commun. 1995; 1623-1624.
11. Garcia Camacho F, Gallardo Rodriguez J, Sanchez Miron A, Ceron Garcia MC, Belarbi EH, Chisti Y, Molina Grima E. Biotechnological significance of toxic marine dinoflagellates. Biotech. Adv. 2007; 25:176-194.
12. Nicholson GM, Lewis RJ. Ciguatoxins: Cyclic polyether modulators of voltage-gated ion channel function. Mar. Drugs 2006; 4:82-118.
13. Oliynyk M., Stark CBW, Bhatt A, Jones MA, Hughes-Thomas ZA, Wilkinson, C, Oliynyk Z, Demydchuk, Y, Staunton J, Leadlay PF. Analysis of the biosynthetic gene cluster for the polyether antibiotic monensin in Streptomyces cinnamonensis and evidence for the role of monB and monC in oxidative cyclization. Mol. Microbiol. 2003; 49:1179-1190.
14. Harvey BM, Hong H, Jones MA, Hughes-Thomas ZA, Goss RM, Heathcote ML, Bolanos-Garcia VM, Kroutil W, Staunton J, Leadlay PF, Spencer JB. Evidence that a novel thioesterase is responsible for polyketide chain release during biosynthesis of the polyether ionophore monensin. ChemBioChem 2006; 7:1435-1442.
15. Bhatt A, Stark CBW, Harvey BM, Gallimore AR, Demydchuk YA, Spencer JB, Staunton J, Leadlay PF. Accumulation of an E,E,E-triene by the monensin-producing polyketide synthase when oxidative cyclization is blocked. Angew. Chem. Int. Ed. 2005; 44:7075-7078.
16. Gallimore AR, Stark CBW, Bhatt A, Harvey BM, Demydchuck Y, Bolanos-Garcia V, Fowler DJ, Staunton J, Leadlay PF, Spencer JB. Evidence for the role of the monB genes in polyether ring formation during monensin biosynthesis. Chem. Biol. 2006; 13:453-460.
17. Hill AM, The biosynthesis, molecular genetics and enzymology of the polyketide-derived metabolites. Nat. Prod. Rep. 2006; 23:256-321.
18. Sun Y, Zhou X, Liu J, Bao K., Zhang G, Tu G, Kieser T, Deng Z. 'Streptomyces nanchangensis', a producer of the insecticidal polyether antibiotic nanchangmycin and the antiparasitic macrolide meilingmycin, contains multiple polyketide gene clusters. Microbiology 2002; 148:361-371.
19. Sun Y, Zhou X, Dong H, Tu G, Wang M, Wang B, Deng Z. A complete gene cluster from Streptomyces nanchangensis NS3226 encoding biosynthesis of the polyether ionophore nanchangmycin. Chem. Biol. 2003; 10:431-441.
20. Liu T, You D, Valenzano C., Sun Y, Li J, Yu Q, Zhou X, Cane DE, Deng Z. Identification of NanE as the thioesterase for polyether chain release in nanchangmycin biosynthesis. Chem. Biol. 2006; 13:945-955.
21. Shen B, Kwon H-J. Macrotetrolide biosynthesis: a novel type ii polyketide synthase. Chem. Rec. 2002; 2:389-396.
22. Nelson ME, Priestley ND. Nonactin biosynthesis: the initial committed step is the condensation of acetate (malonate) and succinate. J. Am. Chem. Soc. 2002; 124:2894-2902.
23. Cox JE, Priestley ND. Nonactin biosynthesis: the product of the resistance gene degrades nonactin stereospecifically to form homochiral nonactate diners. J. Am. Chem. Soc. 2005; 127:7976-7977.
24. Prasad AVK, Shimizu Y. The structure of hemibrevetoxin B: a new type of toxin in the gulf of mexico red tide organism. J. Am. Chem. Soc. 1989; 111:6476-6477.
25. Lee MS, Qin GW, Nakanishi K, Zagoeski MG. Biosynthetic studies of brevetoxins, potent neurotoxins produced by the dinoflagellate Gymnodiniumbreve. J. Am. Chem. Soc. 1989; 111:6234-6241.
26. Smith WC, Xiang L, Shen B. Genetic localization and molecular characterization of the nonS gene required for macrotetrolide biosynthesis in Streptomyces griseus DSM40695. Antimicrob. Agents Chemother. 2000; 44:1809-1817.
Further Reading
For a review on polyether biosynthesis (as well as polyketides and mixed NRPS/PKS metabolites): Hill AM. The biosynthesis, molecular genetics and enzymology of the polyketide-derived metabolites. Nat. Prod. Rep. 2006; 23:256-320.
For a review on polyether biosynthesis in dinoflagellates: Rein KS, Snyder RV. The biosynthesis of polyketide metabolites by dinoflagellates. Adv. Appl. Microbiol. 2006; 50:93-125.
For a review on the isolation and structure elucidation of the marine polyethers: Yasumoto T. The chemistry and biological function of natural marine toxins. Chem. Rec. 2001; 1:228-242.
The first report of cloning polyketide genes from a dinoflagellate: Kubota T, Iinuma Y, Kobayashi J. Cloning of polyketide synthase genes from amphidinolide-producing dinoflagellate Amphidinium sp. Biol. Pharm. Bull. 2006; 29:1314-1318.
For practical information on the manipulation of Streptomyces spp.: Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. 2000. John Innes Foundation, Norwich.
Information on polyether residues in eggs: Too Hard to Crack- Eggs with Residues—Executive Summary. Information Sheet Soil Association. http://www.soilassociation.org/Ubrary.
See Also
Polyketide Biosynthesis, Aromatic Polyketides
Polyketide Biosynthesis, Enediyne Polyketides
Polyketide Biosynthesis, Fungi
Polyketide Biosynthesis, Modular Polyketides
Polyketides as Drugs