CHEMICAL BIOLOGY
Proteins, Chemistry and Chemical Reactivity of
S. David Tilley, Neel S. Joshi, and Matthew B. Francis, Department of Chemistry, University of California, Berkeley, Berkeley, California
doi: 10.1002/9780470048672.wecb493
Protein bioconjugation is a critically important tool for the elucidation of enzyme mechanisms, the tracking of biomolecules in living systems, the improvement of pharmacokinetic properties, and the construction of new materials. All these applications rely on a continually expanding set of chemical reactions that can modify native protein functionality in aqueous solution under mild pH and temperature conditions. To survey these techniques, this article provides an introduction to the chemical reactivity of the amino acid side chains, with an emphasis on the selectivity that can be achieved using a particular reactive strategy. Site-selective techniques that target the unique reactive properties of N-terminal residues are also reviewed, as are native chemical ligation methods for the modification of the C-terminus. Whenever possible, the mechanistic aspects of the reactions are discussed, as these considerations provide the foundation for future reaction development. The article concludes with a brief description of labeling reactions that selectively target unnatural functional groups in the presence of native protein functionality.
Our understanding of the chemical reactivity of proteins lies at the very heart of chemical biology. In concert, the chemical behavior of the amino acid functional groups provides the basis for enzyme catalysis, and the differences between their reactive properties allow chemoselective protein labeling techniques to be developed. The ability to attach new functionality to a protein of interest allows the rich diversity of biomolecular function to be expanded to the limits of one’s imagination, which affords hybrid structures that can enhance our understanding of and interactions with living systems. For example, the biological role of many proteins has been clarified by tracking their cellular localization with attached fluorescent probes (1, 2). In many cases, chemical cross-linking strategies have then facilitated the identification of their binding partners. Radiolabels are commonly introduced to assess protein biodistribution in vivo, and polymers have been attached to improve circulation behavior (3, 4). Surface immobilization offers exceptional promise for the generation of microarrays for proteomic studies, and chemically modified proteins have been used to build new materials that use biological structure to template inorganic crystal growth (5, 6).
At the core of these studies is the expanding set of chemical reactions that can modify biomolecules in buffered aqueous solution. These techniques push organic reactivity to the limits of efficiency and chemoselectivity, often with the ability to modify a single functional site among hundreds of spectator groups. This article focuses on the reactive principles that have led to the development of these strategies, with a particular emphasis on the site selectivity that can be achieved for each. It is hoped that this survey can serve as a useful guide for those individuals wishing to modify a protein of interest and that it can facilitate the design of new strategies for modifying proteins with ever-increasing levels of precision and yield.
General Reactivity and Stability Considerations
Proteins have evolved to catalyze a remarkably diverse set of chemical transformations, largely by employing general acid/base chemistry and radical pathways (7). Although this range of reactivity provides the very reason for our interest in the chemical behavior of proteins, it also limits our ability to make general predictions about how specific reagents and conditions will affect a biomolecular target. As long as we remember that each protein has a unique reactivity “personality,” however, we can formulate a few guidelines that can serve as logical starting points for protein analysis, purification, and modification protocols.
The amide linkages of proteins are generally stable under both acidic and basic conditions, as are the native amino acid functional groups. However, changes in pH will alter the protonation state of many side chains (Fig. 1), which will result in changes in their reactivity and the overall folding state of the protein. To maintain higher-order structure, most modification techniques are conducted at a pH between 6 and 8.5, although many proteins will remain folded outside this range. The three-dimensional structure of most proteins is stable in aqueous solution between room temperature and 37° C, and many proteins (such as those isolated from thermophilic organisms) can be heated to temperatures that are significantly higher before unfolding occurs. Large fractions of organic cosolvents (such as methanol, DMF, and DMSO) are generally disruptive to protein structure, whereas small amounts are often used to solubilize the organic reagents that are used for protein modification. Although it is sometimes possible to refold proteins into their native conformation after denaturation occurs, this refolding can be difficult to achieve in practice because of unwanted aggregation or the absence of chaperones that facilitate the correct folding pathway. Thus, unless a refolding protocol is available or anticipated, it is recommended that proteins be processed under the mildest conditions possible if more biochemical studies are to be pursued.
The side chain functional groups contain a wide range of nucleophiles, including amines, thiolates, carboxylates, phenolates, and electron-rich aromatic rings, but disulfide bonds represent the only appreciably electrophilic functional groups. Therefore, the bulk of protein modification reagents are themselves electrophilic in nature. Amino acid side chains in the active sites of enzymes often show distinct nucleophilicity because of the alteration of their pKa values or other forms of activation. This nucleophilicity allows these positions to be targeted by less-reactive electrophiles that are ineffective for the modification of “ordinary” residues. An excellent review of this technique for the activity profiling of enzymes has recently appeared (8).
Many side chain groups are prone to oxidation, including thiolates, thioethers, and indole rings, but proteins are relatively nonreactive toward reducing agents (disulfide bonds again being the one exception). Although several proteins are known to catalyze reactions through radical pathways, proteins themselves are often unreactive toward radical species in solution [such as polymerization initiators (9)]. The lack of alkene functional groups also precludes the direct participation of proteins in most electrocyclization reactions.
A critical consideration for any protein labeling experiment is the level of site specificity that can be achieved. Largely, the inherent selectivity is dictated by the abundance of the targeted amino acid in the protein sequence. As listed in Fig. 1, the commonly modified amino acids occur with average frequencies ranging from 1.4% (tryptophan) to 6.3% (glutamic acid) of the amino acids overall (10). However, significant variations are commonly observed, as tabulated for the soluble cellulase domain in Fig. 1 (11). The hydrophobicity of the amino acids also influences their relative surface accessibility and, thus, their ability to participate in a particular reaction. The best strategy for the modification of a particular protein, therefore, varies on a case-by-case basis, which makes it important to have a range of modification techniques that can target several different amino acids.
These considerations form the basis for the numerous techniques that are now available for the chemical modification of proteins. The sections that follow will examine these techniques and the reactive principles by which they function. A section describing reactions that display orthogonal reactivity to native protein functional groups has also been included because of the growing importance of these reactions as tools to label proteins in complex mixtures. Because it is not practical to summarize all protein bioconjugation methods here, this information instead is intended to serve as an introduction to the concepts that drive the development of these reactions. Several additional reviews and books on protein modification have been listed in the Further Reading section.

Figure 1. Tabulated data for amino acid side chains commonly targeted in chemical modification reactions. As a specific example, a space-filling model of a soluble celulase domain from C. cellulolyticum (PDB ID 1IA7) shows the relative abundance and surface accessibility of these residues. Examples of commonly used modification reagents are also listed.
Chemical Reactivity of Amino Acid Residues in Proteins
It should be stressed that proteins are more than just the sum of the amino acids that comprise them. Surprises abound when predicting the chemical modification behavior of a particular protein, largely because of the complex interplay between the individual functional groups and the local environments in which they reside. Nevertheless, the first step to understanding the chemical behavior of proteins is to characterize the reactive properties of the individual side chain groups. The following sections describe these aspects, with an emphasis on the chemical techniques that are commonly used to modify these residues for applications in chemical biology.
Chemical reactions of lysine residues
The amino groups of lysine residues are highly nucleophilic in aqueous solution, which makes them the most commonly targeted sites for covalent protein modification. As they must be deprotonated to react, these reactions are typically run at slightly elevated pH (often 8-9). However, many lysine modification reagents undergo competing hydrolysis under alkaline conditions, and thus a balance must be struck to obtain optimal modification yield. Although the average pKa of lysine ammonium groups is 10.5, this value can be altered significantly by interactions with other charged groups on the protein surface. A recent review has described an analysis technique to evaluate these effects (12).
The most common reagents for lysine modification are NHS-esters, which react with amines at rates that are significantly greater than background hydrolysis, Fig. 2a (13). These reagents are most commonly prepared from carboxylic acids and isolated before exposure to the protein substrate. Because of the popularity of this method, dozens of premade NHS-esters are now commercially available. In cases where aqueous solubility is problematic, sulfonated NHS-esters can be used. Because virtually all proteins have many lysine residues on their surface, this reaction affords the most reliable and general method for protein modification, although initial screens are still recommended to determine the required amount of reagent and optimal buffer conditions. It should be noted that the abundance of lysine residues also renders this technique inherently non-site selective, which leads to mixtures of products that vary in both the number and location of the modifications. Although lysine acylation is the principal reaction that occurs with these reagents, the N-terminus can also be acylated, as can a variety of other nucleophilic side chains. In the latter cases, the transient species that result are often cleaved by exposing the protein to hydroxylamine before isolating the product by gel filtration.
Several related reagents are also available for lysine modification, including isocyanates, which are often prepared from acyl azides using the Curtius rearrangement (Fig. 2b); and isothiocyanates (Fig. 2c), which are more stable for long-term storage. Amine arylation can also be accomplished using 2,4-dinitrofluorobenzene (Sanger’s reagent, Fig. 2d). The products of this reaction are fluorescent, which assists their detection in sequencing applications. All of these reactions proceed under similar conditions and are often selected based on the convenience of reagent preparation from the compounds on hand.
As an alternative strategy, lysine residues can be modified through reductive alkylation, Fig. 2e. This method is most frequently carried out by exposing the protein to aldehydes in the presence of hydride-containing agents that reduce the transiently formed imines. NaB(CN)H3 and NaB(OAc)3H are commonly used for this purpose. As an alternative, transfer hydrogenation can be carried out in the presence of an Ir(III)[Cp*]2(bipyridyl) catalyst, which allows imine reduction to occur under mild conditions using buffered formate as the hydride source (14). Relative to acylation strategies, modification via reductive alkylation preserves the overall charge state (and thus the solubility) of the protein. In general, this technique also suffers from poor site selectivity.

Figure 2. Chemical modification strategies for lysine residues.
Chemical reactions of cysteine residues and disulfides
By far the most widely used methods for site-selective protein modification target cysteine residues. In contrast to lysine, cysteine is one of the rarest amino acids (10), and it is unusual to find it in the reduced form on protein surfaces. Thus, it is often possible to introduce a uniquely reactive cysteine residue using genetic methods. Similar to lysine-modifying reagents, many cysteine-reactive small molecules are commercially available because of the success of this overall strategy.
In the reduced state, the sulfhydryl group of cysteine can be deprotonated (pKa ~8) to generate a potent thiolate nucleophile. This species then can be intercepted by “soft” electrophiles, such as iodoacetamides (Fig. 3a), maleimides (Fig. 3b), acrylamides, and vinyl sulfones (Fig. 3c), to result in the formation of a new carbon-sulfur bond. Although lysine residues can also be modified using many of these reagents, cross-reactivity can often be minimized by running the reactions at relatively low pH to encourage protonation of the amino groups. Varying amounts of cysteine-alkylating reagents (from 1 to 1000 equivalents) are used to reach the desired level of modification. Although possible, the acylation of cysteine residues is not commonly used as a modification strategy because the resulting thioesters hydrolyze over time in solution.
Cysteine-alkylating groups have been coupled to small molecules that bind to specific sites on a protein to gain an additional level of labeling selectivity. As one example, a weakly reactive fluoromethyl ketone was fused to pyrrolopyrimidines known to inhibit a series of kinases. When binding to two kinases (RSK1 and RSK2), selective alkylation was observed for a cysteine residue adjacent to the binding site (15). Ligand-receptor interactions have also been implicated in the modification of ion channels. In these studies, maleimides bearing channel-blocking groups were used to alkylate genetically introduced cysteine residues. After modification, the activity of the ion channels could be controlled by the photoisomerization of an attached azo moiety (16).
A second method for the modification of cysteine residues involves the formation of disulfide bonds (Fig. 3d). In this method, reduced cysteine residues exchange with exogenous disulfides, which leads to the formation of new disulfides on the protein surface. This reaction can be driven to completion by mass action using a large excess of disulfide, or its efficiency can be improved by using asymmetric disulfides that possess good leaving groups. Disulfides of 2-thiopyridone (17) and 3-carboxy-4-nitrothiophenol (Ellman’s reagent) are commonly used for this purpose. To assist with the thermodynamic considerations of these reactions, a helpful table of disulfide reduction potentials has been published (18). A unique aspect of this class of cysteine-modification reactions is that they are reversible, which is a feature that could be exploited for drug delivery applications (19).
Disulfides themselves are often modified during protein analysis. Most commonly, this modification is accomplished by reducing them with dithiothreitol, mercaptoethanol, or tris (2-carboxyethyl)phosphine (20) and following this reduction by a subsequent alkylation step using any of the reagents listed above, Fig. 3e (21). An interesting version of this procedure has been developed to link both of the cysteines that result from the reduction step through two consecutive alkylation reactions (22). The ability of this technique to replace a disulfide bond with a more robust link could be used to improve the stability of proteins, hormones, or antibodies for in vivo applications. To distinguish reduced cysteines from disulfides in proteolytic digests, a biotin-based affinity capture strategy has been developed (23).

Figure 3. Chemical modification strategies for cysteine residues.
Chemical reactions of aspartic and glutamic acid residues
The surface of most proteins displays many aspartic and glutamic acid residues, which provides an additional set of locations for protein modification. The carboxylate groups are predominantly deprotonated at neutral pH (pKa = 3.5-4), which makes it generally difficult to distinguish between the many copies that are present. In cases where site selectivity is not a concern, the carboxylate functional groups can be activated through the use of water-soluble carbodiimides, such as N-ethyl-3-N’,N’-dimethylaminopropyl carbodiimide (EDC), Fig. 4. This reagent forms an O-acylisourea intermediate that can react with amines to form amides. This intermediate can also hydrolyze to regenerate the carboxylate anion, and in some cases it will undergo an acyl shift to yield an N-acyl urea. This latter pathway results in a permanent modification of the protein and in a deactivation of the carboxylate group toward more modification. Nucleophilic catalysts (such as NHS and HOBT) have been shown to suppress this pathway (24). When lysine side chains supply the amino groups, this reaction results in protein cross-links that can be used for topology mapping purposes. As one example, this technique has been used to determine the number of individual protein subunits in an ion channel, which affords information about its multimeric state (25).
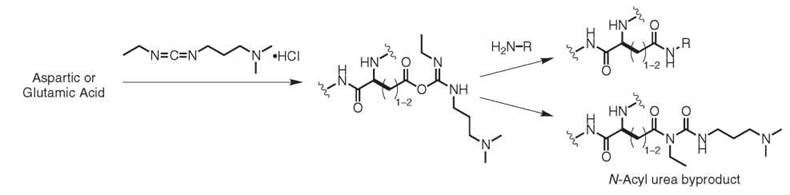
Figure 4. Chemical modification of aspartic and glutamic acid residues with EDC.
Chemical reactions of tyrosine residues
Although the chemical modification of tyrosine residues has enjoyed a long history, this residue remains an underused target for bioconjugation reactions. It is typically modified through electrophilic aromatic substitutions (EAS), which makes its reactivity distinct from other amino acid side chains. This reaction complementarity is particularly useful in cases when cysteine chemistry cannot be used or in combination with cysteine chemistry when multiple labels need to be introduced in specific locations. Although tyrosine is a relatively common amino acid, it is often buried in the interior of the protein. This placement effectively reduces the number of phenolic side chains that can participate in reactions, which often leads to higher site selectivity than can be expected for lysine modification strategies. In general, EAS reactions are selective for the positions adjacent to the phenolic hydroxyl group, and in some cases (such as iodination) two additions can be observed for a single ring. When increased reactivity is needed, reactions are often run at elevated pH (pH 8-10) to access an appreciable population of the more nucleophilic phenolate anion.
The oldest method for the modification of tyrosine residues occurs through the use of diazonium salts, as reported in 1904 (26). This reaction results in the formation of azo compounds that are brightly colored, Fig. 5a. Both tyrosine and histidine participate in these reactions, and it has been reported that aliphatic amino groups can react to form unstable triazens (27, 28). Thus, it should not be assumed that tyrosine is the only modified amino acid without additional confirmation. In the case of tyrosine, these reactions typically require the participation of the phenolate anion and thus are carried out at pH 8-10 at 4° C (29). In most cases, the diazonium salts are generated through the reaction of anilines with sodium nitrite under acidic conditions and are used immediately. This reaction has been used to produce antigens (30) and to elucidate the requirements of tyrosine and histidine in enzyme active sites (31). The azo products have also served as probes of amino acid surface accessibility (29), thus providing an indirect method to determine protein conformation. One recent example has used this reaction to immobilize pig liver esterase on silica to prepare a reusable catalyst for the hydrolytic kinetic resolution of racemic esters (32).
Diazonium coupling reactions have also been applied to the modification of protein assemblies, reaching nearly quantitative conversion for the modification of tyrosine residues that are displayed on MS2 (33) and TMV (34) viral capsids. These proteins are particularly good targets for this reaction, as each capsid monomer possesses a single solvent-exposed tyrosine residue and contains no histidines. In these studies, it was found that electron-withdrawing substituents in the 4 position of the diazonium salt significantly enhanced the efficiency of the reaction. These modification reactions have been used to introduce hundreds to thousands of ketone and aldehyde groups for the subsequent attachment of MRI contrast enhancement agents (35) and water-solubilized carbon nanotubes (36).
Phenol nitration with tetranitromethane (37) (TNM) is typically run at pH 8 at room temperature and is selective for tyrosine residues under these conditions (although some oxidation of cysteine residues has been reported) (38). The product of this reaction could be thought to develop through an electrophilic aromatic substitution reaction, but the mechanism instead has been shown to proceed through a radical coupling pathway (39). The degree of nitration is easily quantified by measuring the absorbance of nitrotyrosine at 428 nm. If the reaction is performed under more rigorous conditions, such as with increased equivalents of TNM, then histidine, tryptophan, and methionine can also react.
The in vivo nitration of tyrosine by peroxynitrite and other reactive nitrogen species is implicated in many disease states and is an area of active research (40). It has recently been proposed that tyrosine nitration, mediated by nitric oxide and superoxide, is a regulated cell signaling pathway that provides quick response to the microenvironment of the cell (41-43).
The iodination of proteins has proven to be invaluable in the area of biomedical research. These techniques allow the biodistribution of biomolecules to be tracked after they have been labeled with radioisotopes, such as Iodine-131. Although early studies used molecular iodine and bromine for this purpose (37), more efficient reagents have since been developed to make better use of small quantities of radioisotopes. An improved iodination method that uses iodine monochloride (ICl) has been exceptionally useful for protein radiolabeling, with efficiencies as high as 60-80% being reported, Fig. 5b (44). Since its original disclosure, several modifications of this reaction have been reported to improve efficiency and reduce handling hazards (45-47). An alternative Chloramine-T based method has also been developed to achieve both low and high levels of specific radioactivity, with iodine use as high as 60-75% (48).
Formaldehyde-induced cross-linking is commonly used for many biochemical applications, from studying protein-protein interactions to immobilizing whole cells on surfaces. Although a detailed early review of the effect of formaldehyde on proteins (49) implicated tyrosine as one residue responsible for cross-linking, a few reports have been written regarding the rigorous chemical characterization of this mechanism with modern analytical techniques. A recent investigation into the nature of these cross-links used extensive enzymatic degradation and MS/MS analysis of insulin that had been exposed to formaldehyde to determine the relative level of participation of different amino acids in cross-linking reactions (50, 51). The researchers concluded that tyrosine was among the most reactive amino acids in this procedure, reacting in an electrophilic aromatic substitution with Schiff bases formed between formaldehyde and the amines of nearby lysine residues, Fig. 5c. It is important to note that all methods for protein cross-linking that use this approach require high (>200 mM) concentrations of formaldehyde and often elevated temperatures (>37° C) that would not be suitable for preserving the native function of most proteins.
A more recent advance in tyrosine modification chemistry has been the development of a Mannich-type reaction that involves the condensation of an aniline derivative with aldehydes, followed by the nucleophilic addition of tyrosine to generate a new carbon-carbon bond, Fig. 5d (52). This modification strategy is operationally straightforward and occurs under mild reaction conditions. In most cases, the protein target is incubated with formaldehyde and an aniline of choice at concentrations that range from 5-25 mM in pH 6.5 aqueous buffer. Importantly, aliphatic amines do not participate in the reaction at these concentrations, which prevents undesired protein cross-linking. The reaction is typically quenched by the addition of hydroxylamine to cleave any imines that have been formed with lysine residues. Proteolytic digests and NMR experiments have confirmed the selectivity of the reaction for tyrosine, although side reactions with tryptophan have also been noted in some cases. Although cross-reactivity is likely to be observed in some cases, this reaction has been reported for the coupling of synthetic peptides to tyrosine residues on intact proteins (53).
Much interest has appeared in the development of techniques that can identify adjacent proteins in large multimeric assemblies. As one example, a tyrosine-based cross-linking strategy has been developed through the use of Ni(II) ions that bind to (His)6 tags on proteins of interest (54, 55). During oxidation to Ni(III) in the presence of persulfate ion or a peracid, the metal ion extracts an electron from a nearby tyrosine residue to create a radical intermediate. This species then couples to additional tyrosine residues and ultimately forms a dityrosine link after abstraction of a hydrogen atom by sulfate radical and re-aromatization, Fig. 5e. In the absence of a nearby tyrosine, a mechanism that involves the addition of a cysteine or lysine is proposed. In a subsequent report, an additional strategy coined “photo-induced crosslinking of unmodified proteins” (PICUP) was described as a technique that did not require a (His)6 tag (56). In this system, ruthenium(II) tris(bipridine), in combination with ammonium persulfate as an electron acceptor, effectively cross-links tyrosine-containing proteins with irradiation times of less than one second. In this reaction, the excited ruthenium complex donates an electron to ammonium persulfate, which generates Ru(III) as the one-electron oxidant. In follow-up studies, it was demonstrated that nickel-catalyzed proximity biotinylation and Ru(II)(bpy)3-mediated oxidative cross-linking can be used effectively to measure the equilibrium dissociation constant and stoichiometry of protein complexes (57). This methodology has also been used to map the large multiprotein complex 20 S proteasome core particle (58) and has provided a valuable tool for the study of pro-amyloid filaments (59). A modified version of this strategy has been used to cross-link tyrosine residues on a viral capsid and to modify them with alkyl halides that were added to the reaction solution (60).
A palladium-based method has been developed for the alkylation of the phenolic oxygen of tyrosine residues, Fig. 5f (61). In this reaction, allylic carbonates, esters, and carbamates are activated by palladium(0) complexes in aqueous solution to form electrophilic pi-allyl complexes. These species react at pH 8-10 with the phenolate anions of tyrosine residues, which results in the formation of an aryl ether and the regeneration of the Pd(0) catalyst. The reaction requires P(m-C6H4SO3-)3 as a water-soluble phosphine ligand. Activated pi-allyl complexes that do not react with tyrosine residues undergo P-hydride elimination under the basic conditions to yield diene by-products. A particularly attractive feature of this method is its ability to use substrates with charged groups in the allylic positions. This ability allows hydrophobic substrates, such as lipids, to be solubilized to facilitate protein modification.
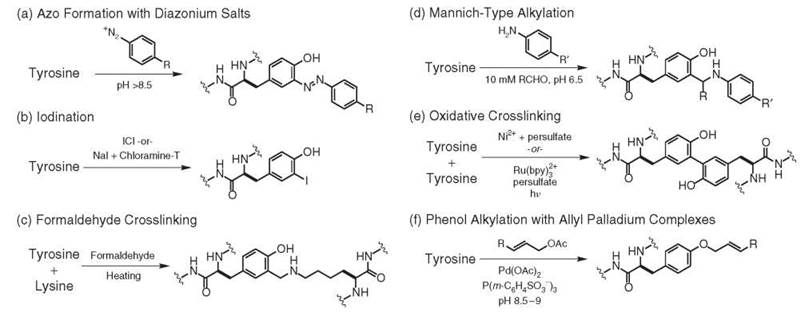
Figure 5. Chemical modification strategies for tyrosine residues.
Chemical reactions of tryptophan residues
Several features of tryptophan residues render them strategic targets for chemical modification. Tryptophan is the most fluorescent of the native amino acids, and the emission wavelength and intensity of this residue are often used to assess changes in the folding state of the protein or to detect ligand binding. Through the chemical alteration of this residue, these properties could be enhanced, shifted, or abrogated to determine the environment of a single tryptophan residue in the presence of others. As tryptophan residues are often hydrophobic contributors in binding sites, the modification of this residue could offer a way to modulate substrate binding. Studies have also revealed the importance of tryptophan side chains as mediators of electron transfer (62), which suggests opportunities to tune the electrical potential of tryptophan side chains through derivatization. As it is the rarest of the amino acids (particularly on protein surfaces), its selective modification could provide a strategic handle for bioconjugation when cysteine chemistry cannot be used. Because of its importance, a growing number of techniques are becoming available for the modification of this residue.
The electron-rich indole ring of tryptophan is susceptible to attack by electrophilic reagents. Although the 3-position is typically the most nucleophilic site of the aromatic ring, the alkyl substituent present in tryptophan residues can direct reactions to occur at the 2-position instead. Electrophilic halogen species, such as N-bromosuccinimide (NBS) (63) and in situ-generated dimethylchlorosulfonium ions, (64) have been used for some time to effect the oxidation of tryptophan residues through the intermediacy of halohydrins, Fig. 6a. These transient species undergo elimination, followed by tautomerization to afford 2-oxindole products. The halogenation conditions can also involve the intramolecular attack of the amide carbonyl on the bromonium ion intermediate, Fig. 6b (65). The resulting iminolactone hydrolyzes readily, which results in peptide bond cleavage. This procedure thus provides a useful alternative to cyanogen bromide cleavage for use in proteomics applications. With NBS, cleavage at tyrosine and histidine residues can also occur through similar pathways. As a more tryptophan-selective alternative, 3-bromo-2-(2-nitrophenylsulfonyl)-3-methylindole (BNPS-skatole) is often used (66).
It was recently reported that the tryptophan residues of proteins could be nitrated by the action of peroxynitrite (67). This reactive nitrogen species (RNS) is generated from the reaction of nitric oxide with superoxide at a rate that is ten times greater than the destruction of superoxide by dismutases. The authors propose that the nitration of tryptophan, although less common than tyrosine nitration, could serve to modulate the function of some proteins. However, at this time the in vivo evidence for tryptophan nitration by RNS has yet to be reported.
One common way to modify tryptophan residues occurs through the addition of 2-hydroxy-5-nitrobenzyl bromide (HNB). Also known as Koshland’s reagent (68), this compound first alkylates the 3-position of the ring and eventually leads to the formation of a product mixture through subsequent intramolecular cyclization pathways, Fig. 6c. This reagent exhibits good tryptophan selectivity, although some levels of cysteine cross-reactivity (about fivefold less) are sometimes observed. Multiple additions to a single tryptophan residue have also been reported (69). Nonetheless, this compound has long served as the reagent of choice to estimate the tryptophan content of proteins (70, 71), and it has been used extensively to probe the role of tryptophan in the active sites of enzymes (72-74).
As an alternative reaction pathway, a recent report has shown that the nitrogen atom of the indole ring can be modified using malonyl dialdehyde derivatives with quantitative conversion, Fig. 6d (75). When carried out under strongly acidic conditions, such as 50% aqueous trifluoroacetic acid, competing Schiff base formation and arginine modification can be avoided. Although the strongly acidic conditions required for this reaction will undoubtedly denature protein substrates, this reaction offers a convenient method for the identification of tryptophan-containing peptides for proteomics applications. The aldehyde group remaining in the product can be derivatized through hydrazine formation, which allows enrichment of tryptophan-containing peptides through solid-phase capture (76).
A transition metal catalyzed reaction has been developed to modify tryptophan residues on proteins with high chemoselectivity, Fig. 6e (77). This reaction involves metallocarbenoid intermediates generated in situ through the degradation of vinyl diazo compounds by rhodium carboxylate salts. The resulting species react with indole rings to form a mixture of N- and 2-substituted products, likely proceeding through direct N-H insertion and cyclopropanation/ring opening pathways, respectively. Although no organic cosolvent is required for the reaction to occur, the addition of small amounts of ethylene glycol can be used to achieve solubilization of the diazo compound. Hydroxylamine hydrochloride was found to promote this reaction significantly, presumably by binding the catalyst and attenuating the reactivity of the metallocarbenoid species. However, the use of this additive requires that the reaction be carried out under low pH conditions (~pH 3.5) that would be expected to denature most proteins. A subsequent study has reported that 4-diazo-1,6-heptadiene-3,5-dione derivatives can be used at neutral pH without the use of this additive (78). This report demonstrated the use of this approach for the attachment of fluorescent probes to a tryptophan residue on beta-lactoglobulin.

Figure 6. Chemical modification strategies for tyrptophan residues.
Chemical reactions of other amino acid residues
Several other amino acids are known to react selectively with chemical reagents. Although they are less commonly targeted for bioconjugation, the modification of these residues is often used for the purposes of analysis or to annihilate catalytic activity. As one example, histidine residues can be acylated using diethylpyrocarbonate (Fig. 7a), a commonly used strategy to deactivate RNase when intact RNA molecules are required. Methionine residues react selectively with cyanogen bromide (CNBr) (79), which results in the formation of cyclic imidates that can be cleaved during acidification (Fig. 7b). This method has long been used to degrade full-sized proteins into smaller peptides for sequencing analysis. Methionine residues are reactive toward oxidants, such as periodate, yielding sulfoxides. Arginine residues can be modified selectively using dicarbonyl compounds (Fig. 7c) (80). Both single- and double-addition products are obtained in these reactions. Serine and threonine residues are generally difficult to modify in aqueous solution unless they are present in the active sites of proteases (8) or at the protein N-terminus, as described below.

Figure 7. Chemical modification strategies of other amino acid residues.
Modification of the N- and C-Termini
As multiple copies of even the rarest amino acids are often present, cases exist in which none of the chemical modification strategies described above can be used to functionalize a single site. To address this challenge, many groups have developed reactions that take advantage of the unique chemical reactivities of the polypeptide termini. In addition to providing improvements in site selectivity, these methods have been used to form native-like peptide links in some instances.
Modification of the N-terminus
Historically, the most important technique to target the N-terminus has been the Edman degradation, Fig. 8a (81). In this sequencing method the N-terminal amino group is first reacted with phenylisothiocyanate to form a thiourea. During subsequent acidification, the sulfur atom attacks the proximal amide bond, which results in the removal of the first amino acid from the polypeptide chain. The initially formed thiazolidinone quickly rearranges under the acidic conditions to form the more stable thiohydantoin. HPLC analysis then is used to identify the liberated residue. The remaining polypeptide chain bears a new N-terminal amino acid that can be removed in a subsequent sequencing round. Peptides up to 50 amino acids in length can be analyzed in this fashion.
The N-terminal amino group is less basic [pKa = 6-8 for the protonated form (82)] than lysine amino groups, which makes it possible to achieve a degree of selective acylation by using NHS-esters at relatively low pH. However, this strategy usually does not yield absolute site selectivity because of the large number of competing lysines that are present. More selective N-terminal modification reactions also involve the side chain functionality of the first amino acid. A common example is the oxidation of beta-amino alcohols of N-terminal serine and threonine residues to yield aldehyde groups for subsequent modification (Fig. 8b) (83, 84). N-terminal serine and cysteine residues can also be condensed with aldehydes to form oxazolines and thiazolidines, respectively (Fig. 8c) (85). Both of these links are stable at high pH, but display varying hydrolysis rates in acidic media. Aldehydes are known to react readily and irreversibly with N-terminal tryptophan residues, yielding high yields of Pictet-Spengler products (Fig. 8d) (86). Finally, N-terminal cysteine residues can participate in native chemical ligations with thioesters, as detailed in the next section (see Fig. 9).
As an alternative to these techniques, reactive functionality can also be introduced at the N-terminus using biomimetic strategies. Early reports by Dixon indicated that reactive functionality can be accomplished by exposing proteins to aldehydes (such as glyoxylic acid) and Ni2+ or Cu2+ ions (87, 88). More recently, a metal-free version of this reaction has been reported using pyridoxal phosphate (PLP) to effect the transamination reaction (89). This method proceeds in buffered aqueous solution at pH 6.5 at 22-50° C. Several N-terminal amino acids, such as glycine and aspartic acid, have been shown to be compatible with this method, whereas tryptophan can react with the aldehyde of PLP through other pathways, as described in the previous paragraph. This technique provides a convenient method for the site-specific modification of several protein targets and has been demonstrated for the attachment of polymerization initiators (9) and surfaces to proteins through well-defined links (90). This method has also been applied to the modification of monoclonal antibodies in sites that are adjacent to the antigen binding regions (91).

Figure 8. Reactive strategies targeting the N-terminus.
Modification of the C-terminus
The native chemical ligation (NCL) has enjoyed tremendous success as one of the few methods for C-terminal protein modification. In its most general form, this approach is a ligation between a peptide that contains a C-terminal thioester and a peptide that contains an N-terminal cysteine, which results in a “native” amide bond (Fig. 9) (92). The full power of this protein synthesis technique is realized in the expressed protein ligation (EPL), a variant of the NCL in which the thioester-containing peptide is produced recombinantly (93). In EPL, the N-terminal portion of the desired peptide is expressed contiguously with an intein domain. The intein catalyzes an N-to-S acyl shift to form a thioester between the peptide target and a cysteine thiol at the end of the intein sequence. During the addition of a functionalized N-terminal cysteine, a transthioesterification occurs to join the two coupling partners and release the intein. A final S-to-N acyl shift forms the native peptide bond to yield the desired product. Additional small-molecule thiols are often added to accelerate this reaction.
A wide variety of biologically relevant molecules have been linked to the C-terminus of proteins using the EPL approach, ranging from naturally occurring biomolecules, such as lipids and polysaccharides, to non-natural probes like fluorophores, affinity tags, and metal chelating agents (94). In combination with solid-phase peptide synthesis (SPPS), EPL has made valuable contributions to proteomics by enabling the synthesis of proteins containing posttranslational modifications that are difficult to introduce (95). This approach has been used to attach fluorophores to proteins for the detection of protein-protein interactions by emission wavelength shifts (96) or through FRET (97). The site-specific incorporation of stable isotopes has been used also to facilitate NMR structural studies. In one example, two intein-splicing events were used to 15N-label an internal region of the 370-residue maltose binding protein (98). In conjugation with the “traceless” Staudinger reaction described below, the NCL technique has also been used to synthesize a functional RNase enzyme from several precursor fragments (99).
In the last few years, several interesting methods have been developed to couple C-terminal thioesters to the N-terminus of synthetic peptides without leaving cysteine residues at the junction site. These techniques instead supply the requisite sulfhydryl group through the use of auxiliaries that are cleaved after the ligation takes place. One notable example of this technique is the total synthesis of the multiply glycosylated erythropoietin alpha (EPO 1), a protein that has found widespread use in the treatment of anemia (100). Applications such as these showcase the versatility of the EPL and its considerable ability to address difficult bioconjugation challenges.

Figure 9. Modification of the C-terminus through native chemical ligation (NCL).
Bioorthogonal Methods for Protein Labeling
Although this review has focused on the reactivity modes that can be used to modify the amino acids directly, it is equally important to be able to predict what will not react with the native functional groups of proteins. Reactions that can proceed in aqueous solution while ignoring the natural amino acids are exceptionally useful for the development of secondary labeling strategies. In these approaches, one reaction described above is used to attach a chemically distinct functional group to a particular amino acid. A second reaction then is used to install a desired label that might have interfered with the chemistry of the first step. The targeting of unique chemical functional groups is also important for the labeling of a single biomolecule that is present among many others, as is the case in cell lysates, living cells, or entire organisms (101).
In particular, the development of “bioorthogonal” labeling reactions has been propelled by the availability of new tools for the direct incorporation of artificial functional groups into biomolecules. For proteins, powerful techniques have been developed for the introduction of new amino acids on the translational level, both through stop codon suppression (102) and amino acid codon reassignment (103). Metabolic engineering techniques have also been developed for the incorporation of artificial groups into carbohydrates (104, 105), and sequence-selective enzymatic labeling techniques have been used to introduce lipids (106) and cofactors (107, 108) that are substituted with new functionality.
The purpose of this section is to provide a brief list of the new chemical strategies that have been developed to target these artificial functional groups because the importance of these techniques will undoubtedly grow in the coming years.
Methods targeting carbonyl groups
The first methods that were used to label non-native functional groups on proteins targeted aldehydes and ketones. These groups can be introduced directly through the periodate oxidation of N-terminal serine residues (83), transamination (89), or carbohydrate oxidation with periodate (109). They can also be installed using metabolic engineering (104) or translational techniques (110). Aldehydes and ketones condense selectively with hydrazine and alkoxyamine derivatives to form hydrazone- and oxime-type derivatives, respectively (Fig. 10a) (110, 104). Both reactions are carried out in aqueous solution using an excess of the ketone-reactive reagents. Early mechanistic studies determined that the rate-limiting step in the reaction is the dehydration of the tetrahedral intermediate that is formed after nucleophilic attack on the carbonyl group (111). Therefore, mildly acidic conditions (e.g., pH 6.5) are typically used to accelerate the dehydration step without fully protonating the nucleophilic reagents. This reaction exhibits excellent chemoselectivity and works well for protein labeling with a diverse array of functional groups. However, the presence of competing ketone and aldehyde metabolites limits its use for protein labeling in crude cell lysates.

Figure 10. Chemoselective modification strategies targeting non-native functional groups.
Methods targeting azides
More recently, azide groups have emerged as popular targets for chemoselective protein modification. They seem to be ignored completely in a variety of biological settings (112), and yet they possess a favorable thermodynamic reaction potential through the loss of nitrogen gas (113). They are also small in size and thus can be incorporated using a variety of enzymatic, metabolic, or translational techniques. The azide group also provides a unique IR chromophore for spectroscopic characterization convenience.
The first technique that was used to modify azide groups in a biological setting was a modified Staudinger ligation (105), which used a pendant ester group to capture the iminophos- phorane intermediate. The resulting amide bond served to link additional functionality on the phosphine to the protein permanently (Fig. 10b). Although the original version of this reaction incorporated the triarylphosphine oxide into the coupling product, more recent “traceless” versions have appeared (114, 115). When used in conjunction with native chemical ligation techniques (see above), this reaction can serve as a means to synthesize full-sized proteins (99). A version of this technique has also been developed for the fluorescent detection of azido groups (116). Perhaps the ultimate testament to the bioorthogonality of this method is its reported use in living animals (112), which renders it a uniquely useful approach for the study of gly- cosylation using in vivo imaging techniques (117). A detailed mechanistic investigation of this reaction has appeared (118).
A second mode of reactivity for azide groups occurs through a [3 + 2] cyclization reaction with alkynes, as originally described by Huisgen (119). Although this reaction occurs thermally in the absence of additional reagents, a critical advance for the targeting of azide groups on biomolecules was the observation that Cu(I) salts catalyzed the formation of the triazole products in aqueous solution at ambient temperature (120, 121) (Fig. 10c). This technique has been used for the labeling of viral capsids (122, 123) and the surface of bacterial cells (124), and it has been used to attach proteins to surfaces (125). Both azide and alkyne groups have been introduced on the translational level to provide the appropriate coupling partners (124, 126). This reaction has been used with particular success as a detection method for the identification of new proteases, which were labeled with azides at active site residues (127). The monumental success of this reaction for the facile construction of diverse molecules in organic synthesis suggests that it will have a very bright future ahead for biomolecule labeling. The mechanism of the copper-catalyzed reaction has been described (128).
For situations in which copper ions are observed to interfere with protein function or to exhibit toxicity, a metal-free version of this reaction has been developed. This strategy uses a strained cyclooctyne as the alkyne component, which readily undergoes the Huisgen cyclization with azides at room temperature (Fig. 10d) (129). Although unsubstituted cyclic alkynes participate in the reaction with relatively slow rates, a subsequent report has shown that the presence of electron-withdrawing fluorine substituents can accelerate the cycloaddition significantly (130). Taken together, this set of reactive pathways renders the azide group one of the most promising targets for bioconjugation reactions in which complete functional group tolerance is required.
Modification through oxidative coupling reactions
An alternative method has been developed for the modification of proteins through the oxidative coupling of aniline groups (131) (Fig. 10e). This method targets aminophenylalanine, which is one of the most successful amino acids to be introduced into protein sequences using the stop codon suppression technique (132). In the presence of oxidants, such as periodate or cerium (IV), this group reacts rapidly with phenylene diamine derivatives to afford adducts that are highly stable toward hydrolysis, reduction, or oxidation. The reaction displays very high chemoselectivity, although the reliance on periodate leads to varying levels of methionine oxidation in addition to the desired products.
Conclusion
Although protein bioconjugation has enjoyed a long and successful history, recent years have witnessed a dramatic increase in the number of available techniques for chemical modification. This increase has developed largely because of the availability of high-resolution mass spectrometry and NMR characterization techniques, which have accelerated the rate with which detailed chemical information can be obtained for the modification products. With the addition of each new reaction, previously inaccessible protein bioconjugates of ever-increasing complexity can be realized. The chemical underpinnings of these reactions also form the basis for future modification strategies and expand our understanding of protein reactivity in living systems.
References
1. Staight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science 2003; 299:1743- 1747.
2. Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science 1998; 281:269-272.
3. Zalipsky S. Adv. Chemistry of polyethylene-glycol conjugates with biologically-active molecules. Adv. Drug Deliv. Rev. 1995; 16:157-182.
4. Zalipsky S, Harris JM. Introduction to chemistry and biological applications of poly(ethylene glycol). Poly(Ethylene Glycol). 1997; 680:1-13.
5. Niemeyer CM. Nanoparticles, proteins, and nucleic acids: biotechnology meets materials science. Angew. Chem. Int. Ed. 2001; 40:4128-4158.
6. Seeman NC, Belcher AM. Emulating biology: building nanostructures from the bottom up. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:6451-6455.
7. McMurry J, Begley TP. The Organic Chemistry of Biological Pathways. 2005. Roberts and Company, New York.
8. Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem. Rev. 2006; 106:3279-3301.
9. Heredia KL, Maynard HD. Synthesis of protein-polymer conjugates. Org. Biomol. Chem. 2007; 5:45-53.
10. Doolittle RF. Redundancies in protein sequences. In: Prediction of Protein Structure and the Principles of Protein Conformation. Fasman GD, ed. 1989. Plenum Press, New York.
11. Parsiegla G, Belaich A, Belaich JP, Haser R. Crystal structure of the cellulase Cel9M enlightens structure/function relationships of the variable catalytic modules in glycoside hydrolases. Biochemistry 2002; 41:11134.
12. Gitlin I, Carbeck JD, Whitesides GM. Why are proteins charged? Networks of charge-charge interactions in proteins measured by charge ladders and capillary electrophoresis. Angew. Chem. Int. Ed. 2006; 45:3022-3060.
13. Hermanson GT. Bioconjugate Techniques. 1996. Academic Press, San Diego.
14. McFarland JM, Francis MB. Reductive alkylation of proteins using iridium catalyzed transfer hydrogenation. J. Am. Chem. Soc. 2005; 127:13490-13491.
15. Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 2005; 308:1318-1321.
16. Szobota S, Gorostiza P, Del Bene F, Wyart C, Fortin DL, Kolstad KD, Tulyathan O, Volgraf M, Numano R, Aaron HL, Scott EK, Kramer RH, Flannery J, Baier H, Trauner D, Isacoff EY. Remote control of neuronal activity with a light-gated glutamate receptor. Neuron 2007; 54:535-545.
17. King TP, Li Y, Kochoumian L. Preparation of protein conjugates via intermolecular disulfide bond formation. Biochemistry 1978; 17:1499-1506.
18. Houk J, Whitesides GM. Structure-Reactivity Relations for Thiol-Disulfide Interchange. J. Am. Chem. Soc. 1987; 109:6825-6836.
19. Zalipsky S, Qazen M, Walker JA, Mullah N, Quinn YP, Huang SK. New detachable poly(ethylene glycol) conjugates: Cysteine-cleavable lipopolymers regenerating natural phospholipid, diacyl phosphatidylethanolamine. Bioconjug. Chem. 1999; 10:703-707.
20. Burns JA, Butler JC, Moran J, Whitesides GM. Selective reduction of disulfides by Tris(2-Carboxyethyl)Phosphine. J. Org. Chem. 1991; 56:2648-2650.
21. Sun MMC, Beam KS, Cerveny CG, Hamblett KJ, Black- more RS, Torgov MY, Handley FGM, Ihle NC, Senter PD, Alley SC. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug. Chem. 2005; 16:1282-1290.
22. Shaunak S, Godwin A, Choi JW, Balan S, Pedone E, Vi- jayarangam D, Heidelberger S, Teo I, Zloh M, Brocchini S. Site-specific PEGylation of native disulfide bonds in therapeutic proteins. Nat. Chem. Biol. 2006; 2:312-313.
23. Yen TY, Joshi RK, Yan H, Seto NOL, Palcic MM, Macher BA. Characterization of cysteine residues and disulfide bonds in proteins by liquid chromatography/electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 2000; 35:990-1002.
24. Schlick TL, Ding ZB, Kovacs EW, Francis MB. Dual-surface modification of the tobacco mosaic virus. J. Am. Chem. Soc. 2005; 127:3718-3723.
25. Maurer JA, Elmore DE, Lester HA, Dougherty DA. Comparing and contrasting Escherichia coli and Mycobacterium tuberculosis mechanosensitive channels (MscL)—new gain of function mutations in the loop region. J. Biol. Chem. 2000; 275:22238-22244.
26. Pauly, H. On the constitution of histidine I: Announcement. Hoppe-Seylers Zeitschrift Fur Physiologische Chemie 1904; 42:508-518.
27. Busch, M, Patrascanu, N, Weber, W. The coupling of D-amino acids with diazonium salts. J. Fur Praktische Chemie-Leipzig 1934; 140:117-128.
28. Howard, AN, Wild, F. Reactions of diazonium compounds with amino acids and proteins. Biochem. J. 1957; 65:651-659.
29. Riordan JF, Vallee BL. Diazonium salts as specific reagents and probes for protein conformation. Methods Enzymol. 1972; 25:521-531.
30. Landsteiner K. The Specificity of Serological Reactions. 1945. Harvard University Press, Cambridge, MA.
31. Fraenkel-Conrat H, Bean RS, Lineweaver H. Essential groups for the interaction of ovomucoid (egg white trypsin inhibitor) and trypsin, and for tryptic activity. J. Biol. Chem. 1949; 177:385-403.
32. Herdan JM, Balulescu M, Cira O. Enantioselective hydrolysis of racemic esters using pig liver esterase. J. Mol. Catal. A: Chem. 1996; 107:409-414.
33. Hooker JM, Kovacs EW, Francis MB. Interior surface modification of bacteriophage MS2. J. Am. Chem. Soc. 2004; 126:3718-3719.
34. Schlick TL, Ding ZB, Kovacs EW, Francis MB. Dual-surface modification of the tobacco mosaic virus. J. Am. Chem. Soc. 2005; 127:3718-3723.
35. Hooker JM, Datta A, Botta M, Raymond KN, Francis MB. Magnetic resonance contrast agents from viral capsid shells: a comparison of exterior and interior cargo strategies. Nano Lett. 2007; 7:2207-2210.
36. Holder PG, Francis MB. Integration of a self-assembling protein scaffold with water-soluble single-walled carbon nanotubes. Angew. Chem. Int. Ed. 2007; 46:4370-4373.
37. Wormall A. The immunological specificity of chemically altered proteins—halogenated and nitrated proteins. J. Exp. Med. 1930; 51:295-317.
38. Riordan JF, Vallee BL. Nitration with tetranitromethane. Methods Enzymol. 1972; 25:521-531.
39. Bruice TC, Gregory JJ, Walters SL. Reactions of tetranitromethane.1. kinetics and mechanism of nitration of phenols by tetranitromethane. J. Am. Chem. Soc. 1968; 90:1612-1614.
40. Wong PSY, van der Vliet A. Diazonium salts as specific reagents and probes of protein conformation. Methods Enzymol. 2002; 359:399-410.
41. Monteiro HP. Signal transduction by protein tyrosine nitration: competition or cooperation with tyrosine phosphorylation-dependent signaling events? Free Radic. Biol. Med. 2002; 33:765-773.
42. Koeck T, Stuehr DJ, Aulak KS. Mitochondria and regulated tyrosine nitration. Biochem. Soc. Trans. 2005; 33:1399-1403.
43. Ischiropoulos H, Gow A. Pathophysiological functions of nitric oxide-mediated protein modifications. Toxicology 2005; 208:299- 303.
44. McFarlane AS. Efficient trace-labelling of proteins with iodine. Nature 1958; 182:53-53.
45. Helmkamp RW, Goodland RL, Bale WF, Spar IL, Mutschler LE. High specific activity iodination of gamma-globulin with iodine-131 monochloride. Cancer Res. 1960; 20:1495-1500.
46. Samols E, Williams HS. Trace-labelling of insulin with iodine. Nature 1961; 190:1211-1212.
47. Helmkamp RW, Contreras MA, Izzo MJ. I131-labeling of proteins at high activity level with I131Cl produced by oxidation of total iodine in NaI131 preparations. Int. J. Appl. Radiat. Isot. 1967; 18:747-754.
48. Hunter WM, Greenwood FC. Preparation of iodine-131 labelled human growth hormone of high specific activity. Nature 1962; 194:495-496.
49. Fraenkel-Conrat H, Olcott HS. Reaction of formaldehyde with proteins.6. Cross-linking of amino groups with phenol, imidazole, or indole groups. J. Biol. Chem. 1948; 174:827-843.
50. Metz B, Kersten GFA, Baart GJE, de Jong A, Meiring H, ten Hove J, van Steenbergen MJ, Hennink WE, Crommelin DJA, Jiskoot W. Identification of formaldehyde-induced modifications in proteins: Reactions with insulin. Bioconjug. Chem. 2006; 17:815-822.
51. Metz B, Kersten GFA, Hoogerhout P, Brugghe HF, Timmermans HAM, de Jong A, Meiring H, ten Hove J, Hennink WE, Crommelin DJA, et al. Identification of formaldehyde-induced modifications in proteins—Reactions with model peptides. J. Biol. Chem. 2004; 279:6235-6243.
52. Joshi NS, Whitaker LR, Francis MB. A three-component Mannich-type reaction for selective tyrosine bioconjugation. J. Am. Chem. Soc. 2004; 126:15942-15943.
53. Romanini DW, Francis MB. Attachment of peptide building blocks to proteins through tyrosine bioconjugation. Bioconjug. Chem. In Press.
54. Kodadek T, Duroux-Richard I, Bonnafous JC. Techniques: oxidative cross-linking as an emergent tool for the analysis of receptor-mediated signaling events. Trends Pharmacol. Sci. 2005; 26:210-217.
55. Fancy DA. Elucidation of protein-protein interactions using chemical cross-linking or label transfer techniques. Curr. Opin. Chem. Biol. 2000; 4:28-33.
56. Fancy DA, Kodadek T. Chemistry for the analysis of protein-protein interactions: rapid and efficient cross-linking triggered by long wavelength light. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:6020-6024.
57. Amini F, Kodadek T, Brown KC. Protein affinity labeling mediated by genetically encoded peptide tags. Angew. Chem.-Int. Ed. 2001; 41:356-359.
58. Denison C, Kodadek T. Toward a general chemical method for rapidly mapping multi-protein complexes. J. Proteome Res. 2004; 3:417-425.
59. Bitan G, Teplow DB. Rapid photochemical cross-linking-a new tool for studies of metastable, amyloidogenic protein assemblies. Acc. Chem. Res. 2004; 37:357-364.
60. Meunier S, Strable E, Finn MG. Crosslinking of and coupling to viral capsid proteins by tyrosine oxidation. Chem. Biol. 2004; 11:319-326.
61. Tilley, SD, Francis MB. Tyrosine-selective protein alkylation using pi-allylpalladium complexes. J. Am. Chem. Soc. 2006; 128:1080-1081.
62. Stubbe J, Nocera DG, Yee CS, Chang MCY. Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem.Rev. 2003, 103:2167-2201.
63. Patchornik A, Lawson WB, Witkop B. Selective cleavage of peptide bonds.2. The tryptophyl peptide bond and the cleavage of glucagon. J. Am. Chem. Soc. 1958; 80:4747-4748.
64. Savige WE, Fontana A. Modification of tryptophan to oxin-dolylalanine by dimethyl sulfoxide-hydrochloric acid. Methods Enzymol. 1977; 47:442-453.
65. Ramachandran LK, Witkop B. N-bromosuccinimide cleavage of peptides. Methods Enzymol. 1967; 11:283-299.
66. Fontana A. Modification of tryptophan with BNPS-skatole (2-(2-nitrophenylsulfenyl)-3-methyl-3-bromoindolenine). Methods Enzymol. 1972; 25:419-423.
67. Yamakura F, Ikeda K. Modification of tryptophan and tryptophan residues in proteins by reactive nitrogen species. Nitric Oxide 2006; 14:152-161.
68. Koshland DE, Karkhanis YD, Latham HG. Environmentally-sensitive reagent with selectivity for tryptophan residue in proteins. J. Am. Chem. Soc. 1964; 86:1448-1450.
69. Strohalm M, Kodicek M, Pechar M. Tryptophan modification by 2-hydroxy-5-nitrobenzyl bromide studied by MALDI-TOF mass spectrometry. Biochem. Biophys. Res. Commun. 2003; 312:811- 816.
70. Barman TE, Koshland DE. A colorimetric procedure for quantitative determination of tryptophan residues in proteins. J. Biol. Chem. 1967; 242:5771-5776.
71. Dasgupta BR, Rothstein E, Boroff DA. Method for quantitative determination of free and peptide-linked tryptophan after reaction with 2-hydroxy-5-nitrobenzyl bromide. Anal. Biochem. 1965; 11:555-565.
72. Amutha B, Khire JM, Khan MI. Active site characterization of the exo-N-acetyl-beta-D-glucosaminidase from thermotolerant Bacillus sp NCIM 5120: involvement of tryptophan, histidine and carboxylate residues in catalytic activity. Biochim. Biophys. Acta 1999; 1427:121-132.
73. Ghosh AK, Naskar AK, Sengupta S. Characterisation of a xylanolytic amyloglucosidase of Termitomyces clypeatus. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 1997; 1339:289- 296.
74. Wink MR, Buffon A, Bonan CD, Valenzuela MA, Sarkis JJF, Battastini AMO. Effect of protein-modifying reagents on ecto-apyrase from rat brain. Int. J. Biochem. Cell Biol. 2000; 32:105-113.
75. Foettinger A, Melmer M, Leitner A, Lindner W. Reaction of the indole group with malondialdehyde: application for the derivatization of tryptophan residues in peptides. Bioconjug. Chem. 2007; 18:1678-1683.
76. Foettinger A, Leitner A, Lindner W. Selective enrichment of tryptophan-containing peptides from protein digests employing a reversible derivatization with malondialdehyde and solid-phase capture on hydrazide beads. J. Proteome Res. 2007; 6:3827-3834.
77. Antos JM, Francis MB. Selective tryptophan modification with rhodium carbenoids in aqueous solution. J. Am. Chem. Soc. 2004; 126:10256-10257.
78. Bao ZJ, Wang SJ, Shi W, Dong SY, Ma HM. Selective modification of Trp19 in beta-lactoglobulin by a new diazo fluorescence probe. J. Proteome Res. 2007; 6:3835-3841.
79. Gross E. The cyanogen bromide reaction. Methods Enzymol. 1967; 11:238-255.
80. Yankeelov JA. Modification of arginine by diketones. Methods Enzymol. 1972; 25:566-579.
81. Edman P. Method for determination of the amino acid sequence in peptides. Acta Chem. Scand. 1950; 4:283-293.
82. Sereda TJ, Mant CT, Quinn AM, Hodges RS. Effect of alpha-amino group on peptide retention behavior in reversed-phase chromatography—determination of the pKa values of the alpha-amino group of 19 different N-terminal amino-acid-residues. J. Chromatogr. 1993; 646:17-30.
83. Geoghegan KF, Stroh JG. Site-directed conjugation of nonpeptide groups to peptides and proteins via periodate-oxidation of a 2-amino alcohol—application to modification at N-terminal serine.Bioconjug. Chem. 1992; 3:138-146.
84. Chen JK, Lane WS, Brauer AW, Tanaka A, Schreiber SL. Biased combinatorial libraries—novel ligands for the Sh3 domain of phosphatidylinositol 3-kinase. J. Am. Chem. Soc. 1993; 115:12591-12592.
85. Tam JP, Yu QT, Miao ZW. Orthogonal ligation strategies for peptide and protein. Biopolymers 1999; 51:311-332.
86. Li XF, Zhang LS, Hall SE, Tam JP. A new ligation method for N-terminal tryptophan-containing peptides using the Pictet-Spengler reaction. Tetrahedron Lett. 2000; 41:4069-4073.
87. Dixon HBF. N-terminal modification of proteins—a review. J. Protein Chem. 1984; 3:99-108.
88. Wu P, Brand L. N-terminal modification of proteins for fluorescence measurements. Methods Enzymol. 1997; 278:321-330.
89. Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. N-terminal protein modification through a biomimetic transamination reaction. Angew. Chem. Int. Ed. 2006; 45:5307-5311.
90. Christman KL, Broyer RM, Tolstyka ZP, Maynard HD. Site-specific protein immobilization through N-terminal oxime linkages. J. Mater. Chem. 2007; 17:2021-2027.
91. Scheck RA, Francis MB. Regioselective labeling of antibodies through N-terminal transamination. ACA Chem. Biol. 2007; 2:247-251.
92. Dawson PE, Muir TW, Clarklewis I, Kent SBH. Synthesis of proteins by native chemical ligation. Science 1994; 266:776-779.
93. Hofmann RM, Muir TW: Recent advances in the application of expressed protein ligation to protein engineering. Curr. Opin. Biotechnol. 2002; 13:297-303.
94. Tolbert TJ, Wong CH. Intein-mediated synthesis of proteins containing carbohydrates and other molecular probes. J. Am. Chem. Soc. 2000; 122:5421-5428.
95. Ottesen JJ, Huse M, Sekedat MD, Muir TW. Semisynthesis of phosphovariants of Smad2 reveals substrate preference of activated T-beta-RI kinase. Biochemistry 2004; 43:5698-5706.
96. Cotton GJ, Ayers B, Xu R, Muir TW. Insertion of a synthetic peptide into a recombinant protein framework: a protein biosensor. J. Am. Chem. Soc. 1999; 121:1100-1101.
97. Cotton GJ, Muir TW. Generation of a dual-labeled fluorescence biosensor for Crk-II phosphorylation using solid-phase expressed protein ligation. Chem. Biol. 2000; 7:253-261.
98. Otomo T, Ito N, Kyogoku Y, Yamazaki T. NMR observation of selected segments in a larger protein: central-segment isotope labeling through intein-mediated ligation. Biochemistry 1999; 38:16040-16044.
99. Nilsson BL, Hondal RJ, Soellner MB, Raines RT. Protein assembly by orthogonal chemical ligation methods. J. Am. Chem.l Soc. 2003; 125:5268-5269.
100. Wu B, Chen JH, Warren JD, Chen G, Hua ZH, Danishefsky SJ. Building complex glycopeptides: development of a cysteine-free native chemical ligation protocol. Angew. Chem. Int. Ed. 2006; 45:4116-4125.
101. Prescher JA, Bertozzi CR. Chemistry in living systems. Nat. Chem. Biol. 2005; 1:13-21.
102. Wang L, Schultz PG. Expanding the genetic code. Chem. Commun. 2002; 1-11.
103. Kiick KL, Tirrell DA. Protein engineering by in vivo incorporation of non-natural amino acids: Control of incorporation of methionine analogues by methionyl-tRNA synthetase. Tetrahedron 2000; 56:9487-9493.
104. Mahal LK, Yarema KJ, Bertozzi CR. Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis. Science 1997; 276:1125-1128.
105. Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science 2000; 287:2007-2010.
106. Kho Y, Kim SC, Jiang C, Barma D, Kwon SW, Cheng JK, Jaunbergs J, Weinbaum C, Tamanoi F, Falck J, Zhao YM. A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:12479-12484.
107. Yin J, Liu F, Li XH, Walsh CT. Labeling proteins with small molecules by site-specific posttranslational modification. J. Am. Chem. Soc. 2004; 126:7754-7755.
108. Chen I, Howarth M, Lin W, Ting AY. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2005; 2:99-104.
109. Hage DS, Wolfe CAC, Oates MR. Development of a kinetic model to describe the effective rate of antibody oxidation by periodate. Bioconjug. Chem. 1997; 8:914-920.
110. Cornish VW, Hahn KM, Schultz PG. Site-specific protein modification using a ketone handle. J. Am. Chem. Soc. 1996; 118:8150-8151.
111. Jencks WP. Studies on the mechanism of oxime and semicarbazone formation. J. Am. Chem. Soc. 1959; 81:475-481.
112. Prescher JA, Dube DH, Bertozzi CR. Chemical remodelling of cell surfaces in living animals. Nature 2004; 430:873-877.
113. Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem.-Int. Ed. 2001; 40:2004-2021.
114. Nilsson BL, Kiessling LL, Raines RT. Staudinger ligation: a peptide from a thioester and azide. Org. Lett. 2000;2:1939-1941.
115. Saxon E, Armstrong JI, Bertozzi CR. A “traceless” Staudinger ligation for the chemoselective synthesis of amide bonds. Org.Lett. 2000; 2:2141-2143.
116. Lemieux GA, de Graffenried CL, Bertozzi CR. A fluorogenic dye activated by the Staudinger ligation. J. Am. Chem. Soc. 2003; 125:4708-4709.
117. Prescher JA, Dube DH, Lo A, Bertozzi CR. Noninvasive imaging of glycosylation in vivo. Glycobiology 2005; 15:1187-1187.
118. Lin FL, Hoyt HM, van Halbeek H, Bergman RG, Bertozzi CR. Mechanistic investigation of the Staudinger ligation. J. Am. Chem. Soc. 2005; 127:2686-2695.
119. Huisgen R. 1,3-Dipolar Cycloaddition Chemistry, Volume I. Padwa A, ed. 1984. Wiley, New York.
120. Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselec- tive “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002; 41:2596-2599.
121. Torn0e CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002; 67:3057-3062.
122. Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003; 125:3192-3193.
123. Sen Gupta S, Kuzelka J, Singh P, Lewis WG, Manchester M, Finn MG. Accelerated bioorthogonal conjugation: a practical method for the Ligation of diverse functional molecules to a polyvalent virus scaffold. Bioconjug. Chem. 2005; 16:1572-1579.
124. Link AJ, Tirrell DA. Cell surface labeling of Escherichia coli via copper(I)-catalyzed [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003; 125:11164-11165.
125. Gauchet C, Labadie GR, Poulter CD. Regio- and chemoselective covalent immobilization of proteins through unnatural amino acids. J. Am. Chem. Soc. 2006; 128:9274-9275.
126. Deiters A, Cropp TA, Mukherji M, Chin JW, Anderson JC, Schultz PG. Adding amino acids with novel reactivity to the genetic code of Saccharomyces cerevisiae. J. Am. Chem. Soc. 2003; 125:11782-11783.
127. Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003; 125:4686-4687.
128. Rodionov VO, Fokin VV, Finn MG. Mechanism of the ligand- free Cu-I-catalyzed azide-alkyne cycloaddition reaction. Angew. Chem. Int. Ed. 2005; 44:2210-2215.
129. Agard NJ, Prescher JA, Bertozzi CR. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of blomolecules in living systems. J. Am. Chem. Soc. 2004; 126:15046- 15047.
130. Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. A comparative study of bioorthogonal reactions with azides. ACS Chem. Biol. 2006; 1:644-648.
131. Hooker JM, Esser-Kahn AP, Francis MB. Modification of aniline containing proteins using an oxidative coupling strategy. J. Am. Chem. Soc. 2006; 128:15558-15559.
132. Mehl RA, Anderson JC, Santoro SW, Wang L, Martin AB, King DS, Horn DM, Schultz PG Generation of a bacterium with a 21 amino acid genetic code. J. Am. Chem. Soc. 2003; 125:935-939.
Further Reading
Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chemical Reviews 2006; 106:3279-3301.
Francis MB. New Chemical tools for protein modification. In: Chemical Biology. Schreiber SL, Kapoor T, Wess G, eds. 2007. Wiley-VCH: Weinheim, Germany. pp. 593-635.
Hermanson GT. Bioconjugate techniques. 1996. Academic Press, San Diego, CA.
McMurry J, Begley TP. The Organic Chemistry of Biological Pathways. 2005. Roberts and Company, New York.
Niemeyer CM, ed. Bioconjugation Protocols: Strategies and Methods (Methods in Molecular Biology). 2004. Humana Press, Totowa, NJ.
See Also
Chemical Modification of Proteins
Amino Acids, Chemical Properties of
Enzyme Catalysis, Chemical Strategies for
Proteins, In Vivo Chemical Modifications of
Peptides, Chemistry of