CHEMICAL BIOLOGY
Ras Oncoproteins, Structure, Biochemistry and Biology of
Emily J. Chenette, Duke Institute for Genome Sciences and Policy, Duke University Medical Center, Durham, North Carolina
Channing J. Der, Department of Pharmacology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina
doi: 10.1002/9780470048672.wecb503
The Ras small GTPase is well known for its role in regulating normal cellular proliferation, as well as in promoting human oncogenesis when activated mutationally. In signal transduction, Ras functions as a guanine nucleotide-regulated ON-OFF switch. Positioned at cellular membranes, Ras relays signals initiated by diverse extracellular stimuli to a complex network of cytoplasmic signaling cascades to affect changes in gene transcription, cell-cycle progression, survival, and differentiation. The intense research into the biologic and chemical nature of Ras has prompted the development of a variety of biologic, pharmacologic, and genetic tools to study Ras signaling. Also, Ras is the founding member of a superfamily of Ras-related and Ras-like proteins. The technical approaches and chemical concepts that have been generated from the study of Ras have aided greatly the studies of Ras superfamily proteins, which revealed the versatile and divergent biologic roles of small GTPases in cell physiology.
The connection between mutations in RAS genes and in human carcinogenesis was first established nearly 25 years ago (1). Research efforts since then have demonstrated that aberrant Ras signaling plays an integral role in the development and malignant growth of many types of human cancers. Ras proteins are simple binary switches that are controlled by a regulated GDP/GTP cycle (2) (see Fig. 1). A diverse spectrum of extracellular stimuli promotes the transient formation of active, GTP-bound Ras. Then, activated Ras interacts with downstream effectors that regulate cytoplasmic signaling cascades to promote changes in cell-cycle progression, actin cytoskeletal organization, cell survival, and gene transcription (3). Whereas wild-type Ras cycles between the GTP-bound and GDP-bound states in a regulated manner, cancer-associated mutations lock Ras in the “ON” position, which renders the protein constitutively activated. Given its role in oncogenesis, the biologic and chemical nature of Ras and the consequence of persistent Ras activation has been the subject of intensive biologic and pharmaceutical research. This article will provide a brief overview of the chemistry, structure, and biology that underlie Ras signaling and will summarize the key assays and reagents that have been developed and applied to study Ras signal transduction and biologic activity.
Ras proteins function as nodal points in signal transduction. Diverse extracellular stimuli act on plasma membrane-bound receptors (which include receptor tyrosine kinases and G protein-coupled receptors) and cause the activation of Ras (see Fig. 1). Accumulating evidence suggests that once activated, Ras may transit from the plasma membrane to endomembranes (4, 5). Alternatively, endomembrane-bound Ras may be activated by a discrete set of upstream activating proteins (4, 6, 7). Activated Ras then regulates myriad cytoplasmic signaling cascades to cause changes in normal cell physiology, which includes the regulation of cell morphology, growth, survival and differentiation, vesicular transport, and gene expression.
In addition to its role in promoting normal cellular growth and differentiation, Ras is perhaps best known as an oncogenic protein for its involvement in human carcinogenesis. Approximately 30% of all human cancers express a mutationally activated Ras protein (1, 8), with the highest incidence of somatic Ras mutation observed in pancreatic (90%), lung (50%), and colon (30%) cancers (9). An extensive body of research has implicated a strong causal role for mutated Ras in cancer development and growth. This relationship has prompted considerable interest and effort in the development of anti-Ras therapeutic strategies for cancer treatment (10). Recently, de novo germline mutation of Ras and Ras signaling proteins has also been associated with a group of human developmental disorders, the Costello, Noonan, and cardio-facio-cutaneous syndromes (11).

Figure 1. Ras GTPases function as regulated GDP/GTP molecular switches. Diverse extracellular signals, for example those received by membrane-bound receptors such as G-protein coupled receptors and receptor tyrosine kinases, can cause Ras GTPase activation at the plasma membrane and endomembranes. Receptor-mediated activation of Ras most commonly involves the activation of RasGEFs, which then cause transient activation of Ras. Activated Ras-GTP adopts a conformation that enhances its affinity for transient binding to and activation of downstream effectors (E).A The activated effectors then regulate distinct cytoplasmic signaling networks that control cellular proliferation, differentiation, and survival. Ras signaling is terminated by RasGAP-mediated stimulation of hydrolysis of bound GTP to GDP, which precludes further Ras-effector interaction. Tumor-associated Ras mutant proteins are insensitive to GAP stimulation.
Chemistry
The following sections describe the sequence and the structure of Ras proteins, and they examine how distinct sequence elements in Ras proteins dictate function, subcellular location, and interaction with regulatory and effector proteins.
Ras sequence and structure
The three human RAS genes encode four highly related 188-189 amino acid Ras proteins (H-Ras, N-Ras, and two KRAS-encoded splice variants, K-Ras4A and K-Ras4B) that share significant sequence identity (90%) and common structural elements (see Fig. 2). RAS genes are conserved in vertebrate and invertebrate evolution, with homologous genes found in C. elegans, Drosophila, and yeast but not in plants or bacteria. RAS genes are the founding members of a greater than 150-member superfamily of RAS-related genes that encode small GTP-binding and hydrolyzing proteins (GTPases) (12, 13).
Ras proteins are composed of two discrete sequence elements: a catalytic G domain and a hypervariable membrane-targeting domain (see Fig. 2). The amino-terminal 166 amino acids of Ras compose the G domain, which contains the consensus sequence motifs found in classic GTP-binding proteins. The carboxyl-terminal 23 to 24 amino acids compose the hypervariable region. The hypervariable region terminates in a consensus CAAX tetrapeptide motif that signals for posttranslational modifications that increase the hydrophobic nature of Ras. This region also contains additional sequences that promote membrane association and direct subcellular localization of Ras.
The three-dimensional structure of the G domain consists of five α-helices, six β-strands, and five loop regions (G1-G5) (see Fig. 2). The five loop regions are involved in protein-protein interaction and nucleotide binding (14). Within the G domain, two switch regions (switch I and II, residues 30-38 and 59-76, respectively) form exposed flexible loops that adopt different conformations depending on the identity of the bound guanine nucleotide. The effector loop or effector domain, which corresponds to G2, is embedded in switch I and forms critical contacts with effector molecules. Therefore, the stereochemical arrangement of the switch I and switch II regions translates Ras activation into a downstream response by diminishing effector binding to GDP-bound Ras and favoring effector binding to GTP-bound Ras. The other G loops contact the guanine base, phosphate groups, and/or Mg2+ ion (15). In addition to mediating effector binding, the two switch regions of Ras interact with proteins that regulate the GDP/GTP cycle: guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs).
Ras possesses a weak GDP/GTP exchange activity. Because the cellular levels of GTP are 20-fold greater than that of GDP, the dissociation of GDP from Ras favors the formation of Ras-GTP. Ras also possesses low intrinsic GTPase enzymatic activity, which stimulates hydrolysis of bound GTP to GDP to cycle Ras back to its inactive GDP-bound state. The intrinsic exchange and hydrolytic activity of Ras is not sufficient to promote rapid GDP/GTP cycling. Ras-specific GEFs and GAPs are critical to accelerate guanine nucleotide cycling and hence to promote a response to extracellular stimuli. RasGEFs (e.g., Sos, RasGRF, or RasGRP) promote the exchange of GDP for GTP and activate Ras signaling activity (16, 18). RasGAPs (e.g., p120 RasGAP, or NF1 neurofibromin) stimulate hydrolysis of GTP to GDP and terminate signaling activity (19). The activity of GEFs and GAPs enable Ras proteins to signal downstream to their effector molecules in a regulated manner.
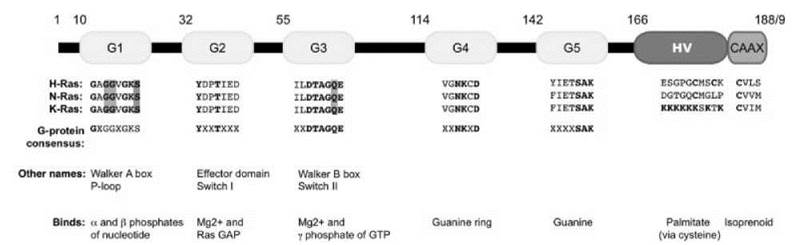
Figure 2. Schematic of Ras small GTP binding and hydrolyzing proteins. The five G boxes (G1 G5, yellow) comprise the cataalytic domain of Ras. The hypervariable region (red) at the carboxyl terminus contains the second signal that is critical for proper subcellular localization. The CAAX box (blue) is post-translationally modified by a 15-carbon isoprenyl lipid and is required for membrane association. The sequence of each motif for H-, N-, and K-Ras4B is given, as well as the consensus sequence found in all G domain-containing proteins. Mutation of residues highlighted in green (at G12, G13, or Q61) results in a constitutively activated protein. Conversely, mutation of the residue highlighted in red (S17) creates a dominant negative protein. Alternative names for some motifs, as well as their main function in Ras biochemistry, are listed as well.
Subcellular localization of Ras
In addition to nucleotide binding, the signaling activity of Ras is governed by subcellular localization. Ras proteins terminate in a tetrapeptide CAAX motif, where C is cysteine, A is any aliphatic amino acid, and X is serine or methionine. Ras proteins are synthesized initially as cytoplasmic proteins that have no signaling activity (see Fig. 3). Ras then undergoes a series of CAAX-directed posttranslational modifications. Farnesyltransferase (FTase) catalyzes a covalent addition of a 15-carbon farnesyl isoprenyl group to the cysteine of the CAAX motif, followed by Ras converting enzyme 1 (Rce1)-catalyzed proteolytic cleavage of the — AAX residues (20), and isoprenylcysteine-directed carboxyl methyltransferase (Icmt)-catalyzed carboxylmethylation of the now terminal prenylated cysteine residue. The prenyl residue inserts into cellular membranes, which tethers Ras to the cytosolic face of cellular membranes (21, 23). Point mutations engineered in the carboxyl terminus of Ras proteins (such as a cysteine to serine substitution in the CAAX box) as well as pharmacologic inhibitors (e.g., FTase inhibitors or FTIs) disrupt this posttranslational processing and cause mislocalization of Ras (24, 27).
The three CAAX-signaled modifications increase the overall hydrophobicity of the carboxyl terminus of Ras and are necessary to promote Ras membrane association. However, these modifications alone are not sufficient to direct full plasma membrane association and signaling activity. At least two additional motifs exist at the carboxyl termini of Ras proteins, which serve to facilitate plasma membrane association and direct Ras proteins to discrete membrane subdomains. These motifs function as secondary signals and are composed either of a stretch of basic amino acids (K-Ras4B) or of cysteine(s) that are palmitoylated (H-Ras, N-Ras, K-Ras4A) and positioned immediately upstream of the CAAX motif (28, 30) (see Fig. 3). The addition of a palmitate fatty acid is catalyzed by a protein acyltransferase, which forms a reversible thioester bond between the cysteine and the palmitoyl group (31, 32).

Figure 3. Ras membrane targeting is a multi-step process.A Nascent Ras protein is found in the cytoplasm of the cell.A The cysteine of the CAAX motif is postranslationally covalently modified by a farnesyl (red) lipid in a reaction catalyzed by farnesyltransferase (FTase). A A series of modification steps catalyzed by the indicated enzymes at the endoplasmic reticulum cleave the AAX residues and modify the now terminal prenylatted cysteine with a carboxymethyl group. A The farnesyl group inserts into cellular membranes, tethering the GTPase to the cytoplasmic face of the membranes. A secondary signal, either lysine residues in the case of K-Ras4B or cysteine bound to palmitate in the case of N-Ras and H-Ras is required for final membrane location. Mutating the cysteine of the CAAX motif to a serine (Ras-SAAX) precludes the addition of an isoprenyl group and all subsequent modifications, resulting in a cytosolic, inactive mutant protein.
Effector interaction
The biologic function of Ras is mediated by its ability to activate downstream cytoplasmic signaling networks through binding to a panel of effector proteins. The Ras-effector interaction is mediated through interactions between the core effector domain (amino acids 32-40) of Ras (see Fig. 2) and the specific residues within the Ras-binding domain (RBD) or Ras-association domain (RA) found in most Ras effector proteins. The RBDs and RA domains of effector proteins do not exhibit primary sequence homology, but instead they share a common tertiary structure that consists of an ubiquitin superfold, which forms critical contacts with Ras (33).
At least 10 distinct families of Ras effector proteins have been identified, with the Raf serine/threonine kinases, phosphatidylinositol 3-kinases (PI3K), and RalGEFs being the most extensively characterized (3) (see Fig. 4). Beyond their shared ability to bind preferentially to Ras-GTP, Ras effectors possess highly divergent biochemical and biologic functions. Raf activates the ERK mitogen-activated protein kinase cascade (Raf-MEK-ERK), whereas PI3K regulates phospholipid metabolism, and RalGEFs serve as activators of Ras-like (Ral) small GTPases. These and other pro-growth effectors (e.g., phospholipase C epsilon or Tiam1) promote Ras-mediated cellular proliferation and differentiation. In contrast, other Ras effector pathways (most notably Nore1 and other RASSF family members) can contribute to apoptosis (34).

Figure 4. Ras activates multiple effector pathways. GTP-bound active Ras binds to and stimulates a myriad of cytoplasmic signaling cascades. The three most extensively characterized effector pathways (RalGEF, Raf, and PI3K) are shown. Signal propagation occurs via phosphorylation of Raf, leading to subsequent phosphorylation and activation of the Elk-1 transcription factor and changes in gene expression. PI3K activation regulates phosphoinositide metabolism and formation of the phosphoinositol 3,4,5-triphosphate (PIP3) second messenger. PIP3 promotes activation of the AKT serine/threonine kinase and other signaling proteins. The phosphatase and tensin homolog (PTEN) tumor suppressor protein antagonizes the activity of PI3K by dephosphorylation of PIP3. RalGEF functions as an activator of the Ras-like (RalA and RalB) small GTPases.A Downstream effectors of Ral GTPases include components of the exocyst complex (e.g., Sec5) and RalBPI. Activation of Sec5 promotes activation of the atypical IkB kinase Tank binding kinase 1 (TBK1). RalBPI exhibits GAP activity towards the Rho family small GTPases Cdc42 and Rac1. Each Ras effector pathway regulates distinct cellular processes, which allows Ras to achieve a nuanced response to extracellular stimuli.
Chemical Tools and Techniques
The research on Ras proteins has resulted in the development of an impressive collection of reagents, assays, and techniques to dissect the Ras function. These techniques include mutants of Ras proteins, biochemical assays to monitor Ras function in vitro and in vivo, pharmacologic and genetic tools to dissect Ras signaling, as well as more recent approaches that apply RNA interference (RNAi) and other gene silencing techniques, and fluorescence-based protein assays to monitor the spatial and the temporal activities of Ras. Tables 1-3 provide a comprehensive list of the tools and reagents available to study Ras biology.
The creation and application of Ras mutants that display gain- or loss-of-function phenotypes has provided the foundation for our knowledge of the basic function and regulation of Ras proteins (see Table 1). By analogy to Ras, similar mutations have been introduced in many Ras superfamily proteins as well as other unrelated GTPases. These mutant proteins, particularly mutants of the Rho family small GTPases, have proven to be powerful reagents for functional studies.
Constitutively active Ras proteins can be generated through substitutions of the conserved glycine at position 12 or glutamine at position 61, for example, with valine (G12V) or leucine (Q61L), respectively (see Fig. 2). Essentially, any amino acid substitution at these two positions impairs the intrinsic and GAP-stimulated GTP hydrolysis rate, which results in the formation of a constitutively GTP-bound and chronically active protein. These activated mutants can be used to determine the downstream signaling activities of Ras proteins.
Conversely, Ras with a substitution of serine at position 17 with asparagine (S17N) binds to and forms nonproductive complexes with Ras GEFs (see Fig. 2). Because each RasGEF can activate several Ras proteins, Ras(S17N) acts as a dominant negative inhibitory protein that inhibits activation of all Ras proteins. This dominant negative reagent can be used to determine whether the biologic activity of a particular extracellular stimulus is mediated through RasGEF-dependent activation of Ras.
Substitution of the cysteine residue of the CAAX motif with a serine prevents all CAAX-signaled, posttranslational modifications, which results in an unprocessed, cytosolic, and inactive protein (see Fig. 3). This mutant is useful to determine whether membrane association is required for a particular cellular function of Ras.
Single amino acid substitutions in the core effector domain of Ras can cause differential loss of binding to a subset of Ras effectors (see Table 1). For example, constitutively active Ras with a point mutation at position T35 (Ras G12V/T35S) binds to and activates Raf but not PI3K or RalGEF. Similarly, Ras(G12V/E37G) activates RalGEF but not Raf or PI3K; Ras(12V/Y40C) can activate PI3K but not RalGEF or Raf. This technique has been applied to tease out the contribution of Ras effector pathways to turmorigenesis in mouse and human cells (35-37). One caveat with this approach is that these point mutants are not necessarily precise because they retain interaction with other Ras effectors.
Table 1. Genetic reagents for activation of Ras and Ras effector pathways
|
Reagent |
Description |
|
Ras |
|
|
Ras(G12V), Ras(Q61L) |
GTPase-deficient, GAP-insensitive, constitutively activated mutants. Based on mutations detected in RAS alleles found in human cancers. |
|
RasGRPl |
RasGEF. Activated by treatment with phorbol esters. |
|
Raf-MEK-ERK Pathway |
|
|
H-Ras(G12V/T35S) |
Effector domain mutant of activated H-Ras that binds to and activates Raf but not PI3K or RalGEF. |
|
Raf-22W |
Truncation of 305 amino-terminal amino acids in human c-Raf-1. Lacks the negative regulatory sequences that lie upstream of kinase domain. |
|
Raf-CAAX |
Human c-Raf-1 chimeric protein with carboxyl-terminal 18 amino acid membrane-targeting sequence of K-Ras(4B). Persistent membrane location and signaling activity. |
|
B-Raf(V600E) |
Human B-Raf with the V600E (formerly V599E) missense mutation observed in most mutated BRAF alleles found in human cancers. |
|
MEK(S218D/S222D) |
Missense substitutions at Raf serine phosphorylation sites with charged amino acids to mimic persistent phosphorylation. |
|
∆MEK-ED |
N-terminally truncated MEK, with missense substitutions at Raf serine phosphorylation sites (S218/S222D) with charged amino acids to mimic persistent phosphorylation. |
|
PI3K-AKT Pathway |
|
|
H-Ras(G12V/Y40C) |
Effector domain mutant of activated H-Ras that binds to and preferentially activates PI3K but not Raf or RalGEF. |
|
p110-CAAX |
p110γ chimeric protein terminating with the C-terminal 18 amino acids plasma membrane targeting sequence of K-Ras(4B). Persistent membrane location and signaling activity. |
|
p110α(E545K), |
Constitutively activated forms of the p110α catalytic subunit. |
|
p110α(H1047R) |
Corresponding gene missense mutations observed in most mutant alleles of the gene encoding p110α (PIK3CA) detected in human cancers. |
|
Interfering RNA for PTEN |
Suppresses PTEN expression, prevents PTEN-mediated conversion the PI3K product phosphoinositol 3,4,5-triphosphate (PIP3) to the PI3K substrate phosphoinositol 4,5-diphosphate (PIP2). |
|
Myr-AKT |
Fusion protein of AKT with an amino-terminal myristoylation signal sequence. This fatty acid modification promotes persistent plasma membrane association and signaling activity. |
|
RalGEF-Ral Pathway |
|
|
H-Ras(G12V/E37G) |
Effector domain mutant of activated H-Ras that binds to and activates RalGEFs but not PI3K or Raf. |
|
Rgl-CAAX |
Mouse Rlf/Rgl2 chimeric protein terminating with the C-terminal 18 amino acids of K-Ras(4B); Rlf lacks the C-terminal 247 residues that contain the Ras association domain. Persistent membrane location and signaling activity. |
|
Ral(G23V), Ral(Q71L) |
Human GTPase-deficient mutants of RalA and RalB. Analogous to the Ras(G12V) and Ras(Q61L) mutants. |
Analyses of Ras GDP/GTP cycling
Various protein-protein interaction methods have been developed to study the GTP binding, exchange, and hydrolytic activities of Ras. The in vitro rate of nucleotide exchange or GTP hydrolysis can be measured with real-time fluorescence-based assays. The fluorescence of an N-methylanthraniloyl derivative of GTP (mant-GTP) increases ~20% when binding to small GTPases, and therefore can be used to monitor the rate of nucleotide exchange and incorporation of mant-GTP into Ras (38). The intrinsic and GAP-stimulated GTPase activities of Ras proteins can be monitored through another fluorescence-based assay, which approximates the hydrolysis rate by measuring the release of P; from nucleotide-bound Ras (39). The assay uses a fluorophore (MDCC)-modified bacterial phosphate binding protein (PBP) that has a high affinity for Pi. The fluorescence of MDCC increases sevenfold when bound to Pi. PBP-MDCC is incubated with recombinant GTP-bound Ras, and fluorescent measurements are made with a standard fluorimeter.
Physiologic levels of total cellular GTP-bound Ras can be detected with pull-down assays (40) (see Table 2). With these assays, cells are lysed with detergent-containing buffers and then incubated with a recombinant fusion protein that contains the isolated RBD of c-Raf-1 fused to glutathione-S-transferase (GST; designated GST-Raf-RBD). The presence of Ras in the
GST-Raf-RBD protein complexes is resolved by Western blotting. High-quality, isoform-specific anti-Ras antibodies, which allow determination of activation of a specific Ras isoform, are widely available (41). Only GTP-bound Ras can bind to GST-Raf-RBD, so this affinity reagent provides a quick and easy way to determine the relative levels of cellular GTP-Ras when compared with the total Ras protein. For example, resting cells contain only ~5% Ras-GTP compared with total Ras, whereas growth factor-stimulated cells may show 50% to 70% Ras-GTP.
Several techniques exist to monitor the spatial and temporal patterns of Ras activation. One such probe is composed of the Raf-RBD fused with green fluorescent protein (GFP). When expressed in quiescent cells where Ras is inactive, the GFP-Raf-RBD probe is distributed homogeneously throughout the cell. When cells are stimulated with a growth factor that causes Ras activation, the probe is redistributed rapidly and transiently to the membrane compartments where Ras is activated (42). Matsuda and colleagues developed a chimeric H-Ras-Raf-RBD fluorescent probe called Ras and interacting protein chimeric unit (Raichu). A productive intermolecular interaction between the H-Ras and the Raf-RBD domains of Raichu occurs when it is stimulated with a growth factor or another agent and can be measured by fluorescent resonance energy transfer (43, 44). Bivona et al. have also developed similar approaches to monitor the spatiotemporal parameters of Ras activation in endomembrane compartments (45).
Table 2. Reagents for biochemical detection of Ras and Ras effector pathway activation
|
Reagent |
Description |
|
Ras GST-Ras-RBD |
Glutathione-S-transferase (GST) fusion protein with the isolated Ras binding domain (RBD) of c-Raf-1. Binds preferentially to activated, GTP-bound Ras proteins. |
|
Raf-MEK-ERK Pathway Phospho-specific MEK1/MEK2 antibody Phospho-specific ERK1/ERK2 antibody |
Antibody for Western blot detection of phosphorylated and activated MEK1/2 when phosphorylated by Raf at S221.
Antibody for Western blot detection of phosphorylated and activated ERK1/2 when phosphorylated by MEK at T202 and Y204. Phosphorylation of these residues activates ERK catalytic function. |
|
PI3K-AKT Pathway Phospho-specific AKT antibody |
Antibody for Western blot detection of phosphorylated and activated AKT when phosphorylated by PDK1 at T308 |
|
RalGEF-Ral Pathway GST-Rlf-RBD |
GST fusion protein with the isolated Ral binding domain (RBD) of the RalBP1 (amino acids 397-518) effector. Binds preferentially to activated, GTP-bound RalA, and RalB proteins. |
Analyses of Ras subcellular localization and posttranslational processing
Proper localization of Ras is essential for function, and several pharmacologic and genetic tools are available to study the posttranslational processing and subcellular localization of Ras. Note that epitope or fluorescent tags must be added at the amino terminus of Ras so as not to disrupt posttranslational processing of the carboxyl terminus. The localization of GFP-Ras can be monitored in live cells through confocal or widefield microscopy. Alternatively, anti-Ras antibodies can be applied to detect the location of endogenous protein in fixed cells. Similarly, antibodies that recognize epitope tags can be used to evaluate the subcellular localization of ectopically expressed, amino-terminal epitope-tagged Ras proteins. These antibodies include the commercially available antibodies that recognize peptide sequences found in the influenza hemagglutinin antigen (YPYDVPDYA), the human c-Myc transcription factor (LDEESILKQE), and the FLAG epitope (DYKDDDDK).
The treatment of cells that express GFP- or epitope-tagged Ras with various pharmacologic inhibitors is also useful to determine which posttranslational modifications are required for Ras membrane association. For example, treatment of cells that express GFP-H-Ras with FTIs causes GFP-H-Ras to mislocalize to the cytoplasm, which indicates that farnesylation is required for membrane association. Compounds that block protein palmitoylation, such as 2-bromopalmitate, can also be used to determine whether palmitoylation is required for Ras-related proteins to associate with the plasma membrane (46-48).
Various assays are available for direct analyses of the posttranslational modification of Ras by lipids. Recombinant FTase together with recombinant Ras can be used to evaluate the far- nesylation of Ras. Rabbit reticulocyte lysate can be used for in vitro transcription/translation analyses of all CAAX-signaled modifications. Direct measure of palmitoylation can be assessed by evaluating incorporation of a tritiated palmitate analog (49) or through a novel method that detects free thiol groups in proteins (50, 51). This assay involves blocking all available free cysteines in a protein and then chemically cleaving thioester bonds that link palmitate to cysteine. Cleavage generates a free thiol group, presumably only at the sites of palmitate linkage. The thiol group can then be detected using a biotinylated reagent.
Pharmacologic inhibitors of the enzymes involved in — AAX proteolysis and carboxylmethylation steps are available. However, as with all pharmacologic inhibitors, these compounds are likely to have off-target activities (52). Instead, genetic approaches have provided more specific means to evaluate the role of these modifications in promoting Ras membrane association. In particular, mouse embryo fibroblast cell lines derived from mice that are deficient in Rcel (RCE1 —/—) or Icmt (ICMT—/—) are useful to determine the role of these two enzymatic modifications for Ras membrane association, signaling, and biologic activity. As mentioned, after protein prenylation, the —AAX residues of Ras are cleaved by Rce1 (20). This step is critical for proper localization; exogenous H-, N-, and K-Ras are mislocalized in RCE1 —/— mouse embryo fibroblasts (53). After proteolytic cleavage of the —AAX residues, the prenylated cysteine residue is carboxymethylated by Icmt. As in RCE—/— cells, all three Ras proteins are mislocalized when expressed in ICMTcells. Interestingly, a recent study suggests that the membrane association of CAAX-containing Ras family GT- Pases that are modified by geranylgeranylation do not require these modifications (53).
Ras trafficking to cellular membranes can be measured by fluorescence recovery after photobleaching (FRAP) and fluorescence loss in photobleaching (FLIP) (54). Both techniques rely on the expression of fluorescent-labeled Ras proteins to monitor different parameters of Ras movement across and between cellular membranes. FRAP involves photobleaching a membrane subdomain and measuring the kinetics of fluorescence recovery—and hence Ras trafficking—into the bleached area. With FLIP, a cellular membrane is photobleached repeatedly and the subsequent intercellular movement of the photobleached area is monitored.
Analyses of Ras effector activation and function
In addition to the use of Ras effector domain mutants, structural mutants of Ras effectors and pharmacologic inhibitors of effector signaling are valuable to study the role of specific effector signaling pathways in Ras function. Tumor-derived, constitutively activated mutants, as well as lab-designed and generated mutants, have been particularly useful reagents for these studies (see Table 1). For example, the addition of the carboxyl-terminal CAAX-containing sequences that target K-Ras4B to the plasma membrane onto c-Raf-1, the RalGEF Rlf, and the p110a catalytic subunit of PI3K has created effector fusion proteins that are constitutively membrane-bound and active. Membrane localization activates effector function because Ras activates effectors, in part, by promoting their association with the plasma membrane. More recently, human disease-derived mutants have provided physiologically relevant activated versions of these effectors, with the identification of missense mutational activation of B-Raf and p110a in human tumors, and activated MEK1 and MEK2 in cardio-facio-cutaneous syndrome (11). Novel activating mutations in Ras and B-Raf, as well as the Sos RasGEF, have also been identified in developmental syndromes. These genetic variants may also be useful reagents to study Ras signaling and function (55, 56). Activated effectors can be used to determine whether the activation of a specific effector pathway alone is sufficient to mediate a specific Ras function.
Various approaches have been useful to block the function of a specific effector and to determine its necessity and role in Ras function. First, catalytically dead mutants of Ras effectors or their substrates function as dominant negative mutants that, when ectopically-expressed, block the function of the endogenous effector (see Tables 2 and 3). These mutants include kinase-dead mutants of c-Raf-1, MEK, and ERK that block the Raf effector pathway. Second, selective pharmacologic inhibitors of the MEK (PD98059 and U0126) and PI3K (LY298002) kinase pathways have been useful to study Raf and PI3K effector signaling. Recently, RNAi has been applied to impair specific effector function selectively (57, 58). However, this approach may be limited by the fact that a majority of effectors also have highly related isoforms that are expressed broadly and may have overlapping functions. Finally, the use of mice that are deficient in effector function, as well as cells derived from these animals, has been very effective to determine the role of specific effectors in Ras-mediated oncogenesis. For example, although genetic loss of Tiam1, PLC epsilon, or Ral-GEF does not impair mouse development, each knockout mouse sustained impaired sensitivity to Ras-induced skin tumor formation when initiated by treatment with chemical carcinogens that cause mutational activation of HRAS (59, 61). Genetic deficiency of some Ras effectors, such as Raf or the p110a catalytic subunit of PI3K, cause embryonic lethality and have prevented similar approaches to study their roles in Ras-mediated oncogenesis. However, a recent study by Gupta et al., which used mice that harbored a Ras binding-deficient mutant of p110a, demonstrated elegantly that this specific PI3K isoform was necessary for Ras-mediated lung and skin oncogenesis (62). Hence, the development of similar mouse models will be useful to evaluate the role of Ras effectors that are normally essential for development, because of their roles in oncogenesis.
Table 3. Pharmacologic and genetic inhibitors of Ras and Ras effector pathways
|
Reagent |
Description |
|
Raf-MEK-ERK Pathway Sorafenib (BAY 43-9006) MCP110 U0126, PD98059, CI-1040 MEK(K97A) MKP-1 |
Cell-permeable ATP-competitive inhibitor of Raf kinase activity. Also a potent inhibitor of a variety of other protein kinases. Cell-permeable inhibitor of Ras interaction with c-Raf-1 and activation of ERK. Cell-permeable, non-ATP competitive inhibitor of MEK-mediated activation of ERK. Mutant of MEK1 with missense mutation of the ATP binding domain, resulting in a kinase-deficient mutant. ERK dual-specificity protein phosphatase; removes phosphorylation of ERK1 and ERK2 at T202 and Y204 residues |
|
PI3K-AKT Pathway Wortmannin, LY294002 PTEN
dominant negative PI3K |
Cell-permeable inhibitors of PI3K family lipid kinases. Lipid phosphatase and antagonist of PI3K. Converts the PI3K product phosphoinositol 3,4,5-triphosphate (PIP3) to the PI3K substrate phosphoinositol 4,5-diphosphate (PIP2). Truncation of p85 regulatory subunit; does not bind to p110. |
|
RalGEF-Ral Pathway Ral(G26A), Ral(S28 N) Ral inhibitory RNA |
Dominant negative inhibitor of RalGEF-mediated activation of Ral. Analogous to dominant negative Ras(G15A) and Ras(S17N) mutants. Oligonucleotide- or expression vector, short hairpin-based interfering RNA. Selective suppression of RalA or RalB expression. |
The decades of research on Ras proteins have yielded impressive insight into the biologic and chemical properties of Ras, and they have established many important paradigms that have defined key concepts in signal transduction. Concurrent with these studies has been the development and application of an armamentarium of reagents, genetic and pharmacologic tools, and assays with which to study Ras. These technologic developments have also aided studies greatly to evaluate the biochemical, structural, and biologic properties of other Ras superfamily proteins. These studies reveal that despite a shared simple fundamental biochemical function as GDP/GTP-regulated binary switches, Ras family proteins display an incredible diversity in the mechanisms by which they are regulated and in how their subcellular localization is controlled. These tools have allowed additional delineation of Ras function and an appreciation of their involvement in virtually all facets of cell physiology and behavior. Our earlier depiction of Ras as a relay switch at the plasma membrane in a simple linear signaling pathway has been revised to reflect the current model: Ras is a central component of a highly complex and dynamic signaling network with discrete functions at several subcellular locations. In summary, whereas great strides have been made to understand Ras activation, localization, and signaling, much need still exists for focused research in each of these areas. New methodologies and technologies that are developed will undoubtedly facilitate additional advances. Because we have been surprised by many recent new discoveries regarding Ras, one must expect the unexpected in Ras signaling and biology.
References
1. Malumbres M, Barbacid M. Ras oncogenes: the first 30 years. Nat. Rev. Cancer 2003; 3:459-465.
2. Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science 2001; 294:1299-1304.
3. Repasky GA, Chenette EJ, Der CJ. Renewing the conspiracy theory debate: does RAF function alone to mediate RAS oncogenesis? Trends Cell. Biol. 2004; 14:639-647.
4. Eungdamrong NJ, Iyengar R. Compartment-specific feedback loop and regulated trafficking can result in sustained activation of ras at the Golgi. Biophys. J. 2007; 92:808-815.
5. Goodwin JS, Drake KR, Rogers C, Wright L, Lippincott-Schwartz J, Philips MR, Kenworthy AK. Depalmitoylated ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell Biol. 2005; 170:261-272.
6. Arozarena I, Matallanas D, Berciano MT, Sanz-Moreno V, Calvo F, Munoz MT, Egea G, Lafarga M, Crespo P. Activation of h-ras in the endoplasmic reticulum by the RASGRF family guanine nucleotide exchange factors. Mol. Cell Biol. 2004; 24:1516-1530.
7. Caloca MJ, Zugaza JL, Bustelo XR. Exchange factors of the rasgrp family mediate ras activation in the Golgi. J. Biol. Chem. 2003; 278:33465-33473.
8. Bos JL. Ras oncogenes in human cancer: a review. Cancer Res 1989; 49:4682-4689.
9. Li D, Firozi PF, Zhang W, Shen J, DiGiovanni J, Lau S, Evans D, Friess H, Hassan M, Abbruzzese JL. DNA adducts, genetic polymorphisms, and K-RAS mutation in human pancreatic cancer. Mutat. Res. 2002; 513:37-48.
10. Roberts PJ, Der CJ. Targeting the raf-mek-erk mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007; 26:3291-3310.
11. Gelb BD, Tartaglia M. Noonan syndrome and related disorders: dysregulated ras-mitogen activated protein kinase signal transduction. Hum. Mol. Genet. 2006; 15(Spec No 2):R220-226.
12. Colicelli J. Human ras superfamily proteins and related GTPases. Sci. STKE. 2004; (250):RE13.
13. Wennerberg K, Rossman KL, Der CJ. The ras superfamily at a glance. J. Cell Sci. 2005; 118(Pt 5):843-846.
14. Paduch M, Jelen F, Otlewski J. Structure of small G proteins and their regulators. Acta Biochim. Pol. 2001; 48:829-850.
15. Sprang SR. G protein mechanisms: insights from structural analysis. Annu. Rev. Biochem. 1997; 66:639-678.
16. Quilliam LA, Rebhun JF, Castro AF. A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family gtpases. Prog. Nucleic Acid Res. Mol. Biol. 2002; 71:391-444.
17. Rossman KL, Der CJ, Sondek J. Gef means go: turning on rho GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005; 6:167-180.
18. Schrader JW, Schallhorn A, Grill B, Guo X. Activation of small GTPases of the RAS and rho family by growth factors active on mast cells. Mol. Immunol. 2002; 38:1181-1186.
19. Bernards A, Settleman J. Gap control: regulating the regulators of small GTPases. Trends Cell Biol. 2004; 14:377-385.
20. Otto JC, Kim E, Young SG, Casey PJ. Cloning and characterization of a mammalian prenyl protein-specific protease. J. Biol. Chem. 1999; 274:8379-8382.
21. Casey PJ, Seabra MC. Protein prenyltransferases. J. Biol. Chem. 1996; 271:5289-5292.
22. Fu HW, Casey PJ. Enzymology and biology of caax protein prenylation. Recent Prog Horm Res. 1999; 54:315-342; discussion 342-313.
23. Kato K, Cox AD, Hisaka MM, Graham SM, Buss JE, Der CJ. Isoprenoid addition to RAS protein is the critical modification for its membrane association and transforming activity. Proc. Natl. Acad. Sci. USA. 1992; 89:6403-6407.
24. Bergo MO, Ambroziak P, Gregory C, George A, Otto JC, Kim E, Nagase H, Casey PJ, Balmain A, Young SG. Absence of the caax endoprotease rce1: effects on cell growth and transformation. Mol. Cell Biol. 2002; 22:171-181.
25. Bergo MO, Gavino BJ, Hong C, Beigneux AP, McMahon M, Casey PJ, Young SG. Inactivation of icmt inhibits transformation by oncogenic K-RAS and B-RAF. J. Clin. Invest. 2004; 113:539-550.
26. Kim E, Ambroziak P, Otto JC, Taylor B, Ashby M, Shannon K, Casey PJ, Young SG. Disruption of the mouse rce1 gene results in defective ras processing and mislocalization of ras within cells. J. Biol. Chem. 1999; 274:8383-8390.
27. Lerner EC, Qian Y, Blaskovich MA, Fossum RD, Vogt A, Sun J, Cox AD, Der CJ, Hamilton AD, Sebti SM. Ras caax peptidomimetic fti-277 selectively blocks oncogenic ras signaling by inducing cytoplasmic accumulation of inactive RAS-RAF complexes. J. Biol. Chem. 1995; 270:26802-26806.
28. Hancock JF, Cadwallader K, Paterson H, Marshall CJ. A caax or a caal motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 1991; 10:4033-4039.
29. Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the caax motif to localize p21ras to the plasma membrane. Cell 1990; 63:133-139.
30. Jackson JH, Li JW, Buss JE, Der CJ, Cochrane CG. Polylysine domain of k-RAS 4b protein is crucial for malignant transformation. Proc. Natl. Acad. Sci. USA. 1994; 91:12730-12734.
31. Mitchell DA, Vasudevan A, Linder ME, Deschenes RJ. Protein pamitoylation by a family of dhhc protein s-acyltransferases. J. Lipid Res. 2006; 47:1118-1127.
32. Swarthout JT, Lobo S, Farh L, Croke MR, Greentree WK, Deschenes RJ, Linder ME. Dhhc9 and gcp16 constitute a human protein fatty acyltransferase with specificity for h- and n-RAS. J. Biol. Chem. 2005; 280:31141-31148.
33. Herrmann C. Ras-effector interactions: after one decade. Curr. Opin. Struct. Biol. 2003; 13:122-129.
34. Cox AD, Der CJ. The dark side of ras: regulation of apoptosis. Oncogene. 2003; 22:8999-9006.
35. Khosravi-Far R, White MA, Westwick JK, Solski PA, Chrzanowska-Wodnicka M, Van Aelst L, Wigler MH, Der CJ. Oncogenic ras activation of raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol. Cell Biol. 1996; 16:3923-3933.
36. Ward Y, Wang W, Woodhouse E, Linnoila I, Liotta L, Kelly K. Signal pathways which promote invasion and metastasis: critical and distinct contributions of extracellular signal-regulated kinase and ral-specific guanine exchange factor pathways. Mol. Cell Biol. 2001; 21:5958-5969.
37. White MA, Nicolette C, Minden A, Polverino A, Van Aelst L, Karin M, Wigler MH. Multiple RAS functions can contribute to mammalian cell transformation. Cell 1995; 80:533-541.
38. Leonard DA, Evans T, Hart M, Cerione RA, Manor D. Investigation of the GTP-binding/GTPase cycle of cdc42hs using fluorescence spectroscopy. Biochemistry. 1994; 33:12323-12328.
39. Shutes A, Der CJ. Real-time in vitro measurement of GTP hydrolysis. Methods 2005; 37:183-189.
40. Taylor SJ, Resnick RJ, Shalloway D. Nonradioactive determination of RAS-GTP levels using activated ras interaction assay. Methods Enzymol. 2001; 333:333-342.
41. Major SM, Nishizuka S, Morita D, Rowland R, Sunshine M, Shankavaram U, Washburn F, Asin D, Kouros-Mehr H, Kane D, Weinstein JN. Abminer: a bioinformatic resource on available monoclonal antibodies and corresponding gene identifiers for genomic, proteomic, and immunologic studies. BMC Bioinformat. 2006; 7:192.
42. Bivona TG, Philips MR. Analysis of ras and rap activation in living cells using fluorescent ras binding domains. Methods 2005; 37:138-145.
43. Mochizuki N, Yamashita S, Kurokawa K, Ohba Y, Nagai T, Miyawaki A, Matsuda M. Spatio-temporal images of growth-factor-induced activation of ras and rap1. Nature 2001; 411:1065-1068.
44. Walker SA, Lockyer PJ. Visualizing ras signalling in real-time. J. Cell Sci. 2004; 117:2879-2886.
45. Bivona TG, Perez De Castro I, Ahearn IM, Grana TM, Chiu VK, Lockyer PJ, Cullen PJ, Pellicer A, Cox AD, Philips MR. Phospholipase cgamma activates ras on the Golgi apparatus by means of RASGRP1. Nature 2003; 424:694-698.
46. Michaelson D, Silletti J, Murphy G, D’Eustachio P, Rush M, Philips MR. Differential localization of rho gtpases in live cells: regulation by hypervariable regions and rhogdi binding. J. Cell Biol. 2001; 152:111-126.
47. Varner AS, Ducker CE, Xia Z, Zhuang Y, De Vos ML, Smith CD. Characterization of human palmitoyl-acyl transferase activity using peptides that mimic distinct palmitoylation motifs. Biochem. J. 2003; 373:91-99.
48. Webb Y, Hermida-Matsumoto L, Resh MD. Inhibition of protein palmitoylation, raft localization, and t cell signaling by 2-bromopalmitate and polyunsaturated fatty acids. J. Biol. Chem. 2000; 275:261-270.
49. Jones TL. Role of palmitoylation in rgs protein function. Methods Enzymol. 2004; 389:33-55.
50. Chenette EJ, Abo A, Der CJ. Critical and distinct roles of amino- and carboxyl-terminal sequences in regulation of the biological activity of the chp atypical rho GTPase. J. Biol. Chem. 2005; 280:13784-13792.
51. Drisdel RC, Green WN. Labeling and quantifying sites of protein palmitoylation. Biotechniques 2004; 36:276-285.
52. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000; 351:95-105.
53. Michaelson D, Ali W, Chiu VK, Bergo M, Silletti J, Wright L, Young SG, Philips M. Postprenylation caax processing is required for proper localization of RAS but not rho GTPases. Mol. Biol. Cell. 2005; 16:1606-1616.
54. Goodwin JS, Kenworthy AK. Photobleaching approaches to investigate diffusional mobility and trafficking of ras in living cells. Methods. 2005; 37:154-164.
55. Schubbert S, Shannon K, Bollag G. Hyperactive ras in developmental disorders and cancer. Nat. Rev. Cancer. 2007; 7:295-308.
56. Shannon K, Bollag G. Sending out an SOS. Nat. Genet. 2007; 39:8-9.
57. Chien Y, White MA. Ral gtpases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep. 2003; 4:800-806.
58. Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, Der CJ, Counter CM. Activation of rala is critical for RAS-induced tumorigenesis of human cells. Cancer Cell. 2005; 7:533-545.
59. Bai Y, Edamatsu H, Maeda S, Saito H, Suzuki N, Satoh T, Kataoka T. Crucial role of phospholipase cepsilon in chemical carcinogen-induced skin tumor development. Cancer Res. 2004; 64:8808-8810.
60. Gonzalez-Garcia A, Pritchard CA, Paterson HF, Mavria G, Stamp G, Marshall CJ. Ralgds is required for tumor formation in a model of skin carcinogenesis. Cancer Cell. 2005; 7:219-226.
61. Malliri A, van der Kammen RA, Clark K, van der Valk M, Michiels F, Collard JG. Mice deficient in the rac activator tiam1 are resistant to RAS-induced skin tumours. Nature 2002; 417:867-871.
62. Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J. Binding of RAS to phosphoinositide 3-kinase p110alpha is required for RAS-driven tumorigenesis in mice. Cell 2007; 129:957-968.
Further Reading
Chenette EJ, Repasky GA, Der CJ. Effectors of Ras-mediated oncogenesis. In: Small GTPases: Ras Family. Der CJ, ed. 2006. Dordrecht: Kluwer Academic Publishers.
Roberts PJ, Der CJ Ras stories: the state-of-the art. In: Small GTPases: Ras Family. Der CJ, ed. 2006. Dordrecht: Kluwer Academic Publishers.
See Also
Fluorescence Techniques: Proteins
Gene Silencing Techniques
Mitogen-Activated Protein Kinases (MAPKs): ERK
Protein-Protein Interactions, Tools to Study
Ras-Like Proteins