CHEMICAL BIOLOGY
The Biology and Biochemistry of Steroid Hormones
S. Stoney Simons, Jr., Steroid Hormones Section, NIDDK/CEB, NIH, Bethesda, Maryland
doi: 10.1002/9780470048672.wecb563
Five categories of steroid hormones exist in humans, including androgens, estrogens, glucocorticoids, mineralocorticoids, and progestins. These hormones affect virtually every tissue and organ in the human body and play major roles in the development, differentiation, and homeostasis of normal individuals. Antisteroids usually possess nonsteroidal structures but still block the actions of the steroid hormones and are important tools in endocrine therapies of pathologic conditions. Therefore, how the body regulates where, when, and how much a response to steroids occurs is of major importance. Here we survey what is known about the genomic responses to steroid hormones, each of which is mediated by a unique intracellular receptor protein that interacts with the cellular DNA to modify the rates of gene transcription. These receptors are members of a much larger superfamily of steroid/nuclear receptors, most of which bind either nonsteroidal ligands or no known ligand. Nongenomic (i.e., pathways without initial involvement of genomic DNA) and secondary responses (i.e., changes that require protein synthesis to alter gene transcription) are additional important effects of steroid hormones but are not discussed here. The emphasis is on the biochemistry of the five classes of steroid hormones, the techniques used to study steroid hormone action, and the basic mechanistic steps by which steroids alter gene expression.
Steroid hormones can increase and decrease the level and/or activity of a large number of proteins in eukaryotes. Steroid hormones were first discovered in humans, where they play essential roles in development, differentiation, homeostasis, and endocrine therapies. However, current interest in steroid hormones is increasing because they constitute excellent model systems for examining the control of gene expression. Many human pathologies result from the inappropriate expression of protein(s). Thus, to treat disease states, it is critical to understand the normal processes governing how, when, and how much of the information encoded in the DNA of cells is transcribed to mRNAs and eventually into proteins, which perform most of the functions of cells. Steroid hormones provide excellent model systems with which to address these clinically relevant questions.
Biochemistry of Steroid Hormones
Five classes of steroid hormones are produced in humans: androgens, estrogens, glucocorticoids, mineralocorticoids, and progestins. The structures of several steroid hormones were elucidated in the 1930s. The common feature among each compound is the fused four-ring structure (Fig. 1a) or steroid ring structure that is the defining characteristic of all steroids. The first synthetic steroid, the estrogen equilenin, was prepared in 1939 (1). The numerous synthetic schemes for preparing steroid hormones will not be addressed here. The reader is directed to the article on the Chemistry of Steroid Hormones elsewhere in this reference for this material.
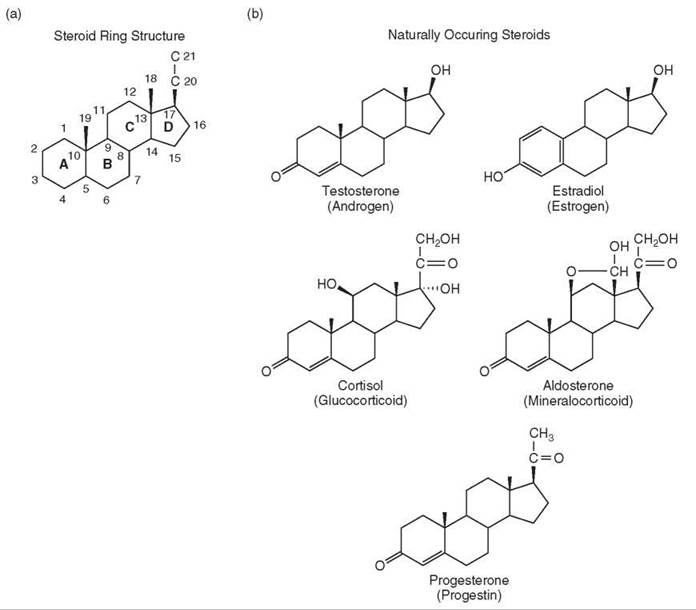


Figure 1. Steroid structures. (a) Basic ring structure and position numbering of steroids with the four rings (a-d). (b) Naturally occurring steroids in humans. (c) Common synthetic agonist steroids. (d) Common synthetic antisteroids.
Steroid hormones bind to receptor proteins
The discovery that steroid hormones bind to unique proteins with high affinity and selectivity commenced with the demonstration by Jensen and Jacobson that [3H]estradiol bound tightly to species found only in the target tissues of estrogens (2). These binders were discovered to be intracellular proteins. In general, the protein that bound a given steroid with the highest affinity was called the cognate receptor for that steroid—i.e., androgen receptor (AR), estrogen receptor (ER), glucocorticoid receptor (GR), mineralocorticoid receptor (MR), and progesterone receptor (PR). More recently, these steroid receptors have been found to be members of a larger superfamily of about 150 members in over 30 subfamilies across a range of species (3). Humans contain 48 receptors. Each receptor protein shares a highly homologous domain of about 66 amino acids that contains two “zinc finger” motifs, each of which includes four cysteine residues (4). The homology between the rest of the receptor proteins is much less. Many members of this superfamily do not bind any known ligand and are thus called orphan receptors. Other members bind ligands that are not steroids, such as triiodothyronine (T3), retinoic acid, and vitamin D, which is not a steroid although its precursor is a steroid. The receptors for these later ligands are found almost exclusively in the nucleus of cells, in either the presence or the absence of ligand, and are therefore collectively called nuclear receptors. These receptors are discussed in the article entitled the Chemistry of Nuclear Receptors. This article will cover only the receptors of the five steroid hormones of Fig. 1b.
Structure of steroid hormones
Agonist steroids
The structures of the major steroid hormones in humans are shown in Fig. 1b. Each of these steroids elicits the maximal response from their cognate receptor and are called agonist steroids. The years after the structural elucidation of these steroids witnessed an explosion of synthetic activity aimed at making “better” steroids. These synthetic steroids (e.g., Fig. 1c) have largely supplanted the naturally occurring steroids both in clinical applications, which will not be covered here, and in research. The structures of the synthetic steroid hormones can be different from those of the natural steroids (e.g., DES and DAC in Fig. 1c) (5). These synthetic ligands are all active at lower concentrations than the natural steroids of Fig. 1b, and thus, they exhibit a greater sensitivity, or potency but have equal efficacy. More precisely, full agonists for a given receptor induce (or repress) gene expression to the same level. However, the concentration of these synthetic agonists required for half of the maximal response (=EC50) is lower than that of the natural steroids. The synthetic ligands are more stable to metabolic degradation, and thus, they are longer acting, and generally retain more activity when taken orally. They also possess greater specificity than the natural steroids so that there is less interaction with noncognate receptors (5).
Antisteroids
Not all compounds that bind to a given receptor give full agonist activity, and some give no activity. These agents also antagonize the actions of the above agonist steroids and are therefore called antagonists or antisteroids. The amount of residual agonist activity of an antisteroid, or the partial agonist activity, is expressed as percent of maximal activity of a full agonist steroid. Thus, a molecule with 15% partial agonist activity will be a more effective antagonist than a different compound with 50% partial agonist activity. Some naturally occurring steroids are antisteroids for other receptors, such as progesterone, which is an antagonist for glucocorticoid and mineralocorticoid receptors. Most antisteroids are synthetic and have very different structures (Fig. 1d). RU486 (RU38,486 or mifepristone) is of interest as it is an efficient antagonist for PRs, GRs, and ARs (6) but not MRs (7) or ERs. Unfortunately, it is currently not possible to predict either what kind of activity a new steroid will have or even to which receptor it will bind. This issue will be discussed in greater detail below.
Current scope of steroid hormones chemistry
Initially, the study of steroid hormones involved relatively unmanipulatable biologic systems and was the purview of biologists. Over the years, new techniques in molecular biology such as cloning, protein identification, and protein purification have made it possible to alter selected macromolecules in cells and even in organisms. Thus, studies of steroid hormone action are now amenable to many of the more precise experimental manipulations expected by chemists, many of whom have merged with the biologists. Indeed, a complete understanding of steroid hormone action will be achieved only when all components are identified and characterized in the manner of more conventional chemical reactions.
Biologic Background of Steroid Hormones
In several instances, the steroid hormones were named for their most prevalent biologic activity. Thus, the steroids found to cause mobilization of glucose were called glucocorticoids. Min- eralocorticoids were those steroids that regulated the balance of the minerals sodium and potassium.
Source of steroids
All steroids are synthesized in humans from cholesterol. The adrenal gland can synthesize all endogenous steroid hormones and is the major source of glucocorticoids and mineralocorticoids. Most androgens are secreted from the testes, whereas most estrogens and progestins derive from the ovaries. In postmenopausal women and in men, a variety of sources, such as adipose tissue, are the major source of estrogens, although in much lower amounts (8). The environment contributes many compounds, called xenobiotics and endocrine disrupters, that can compete with the actions of endogenous steroids with often incompletely documented effects (9, 10), whereas plants are the source of phytoestrogens that are ingested both intentionally and unintentionally (11).
Transport of steroids
Almost all steroids reach the target organs through the circulatory system, which is the definition of an endocrine system. A target organ, or cell, for a steroid is one that contains the cognate receptors. Given the hydrophobic properties and relatively low solubility in water of most steroids, their concentration in the circulatory system is increased by their complexation with serum-binding proteins such as corticosteroid binding globulin (CBG). Although these proteins were initially thought to be passive carriers of steroid, evidence now exists that they play a more active role in steroid hormone action (12). In general, steroids enter cells by passive diffusion, although multi-drug resistance transporters can facilitate the depletion of intracellular steroids (13).
Basic features of steroid hormone action
The general mechanism of receptor-mediated action of steroid hormones is very similar for each of the five classes. Briefly, the steroid enters the cell and binds to an intracelluar protein. The resulting receptor-steroid complex is converted in a poorly understood step called activation to a form that binds to specific, biologically active DNA sequences (called hormone response elements, or HREs) of the nuclear chromatin. These HRE-bound receptor-steroid complexes recruit various cofactors and then interact with the transcription complex containing RNA polymerase II to modify the rates of transcription of a nearby DNA sequence coding for an expressed protein (Fig. 2). This alteration of transcription rate is typically fast (15-30 min) (14).
The mechanism of steroid receptor-mediated changes in transcription has occupied center stage over the last 40 years. Serious attention is only just beginning to be paid to the role of steroid receptors in other, often more rapid, processes that occur within minutes (15, 16), such as the estradiol-mediated activation of eNOS that proceeds in about 5 min (15). These types of responses are frequently called “nongenomic” to indicate that the steroid receptor does not interact with genomic DNA. Steroid receptors have been reported in mitochondria (17) and cell membranes, although it is not yet clear whether all of these receptors are the same as the intracellular steroid receptors (16, 18-22 vs. 23). Some of the membrane-bound receptors for steroids are G protein-coupled receptors (24-27). A recent report suggests that membrane-bound steroid receptors can interact with, and augment the transcriptional activity of, the intracellular receptors (24). Finally, steroids can bind to nonreceptor molecules such as enzymes and transport proteins (see above), which may have yet undiscovered consequences.
The “nongenomic” and “nonreceptor” responses to steroids will not be covered here. Instead, we will concentrate on the mechanisms by which the classic steroid hormones alter gene transcription via their intracellular cognate receptor protein. We will discuss only the primary effects of steroid hormones, which are those rapid (15-30 min) events that lead to changes in gene transcription without any requirement for protein synthesis. It should be remembered that at a sufficiently precise level, the mechanism of action of each class of steroid hormone is different from that of the others.

Figure 2. General steps in steroid hormone action and their assays. The basic model depicts steroid (S) binding to its receptor molecule (R) to form receptor-steroid complexes (RS), which attach to biologically active DNA binding sites (HRE) to eventually produce changes in the levels of specific proteins. Experimental techniques to follow R at various stages in this pathway are indicated at the first point that each method can detect a signal. Most methods can also be used to detect receptors at any step downstream of the one for which it is first used.
Methods for Study of Steroid Hormone Action
Most readers are probably familiar with the use of radioactive steroids, Scatchard plots, affinity labeling, sodium molybdate, immunofluorescence, gel shift assays, and Western and Northern blots to study steroid receptors. Over the last approximately 10 years, molecular biology has introduced many new techniques that are directed toward detecting receptors at various stages in their mechanism of action (Fig. 2). Two hybrid assays were developed to detect interacting proteins (screening done in yeast) and then to characterize these interactions (usually performed in mammalian cells) (28, 29). Cell-free pulldown assays with one partially purified protein offer more definitive evidence for a direct interaction under cell-free conditions because many potential adapter proteins have been removed. This removal can be accomplished either by attaching an immunogenic or high affinity tag on one protein (30) or, in the case of steroid receptors, using the DNA binding properties of receptors to immobilize the receptor and associated proteins (31). Coimmunoprecipitation (co-IP) assays demonstrate that two or more molecules from intact cells are present in the same complex. Mass spectral identification of copurified proteins (by binding or coIP) offer a more rapid method of identifying a large number of potential associated proteins, but time usually limits the number of subsequently examined proteins to those that seem to be particularly interesting (32). Fluorescent-tagged receptors are excellent for observing the real-time location of receptors, with FLIP and FRAP being methods of determining the rate of protein movement (33). Unfortunately, these methods are not yet sensitive enough to see receptors at a single copy of responsive gene. Chromatin immunoprecipitation (ChIP) assays reveal the presence and kinetics of molecules binding to small regions of responsive genes, like the HREs (34). The advantage of ChIP assays is their ability to interrogate endogenous genes, even if the resolution is low (~500 bp). Chromatin conformation capture (CCC) assays detect two separated DNA sequences that are brought together because of the binding of one or more proteins (35). The use of transiently transfected siRNAs in tissue culture cells offers a much simpler method for blocking the expression of selected genes, or at least for reducing the level of translation from the corresponding mRNA, than for preparing gene knockouts in whole animals (36). Microarrays, in which the level of tens of thousands of mRNAs can be determined, offer an unprecedented ability to determine the effects of steroid hormones on essentially the entire genome of a tissue (37). ChIP-on-chip assays use the microarray technology to determine to which regions of the genome receptors (and other proteins) are bound in ChIP assays (37). Systems biology approaches, and the associated model building, provide a powerful method of identifying testable mechanistic hypotheses for which no experimental evidence previously existed (38).
Molecular Biology/Chemistry of Steroid Hormone Action
Structure of steroid receptors
All members of the steroid/nuclear receptor superfamily have the same overall structural organization (Fig. 3) (39). The 66 amino acid DNA binding domain (DBD) is in the middle and is the criterion for superfamily membership because of the high amount of conserved sequence (4). The amino-terminal domain containing the activation function 1 (AF1) displays the greatest variability in length (~200 to 600 amino acids) and possesses <15% homology between receptors. A small hinge region separates the DBD from the multifunctional ligand binding domain (LBD) (40), which includes the second activation function (AF2) and is 245 residues long for GRs (41). High amounts of homology exist between the LBDs of several steroid receptors, which accounts for the fact that ligand binding is rarely totally specific (5). Given the large number of biologic activities that are mediated by the LBD (see below and Reference 40), it is not surprising that most mutations are loss of function or neutral. Gain of function mutations that augment receptor properties are rare (42, 43), although many more mutations alter the binding properties of receptors (44-46).
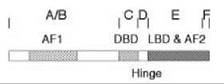
Figure 3. Schematic diagram of functional domains of steroid receptors. The initially labeled domains of A-F are also known by the various activities in each segment of the receptor: activation function 1 (AF1) in domains A&B, DNA-binding domain (DBD) in domain C, hinge region (Hinge) in domain D, ligand-binding domain (LBD) and activation function 2 (AF2) in domain E, and a carboxy-terminal domain F.
Receptor isoforms and variants
Variations in the translational start site, and in mRNA splicing, are two common ways in which receptor isoforms can be formed. Isoforms with identical LBDs and different amino termini occur because of different transcriptional or translational start sites, such as the long and short forms of PR (PR-B and PR-A, respectively), a variety of ERs (47), and multiple forms of GR (48). These isoforms bind ligand with relatively unchanged affinity but usually display different biologic activities. Splicing isoforms can have dramatically altered properties, such as GRβ, which no longer binds steroid. GRβ is found in humans (49) but not in mice (50) and contains a different and smaller sequence for the C-terminal 50 amino acids than the full-length GRα. The sole steroid receptor encoded by two different genes is ER. The classic ER is ERα. The closely related ERP was discovered only in 1996 (51) and often has very different biologic activities (52). The properties of these interesting additional forms of the steroid receptors, including post-translationally modified receptors, are beyond the scope of this article and will not be covered.
Intracellular localization of receptors
In contrast to most receptors, most ligand-free steroid receptors are not membrane bound. They can be cytoplasmic or nuclear, depending on the receptor and the time scale. At a given instant, ERs are mostly nuclear, like the nuclear receptors. For the other receptors, the amount of nuclear localization is PR > MR > AR and GR. However, the dynamic picture is that receptors are “shuttling” back and forth across the nuclear membrane (53) in a manner that is influenced by various factors, including the phosphorylation of receptors (54). The biologic consequences of changing the equilibrium position of the shuttling reaction is not yet clear, and all receptors become strongly localized to the nucleus after binding steroid.
Proteins associated with unactivated receptors
All steroid-free receptors are associated with other proteins regardless of their whole cell localization. Detailed experiments with PRs and GRs have uncovered the workings of a complex of five proteins (hsp90, hsp70, Hop, hsp40, and p23), often called chaperone proteins, which participate in the assembly of newly synthesized receptors to the form capable of binding steroid (reviewed in Reference 55). Although hsp90 is required for the “maturation” of receptors, it does not seem to be required for the de novo folding of proteins (56). This “maturation” consists of both promoting cleft opening (to allow high affinity steroid binding) and to limit excessive cleft opening (to prevent receptors from being targeted for degradation) (57). The final assembled complex retains hsp90 and p23 along with one of four tetratricopeptide repeat (TPR) proteins: three immunophilins (FKBP51, FKBP52 = hsp56, and Cyp40) or protein phosphatase 5 (PP5). This heterogeneity of ligand-free receptors theoretically could lead to a diversity of biologic responses but that has not yet been documented. Intriguingly, it seems that chaperone proteins such as p23 and hsp90 may also facilitate the disassembly of receptor-containing transcriptional complexes (58, 59). The nuclear receptor RXR is a ubiquitous heterodimerizing partner of other nuclear receptors. The only report of a steroid receptor interacting with RXR is for ER, which in several respects is closer to the nuclear receptors than to the other classic steroid receptors, but the ER/RXR heterodimers did not bind to an estrogen response element or inhibit DNA binding of ERs (60).
Steroid-induced changes in receptor structure
The first biochemical evidence that steroid binding modifies receptor structure was that protease digestion produced different sized fragments (61). The ability of this simple technique to discriminate between agonist and antagonist ligands has met with limited success (62, 63 vs. 64, 65). Hydrogen/deuterium exchange mass spectroscopy has been used to distinguish between agonist and antagonist binding (66). Nuclear magnetic resonance permits a more thorough examination of receptor structure, and especially protein motion, but the current methods of signal acquisition limit the size of molecules studied to <50kDa. However, new techniques may be able to increase the size limitations to include intact steroid receptors (67).
X-ray structures of receptors
X-ray crystallography provides the most complete structural data of proteins. Unfortunately, no X-ray structure of an intact receptor is yet available. Numerous structure determinations of receptor LBDs exist (see below for DBD structures), and they all show the same basic feature of an “α-helical sandwich” composed of 12 α-helices and 2 β-sheets (Fig. 4) (68, 69), although the number of α-helices (10-13) and β-sheets (2-4) can vary across the entire family of steroid/nuclear receptors. Significant differences were noted between agonist- and antagonist-bound receptors because of induced-fit changes in protein structure, but they are not yet predictable (70) because small changes in ligand-binding position can yield much larger effects on receptor structure (71).
The first X-ray structures of ligand-free and ligand-bound receptors were not of the same receptor. Nevertheless, they suggested an attractive model in which the C-terminal helix (helix 12) was triggered (like a “mousetrap”) to close over the ligand-binding pocket upon steroid binding (68). The generality of this model is unclear, though, because no repositioning of helix 12 was observed in ligand-free and ligand-bound forms of two nuclear receptors [PPARy (72) and PXR (73)] and because residues C-terminal of helix 12 seem to be important for steroid binding to PRs (74), GRs (65), and ARs (75). An alternative path for ligand binding to and dissociation from thyroid receptors (a nuclear receptor) has been proposed to occur through an opening caused by a proline that creates a kink in helix 3 (76). Again, this may not be general as all steroid receptors are lacking the comparable proline residue (68). Thus, multiple binding mechanisms with attending conformational changes may exist.

Figure 4. X-ray structure of the GR LBD/Dex/TIF2 complex. Two 90-degree views are shown. The bulk of the LBD is shown in yellow with p strands in pink. The bound TIF2 peptide is in purple. Regions of the LBD that play major roles in TIF2 binding are shown in red (the AF-2 helix = helix 12) and blue. The bound dexamethasone molecule is in space-filling representation with carbon, oxygen, and hydrogen colored in green, red, and white, respectively. Reprinted from Reference (69) with permission from Elsevier.
Activation
Regardless of whether the initially formed receptor-steroid complex is cytoplasmic or nuclear, a still poorly understood process called activation (or transformation) converts the complex into a species with increased affinity for DNA and for nuclei. The term “activation” was defined before it was possible to examine receptor binding to the promoter regions of endogenous genes. This definition may need revision because it is now clear that ligand-free steroid receptors can bind to the HREs of regulated genes, albeit with little observable transcriptional activity (77, 78, 79). This finding should be contrasted with the promoter binding of ligand-free nuclear receptors, which usually decrease gene transcription. In all cases, however, agonist steroid binding initiates events that alter gene transcription. Activation for DNA binding is affected by heat, salt, dilution, ATP, RNAse, and high pH (80), and it is blocked by the salts of molybdate, vanadate, and tungstate (81). It is also accompanied by the loss and/or increased rate of dissociation of many associated non-receptor proteins (57). This step is not microscopically reversible, but unactivated GR and PR can be regenerated in an ATP-dependent, reticulocyte lysate system (reviewed in Reference 55).
Nuclear translocation and DNA binding
The only way into the nucleus is through the nuclear pores. The entry of all molecules larger than ≈60 kDa, including all steroid receptors, is mediated by two nuclear translocation sequences in the hinge region and LBD of the receptors and proceeds via a two-step process. The first step, binding to nuclear pores, is followed by active transport of the receptor through the pores (82). Once in the nucleus, the activated receptor-steroid complexes readily bind to naked DNA, both biologically active sequences (or HREs) and nonspecific DNA. Whether receptors bind to DNA as monomers or preformed dimers is still debated. Recent studies with progesterone receptors (83) and other DNA-binding proteins (84) suggest that preexisting dimers dissociate and bind to DNA cooperatively as monomers. Nonspecific DNA binding is thought to be important both for buffering the binding to HREs and for facilitating the search of the HREs within the cellular genome (85). Each receptor binds to an HRE either as a homodimer or as a heterodimer with a closely related receptor [e.g., PR-A/PR-B (86), ERa/ERP (87), and GR/MR heterodimers (88)]. In contrast with the nuclear receptors, no functional heterodimer of RXR with a steroid receptor has been reported. The HRE has the features of an inverted palindrome of six nucleotides on either side of a 3-nucleotide spacer. Considerable variation in HRE sequence is tolerated by GRs (89) and PRs (86), and maximal gene induction can be achieved with suboptimal in vitro DNA binding sequences. Surprisingly, ARs, GRs, MRs, and PRs, but not ERs, can all recognize many of the same HRE sequences. The HRE sequence recognized by ERs is much closer to that of the nuclear receptors (4). The binding of ARs, GRs, MRs, and PRs to the same HRE would cause much of the specificity that is gained by having separate receptors for each steroid to be lost. Among the many mechanisms that may restore specificity are differential responses of cofactor association with receptors (90) and effects of flanking and spacer DNA (91, 92).
All HREs have the properties of an enhancer in that their activity to induce gene transcription is position and orientation independent. HREs are commonly found within the 2 kb upstream of the promoter of regulated genes but can be much further upstream (93, 94) and even downstream (95) of the start of transcription. Those genes that are repressed by steroid receptors usually do not contain the same HRE sequences (96) because the receptors are often bound to another protein that directly contacts other DNA sequences (97, 98) but see (99). Gene repression can also occur by preventing other factors from binding to their regulatory sites in responsive genes (100, 101).
X-ray structures of DNA-bound receptors
Almost all X-ray structures to date are of the DBD complexed with DNA. They all display several common features, including a dimerization of the DNA-bound DBDs, which is consistent with the highly conserved amino acid sequence of the DBDs, including two “zinc fingers,” each of which contains four cysteine residues that complex one Zn++ ion (Fig. 5). The right-hand “knuckle” of the first zinc finger, or p-box, contains part of the recognition helix for DNA binding and contacts the major groove of the HRE double helical sequence. The recognition helix is dominated by three amino acids of the p-box: GS--V for AR, GR, MR, and PR and EG--A for ER. The identical sequence in AR, GR, MR, and PR explains why they all can bind to the same HREs. The sequence in ER is very similar to the EG--G of the nuclear receptors. Interestingly, despite the critical role of these residues in determining the specificity of DNA binding, only one amino acid contacts the DNA of the HRE (4, 102). The first X-ray structure of a steroid receptor DBD, including the C-terminal extension (CTE) of the DBD, is that for PR. This structure shows that, like the nuclear receptors (4), additional contacts are made by the CTE in the minor groove flanking the PRE sequence, thus extending the size of the PR binding site (92).

Figure 5. Receptor DBD binding to HRE. (Top) General structure of DBD of steroid receptors. The two zinc fingers, each containing four cysteines coordinating one Zn++ ion, have different functions. The p-box and downstream 8 amino acids participate in binding to the major grove of the HRE, whereas the d-box mediates DBD dimer formation. The numbers correspond to the specific cysteine residues of ER. (Bottom) X-ray structure of ER DBD/ERE complex. Shown is the view looking down the recognition helices of the DBD dimer that contact the major groove of the ERE DNA. Gray spheres are the zinc atoms. The overlap at the top of the structure is the d-box of each ER DBD monomer.
Receptor binding to chromatin
Most DNA in cells is not present as naked DNA, waiting for receptors to bind. Instead, a large assortment of proteins decorates the DNA to give chromosomal DNA. The most abundant proteins are the histones, which form nucleosomes and obscure many of the intrinsic binding sites of proteins. In this manner, most regions of the genome are rendered transcriptionally inactive. In many cases, chromatin architecture greatly reduces the basal level of gene expression and steroids increase the efficiency of transcription by altering chromatin structure. In other cases, often where the basal transcription levels are much higher, there is less need for chromatin remodeling and the fold-increases in gene induction are correspondingly less (103). In several GR-regulated genes, the nucleosomes in the region of the HREs are phased so that the HRE sequence is facing away from the center of the nucleosome and is accessible to activated receptor-steroid complexes (104).
After a receptor binds to the HRE, a host of other factors are thought to be recruited (78). One group of proteins is involved in chromatin remodeling. These proteins fall into two subgroups: species like pCAF and CARM1, which have kinase, acetylase, or methylase activity and covalently modify histones (reviewed in Reference 105), and ATP-dependent complexes, such as SWI/SNF and NURD (reviewed in References 106 and 107; see also the article on Chromatin Remodeling). Elegant real-time, whole-cell fluorescence studies indicate that GR binding to chromosomal GREs is much more rapid, transient, and readily exchangeable than previously suspected (33). These results suggest that GR presence is not needed to perpetuate the initial activating signal. This process could occur by cofactors (e.g., CBP/p300 and CARM1) covalently modifying histones to facilitate transcription initiation, with subsequent rounds occurring in the absence of the initially bound receptor-steroid complex. Alternatively, the entire cycle of receptor binding and regulated gene transcription could be rapid (108).
Control of transcription—induction vs. repression
Regardless of whether the receptor is mediating gene induction or repression, the same two domains are involved. These domains, called activation function (AF) 1 and 2, are located in the N- and C-terminal regions of the receptor, respectively. The N-terminal AF1 domain is usually the most active and displays intrinsic transactivation activity independent of the rest of the receptor protein. No canonical activation sequences, such as acidic blobs, glutamine-rich regions, and amphipathic helices (109), have been identified in the steroid receptors. In fact, the AF1 domain, and the N-terminal half of steroid receptors, is generally unstructured but is induced to fold into a more regular structure both when exposed to helix-stabilizing environments (110, 111) and upon interacting with other transcription cofactors (112). The AF2 domain commonly has much less intrinsic activity and is comprised of helices 3, 4, 5, and 12. These helices form a hydrophobic pocket that binds coactivators, such as the p160 coactivators SRC-1, TIF2/GRIP1, and AIB1/pCIP/ACTR/RAC3/TRAM1 (Fig. 4), and corepressors, such as NCoR and SMRT. Coactivator binding is mediated by α-helical structures with the sequence LxxLL, were L is leucine and x is any amino acid. Corepressor binding to an overlapping, but not identical, region of the receptor involves a related sequence of LxxI/HIxxxI/L (113). This sequence, and region of corepressors, was initially defined from interactions with nuclear receptors but has been confirmed for the steroid receptors (114, 115). However, not all LxxLL or LxxI/HIxxxI/L sequences in coactivators or corepressors (or other molecules) are sufficient for binding to receptors as adjacent residues also make contributions (116, 117). Given the partial overlap of the binding sites, it is not surprising that the association of coactivators and corepressors to a given receptor-steroid complex displays competitive inhibition in a manner that is controlled, at least in part, by the ratio of coactivators to corepressors (114, 118, 119). These receptor-associated cofactors are then thought to recruit a burgeoning array of additional factors, although probably not at the same time (78, 120, 121; see also the articles on Transcriptional Control and on Activators and Repressors of Transcription). The mechanism of action of most of these factors remains poorly understood.
Steroid hormones both increase and decrease the levels of gene expression to give induction and repression, respectively. However, it is not yet clear whether all HRE-bound receptor-agonist complexes are transcriptionally active (77). A major unanswered question is how superficially similar steps can cause opposite responses. As described, the HREs for gene induction and repression are different. Repression is often achieved via receptors that are tethered to a different DNA-bound protein (97, 98) but see (99) as opposed to the receptor binding directly to DNA, as observed in induction. Tethering is not per se contraindicated for gene induction, as observed by the ability of GR sequences fused to the GAL4 DBD to bind via the GAL-DBD to a GAL4 upstream activating sequence and still give gene induction that is augmented by added coactivators (31, 122). Also, the coactivators TIF2 and STAMP still augment the activity of GR-agonist complexes in both GR-mediated induction and repression (123). Hence, the ability of the same receptor-agonist steroid complex to cause increased or decreased gene expression implicates the importance of other processes such as DNA-induced conformational changes (124, 125) and the ability of nearby DNA-bound factors to influence the recruitment of and/or interaction with additional cofactors (91, 92, 99).
The opposite effect of agonist steroids in induction versus repression also causes some confusion regarding the role of coactivators and corepressors. In the absence of precise mechanistic data, we currently must rely on phenomenological descriptions. The original definition of a coactivator was a factor that increases the activity of an agonist steroid (126). Accordingly, a protein that is labeled a coactivator because it increases steroid-regulated induction would be expected to enhance steroid-mediated repression and afford less gene expression (123). Therefore, the apparently opposing effects of a coactivator during induction and repression simply reflect the opposite and currently unknown actions of the agonist steroid.
Virtually all studies of gene induction involve averages from populations of cells, which led to the assumption that all cells respond equally to the same concentration of steroid. However, starting with the demonstration that two daughter cells can display drastically different levels of response to a common steroid concentration (127), it has become increasingly clear that transcription is a stochastic event for which probabilistic events play an important role (128). The random nature of gene transcription versus receptor binding to the gene was recently described in a real-time, single-cell study of the binding of green fluorescent, protein-labeled GRs to an integrated 200-copy tandem array of a reporter gene under the control of the mouse mammary tumor virus promoter (129).
Modulation of the parameters of receptor-mediated gene expression
Two distinctive parameters for gene induction (or repression) are the total activity (equivalent to Vmax in enzyme kinetics) and the steroid concentration required for half-maximal activity, or EC50. Most factors represented in the current model of steroid hormone action (e.g., Fig. 2) were identified on the basis of their ability to increase, or decrease, the total transcriptional activity (reviewed in References 113 and 130). The determinants of the EC50 have usually not been considered, partially because it has long been considered to be controlled predominantly by the affinity of steroid binding to receptor (131). However, this predicts that all genes regulated by a given steroid hormone will be induced (or repressed) with the same EC50 In contrast, it is well known that EC50s of multiple genes induced by the same receptor-steroid complex are not the same, even within the same cell (132, 133 and reviewed in Reference 134). Furthermore, the EC50 for gene repression is often much lower than for gene induction (reviewed in References 119 and 135). The circulating concentrations of steroids (e.g., cortisol ≈ 0.4 μM, estradiol ≈ 0.1 nM, or progesterone ≈ 5 nM) are in the region of half-maximal induction of many regulated genes. Consequently, genes with different EC50s will display different extents of response to the single circulating concentration of each steroid that is present at any one time. In many instances, the levels of steroid change, such as for glucocorticoids during the normal 24-hr cycle and during stress or for estrogens and progestins during the female menstrual cycle and pregnancy. Under these conditions, the transcriptional levels of those genes with EC50s near the average of the initial and final steroid concentration will be altered more than the levels of other genes.
Antisteroids are widely used in endocrine therapies to block the action of endogenous steroids, such as androgen-dependent prostate cancer and estrogen-dependent breast cancer. A very important parameter under these conditions is the amount of residual agonist activity, or partial agonist activity, of the antisteroid. The expectation from the general model of steroid action that the amount of partial agonist activity of an antisteroid will be the same with all responsive genes has not been experimentally verified. Instead, as above with the EC50, it was found that the amount of partial agonist activity usually varies with the gene (136, 137). Indeed, for reasons that are not understood, there seems to be an inverse correlation between these two parameters. Thus, for receptor-mediated induction of a given gene, the partial agonist activity of an antisteroid invariably increases when the EC50 of an agonist decreases and vice versa (reviewed in References 119 and 135 and 136). It was then realized that these gene-specific differences in partial agonist activity were desirable and offered a theoretical means of blocking only those genes responsible for the undesired pathology while retaining nearly normal levels of other regulated genes. A prime example is the ability of the antiestrogen raloxifene to block estrogen actions in breast cancer but not in bone (138). In recognition of the importance of having the amount of partial agonist activity of a steroid vary in a gene- and cell-specific manner, the term “selective receptor modulator” (SRM) is increasingly used instead of antagonist (139, 140). Thus, antiestrogens are often referred to as SERMs (selective estrogen receptor modulators) and so forth.
Elucidating the mechanisms driving these changes in EC50 for gene induction, and in partial agonist activity of antisteroids or SRMs, may greatly expand the available therapeutic targets for treatment of a variety of human pathologies. Initial progress has been made with the findings that these changes can be reproduced by varying the concentration of steroid receptor, coactivator, corepressor, and other transcriptional cofactors such as CBP, pCAF, Ubc9, (reviewed in References 119 and 135) and a new protein STAMP (123). The physiologic relevance of these fluctuations is strengthened by the report that the levels of GR mRNA display circadian rhythms in several mouse tissues (141). The EC50 for glucocorticoid killing of thymocytes was lowered 10-fold in transgenic mice containing a 2-fold increase in GR gene dosage (142). Also, increasing the concentration of ERβ 10-fold in human breast cancer cells increases the percentage of those genes that are induced by ERβ from 32% to 61%, and those that are repressed by ER|3 from 11% to 46%, (52) presumably by lowering the EC50 of the additional genes into the range of estradiol used in the assay. Similarly, in isogenic prostate cancer xenograft models, increased AR protein was the only correlate with androgen insensitivity in prostate tumors and caused both a left-shift in the dose-response curve for gene induction by agonists and increased partial agonist activity for antiandrogens (136). Coactivators and corepressors display competitive equilibrium binding to GRs and probably the other steroid receptors. Therefore, changing the ratio of coactivators to corepressors, and/or the concentrations of other factors, provides a means of adjusting, like with a rheostat, the EC50, and partial agonist activity, to a continuum of values (114). A recent systems biology approach with model building suggests that many factors and pathways can alter the EC50, and partial agonist activity, of steroid regulated gene expression under the appropriate conditions (38).
Disassembly and deinduction: turning off steroid hormone action
Many steroid-regulated genes are only transiently induced or repressed, which is especially true for GR- and MR-responsive genes as the concentration of the activating hormone varies daily. Thus, cells need to be able to turn off steroid-controlled responses reasonably rapidly. Regrettably, very little research has been focused in this direction and even less is known. Interestingly, the chaperone proteins p23 and hsp90, which are required for the proper assembly of functional receptors (see above), may also promote the disassembly of the receptor transcriptional complex (58, 59 vs. 143). Over the last few years, evidence has emerged that the proteolysis of transcription factors and the 26 S proteosome seem to be linked to active transcription (77, 143, 144). More recent results, however, suggest that the role of the proteasome may be independent of proteolysis (145). Furthermore, the proteasome inhibitor MG132 has been reported to both increase and decrease gene transcription (106). Additional work is clearly required to determine the generality of this interesting mechanism for downregulating steroid hormone action.
Summary
Much has been learned in the last 40+ years since steroid hormones were found to act via soluble intracellular proteins. This progress has been made under the guise of different labels, each of which is appropriately a discipline of chemical biology. First, better, longer lasting, and more specific steroids were prepared by chemists. Next, biochemists and biologists uncovered the basic steps by which steroid hormones affect the functioning of many cells and tissues of organisms. Biochemists and molecular endocrinologists discovered that the steroid receptors were a small subgroup of a much larger superfamily of related proteins that share many of the same features when inducing or repressing gene transcription. Molecular biologists have uncovered numerous additional factors that participate in steroid receptor regulation of gene transcription. Molecular physiologists are currently using powerful new techniques to determine the extent to which those mechanisms observed in isolated cell systems are employed in intact organisms for endogenous genes. System biologists are constructing mathematical models incorporating the burgeoning number of relevant factors in an effort to predict the effects of changing one or more components of the system. Eventually, clinicians will employ this wealth of information to correct selectively numerous pathologic conditions of the endocrine system. It is now clear that the specifics of steroid hormone action vary with the gene being regulated. The challenge ahead will be to gather sufficient gene-specific information to limit the effects of steroids to selected target genes and tissues, thereby increasing the desired therapeutic outcomes while reducing the number of unwanted responses.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH and NIDDK. The critical review of this article by John Funder (Prince Henry’s Institute of Medical Research, Australia), William Pratt (University of Michican Medical School), Brad Thompson (University of Texas Medical Branch, Galveston), Douglas Forrest (NIH), members of the Steroid Hormones Section, NIDDK/NIH, and the referees is greatly appreciated. We regret that space constraints prevented mentioning many other excellent and informative studies.
References
1. Bachmann WE, Cole W, Wilds AL. Total synthesis of the sex hormone equilenin. J. Amer. Chem. Soc. 1939; 61:974-975.
2. Jensen EV, Jacobson HI. Fate of steroidal estrogens in target tissues. In Biological Activities of Steroids in Relation to Cancer. Pincus G, Vollmer EP, eds. 1960. Academic Press, New York, pp. 161-174.
3. Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell 1995; 83:841-850.
4. Rastinejad F. Structure and function of the steroid and nuclear receptor DNA binding domain. In The Molecular Biology of Steroid and Nuclear Hormone Receptors. Freedman LP, ed. 1998. Birkhauser, Boston, pp. 105-131.
5. Ojasoo T, Dore J-C, Gilbert J, Raynaud J-P. Binding of steroid to the progestin and glucocorticoid receptors analyzed by correspondence analysis. J. Med. Chem. 1988; 31:1160-1169.
6. Song LN, Coghlan M, Gelmann EP. Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol. Endocrinol. 2004; 18:70-85.
7. Massaad C, Lombes M, Aggerbeck M, Rafestin-Oblin ME, Barouki R. Cell-specific, promoter-dependent mineralocorticoid agonist activity of spironolactone. Mol. Pharmacol. 1997; 51:285-292.
8. Simpson ER. Sources of estrogen and their importance. J. Steroid Biochem. Mol. Biol. 2003; 86:225-230.
9. Gray LEJ, Wilson VS, Stoker T, Lambright C, Furr J, Noriega N, Howdeshell K, Ankley GT, Guillette L. Adverse effects of environmental antiandrogens and androgens on reproductive development in mammals. Int. J. Androl. 2006; 29:96-104; discussion 105-108.
10. Tabb MM, Blumberg B. New modes of action for endocrine-disrupting chemicals. Mol. Endocrinol. 2006; 20:475-482.
11. Usui T. Pharmaceutical prospects of phytoestrogens. Endocr. J. 2006; 53:7-20.
12. Breuner CW, Orchinik M. Plasma binding proteins as mediators of corticosteroid action in vertebrates. J. Endocrinol. 2002; 175:99-112.
13. Webster JI, Carlstedt-Duke J. Involvement of multidrug resistance proteins (MDR) in the modulation of glucocorticoid response. J. Steroid Biochem. Mol. Biol. 2002; 82:277-288.
14. Ringold GM, Yamamoto KR, Bishop JM, Varmus HE. Glucocorticoid-stimulated accumulation of mouse mammary tumor virus RNA: increased rate of synthesis of viral RNA. Proc. Natl. Acad. Sci. U.S.A. 1977; 74:2879-2883.
15. Cato AC, Nestl A, Mink S. Rapid actions of steroid receptors in cellular signaling pathways. Sci. STKE. 2002; 2002:RE9.
16. Bartholome B, Spies CM, Gaber T, Schuchmann S, Berki T, Kunkel D, Bienert M, Radbruch A, Burmester GR, Lauster R, Scheffold A, Buttgereit F. Membrane glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB J. 2004; 18:70-80.
17. Sionov RV, Cohen O, Kfir S, Zilberman Y, Yefenof E. Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. J. Exp. Med. 2006; 203:189-201.
18. Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-alpha, hER-alpha 36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:9063-9068.
19. Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:19204-19207.
20. Falkenstein E, Meyer C, Eisen C, Scriba PC, Wehling M. Full-length cDNA sequence of a progesterone membrane-binding protein from porcine vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1996; 229:86-89.
21. Bayaa M, Booth RA, Sheng Y, Liu XJ. The classical progesterone receptor mediates Xenopus oocyte maturation through a nongenomic mechanism. Proc. Natl. Acad. Sci. U.S.A. 2000; 97: 12607-12612.
22. Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol. Endocrinol. 2006; 20:1996-2009.
23. Warner M, Gustafsson JA. Nongenomic effects of estrogen: why all the uncertainty? Steroids 2006; 71:91-95.
24. Karteris E, Zervou S, Pang Y, Dong J, Hillhouse EW, Randeva HS, Thomas P. Progesterone signaling in human myometrium through two novel membrane G protein-coupled receptors: potential role in functional progesterone withdrawal at term. Mol. Endocrinol. 2006; 20:1519-1534.
25. Zhu Y, Rice CD, Pang Y, Pace M, Thomas P. Cloning, expression, and characterization of a membrane progestin receptor and evidence it is an intermediary in meiotic maturation of fish oocytes. Proc. Natl. Acad. Sci. U. S.A. 2003; 100:2231-2236.
26. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005; 307:1625-1630.
27. Funakoshi T, Yanai A, Shinoda K, Kawano MM, Mizukami Y. G protein-coupled receptor 30 is an estrogen receptor in the plasma membrane. Biochem. Biophys. Res. Commun. 2006; 346:904-910.
28. Aronheim A. Improved efficiency sos recruitment system: expression of the mammalian GAP reduces isolation of Ras GTPase false positives. Nucleic Acids Res. 1997; 25:3373-3374.
29. Fields S. High-throughput two-hybrid analysis. The promise and the peril. FEBS J. 2005; 272:5391-5399.
30. Cavailles V, Dauvois S, Danielian PS, Parker MG. Interaction of proteins with transcriptionally active estrogen receptors. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:10009-10013.
31. Cho S, Blackford JA Jr, Simons SS Jr. Role of activation function domain 1, DNA binding, and coactivator in the expression of partial agonist activity of glucocorticoid receptor complexes. Biochemistry 2005; 44:3547-3561.
32. Graves PR, Haystead TA. A functional proteomics approach to signal transduction. Recent Prog. Horm. Res. 2003; 58:1-24.
33. McNally JG, Muller WG, Walker D, Wolford R, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 2000; 287:1262-1265.
34. Das PM, Ramachandran K, vanWert J, Singal R. Chromatin immunoprecipitation assay. Biotechniques 2004; 37:961-969.
35. Magee JA, Chang LW, Stormo GD, Milbrandt J. Direct, androgen receptor-mediated regulation of the FKBP5 gene via a distal enhancer element. Endocrinology 2006; 147:590-598.
36. Pei Y, Tuschl T. On the art of identifying effective and specific siRNAs. Nat. Methods 2006; 3:670-676.
37. Tavera-Mendoza LE, Mader S, White JH. Genome-wide approaches for identification of nuclear receptor target genes. Nucl. Recept. Signal. 2006; 4:e018.
38. Kim Y, Sun Y, Chow C, Pommier YG, Simons SS Jr. Effects of acetylation, polymerase phosphorylation, and DNA unwinding in glucocorticoid receptor transactivation. J. Steroid Biochem. Molec. Biol. 2006; 100:3-17.
39. Evans RM. The steroid and thyroid hormone receptor superfamily. Science 1988; 240:889-895.
40. Simons SS Jr. Structure and function of the steroid and nuclear receptor ligand-binding domain. In The Molecular Biology of Steroid and Nuclear Hormone Receptors. Freedman LP, ed. 1998. Birkhauser, Boston, pp. 35-104.
41. Xu M, Chakraborti PK, Garabedian MJ, Yamamoto KR, Simons SS Jr. Modular structure of glucocorticoid receptor domains is not equivalent to functional independence. Stability and activity of the steroid binding domain are controlled by sequences in separate domains. J. Biol. Chem. 1996; 271:21430-21438.
42. Chakraborti PK, Garabedian MJ, Yamamoto KR, Simons SS Jr. Creation of “super” glucocorticoid receptors by point mutations in the steroid binding domain. J. Biol. Chem. 1991; 266:22075- 22078.
43. Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol. Endocrinol. 1996; 10:1388-1398.
44. Tora L, Mullick A, Metzger D, Ponglikitmongkol M, Park I, Chambon P. The cloned human oestrogen receptor contains a mutation which alters its hormone binding properties. EMBO J. 1989; 8:1981-1986.
45. Allen A, Parikh G, McPhaul MJ. Doris Duke Clinical Research Fellowship: report of the second annual meeting. J. Investig. Med. 2003; 51:330-340.
46. Bohl CE, Miller DD, Chen J, Bell CE, Dalton JT. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol. Chem. 2005; 280:37747-37754.
47. Hirata S, Shoda T, Kato J, Hoshi K. Isoform/variant mRNAs for sex steroid hormone receptors in humans. Trends Endocrinol. Metab. 2003; 14:124-129.
48. Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol. Cell. 2005; 18:331-342.
49. Charmandari E, Chrousos GP, Ichijo T, Bhattacharyya N, Vottero A, Souvatzoglou E, Kino T. The human glucocorticoid receptor (hGR) beta isoform suppresses the transcriptional activity of hGRalpha by interfering with formation of active coactivator complexes. Mol. Endocrinol. 2005; 19:52-64.
50. Otto C, Reichardt HM, Schutz G. Absence of glucocorticoid receptor-beta in mice. J. Biol. Chem. 1997; 272:26665-26668.
51. Kuiper GGJM, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson J-A. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:5925- 5930.
52. Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology 2006; 147:4831-4842.
53. Madan AP, DeFranco DB. Bidirectional transport of glucocorticoid receptors across the nuclear envelope. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:3588-3592.
54. Qiu M, Olsen A, Faivre E, Horwitz KB, Lange CA. Mitogen-activated protein kinase regulates nuclear association of human progesterone receptors. Mol. Endocrinol. 2003; 17:628-642.
55. Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. 2003; 228:111-133.
56. Nathan DF, Vos MH, Lindquist S. In vivo function of the Saccharomyces cerevisiae hsp90 chaperone. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:12949-12956.
57. Pratt WB, Morishima Y, Osawa Y. The Hsp90 chaperone machinery acts at protein folding clefts to regulate both signalling protein function and protein quality control. Heat Shock Prot. Cancer. In press.
58. Liu J, DeFranco DB. Chromatin recycling of glucocorticoid receptors: implications for multiple roles of heat shock protein 90. Mol. Endo. 1999; 13:355-365.
59. Freeman BC, Yamamoto KR. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 2002; 296:2232-2235.
60. Lee SK, Choi HS, Song MR, Lee MO, Lee JW. Estrogen receptor, a common interaction partner for a subset of nuclear receptors. Mol. Endo. 1998; 12:1184-1192.
61. Reichman ME, Foster CM, Eisen LP, Eisen HJ, Torain BF, Simons SS Jr. Limited proteolysis of covalently labeled glucocorticoid receptors as a probe of receptor structure. Biochemistry 1984; 23:5376-5384.
62. Allan GF, Leng X, Tsai SY, Weigel NL, Edwards DP, Tsai M-J, O’Malley BW. Hormone and antihormone induce distinct conformational changes which are central to steroid receptor activation. J. Biol. Chem. 1992; 267:19513-19520.
63. Kuil CW, Berrevoets CA, Mulder E. Ligand-induced conformational alterations of the androgen receptor analyzed by limited trypsinization. J. Biol. Chem. 1995; 270:27569-27576.
64. Allan GF, Lombardi E, Haynes-Johnson D, Palmer S, Kiddoe M, Kraft P, Campen C, Rybczynski P, Combs DW, Phillips A. Induction of a novel conformation in the progesterone receptor by ZK299 involves a defined region of the carboxyl-terminal tail. Mol. Endocrinol. 1996; 10:1206-1213.
65. Modarress KJ, Opoku J, Xu M, Sarlis NJ, Simons SS Jr. Steroid-induced conformational changes at ends of the hormone binding domain in the rat glucocorticoid receptor are independent of agonist vs. antagonist activity. J. Biol. Chem. 1997; 272:23986-23994.
66. Frego L, Davidson W. Conformational changes of the glucocorticoid receptor ligand binding domain induced by ligand and cofactor binding, and the location of cofactor binding sites determined by hydrogen/deuterium exchange mass spectrometry. Protein Sci. 2006; 15:722-730.
67. Bax A, Torchia DA. Structural biology: molecular machinery in action. Nature. 2007; 445:609.
68. Wurtz J-M, Bourguet W, Renaud J-P, Vivat V, Chambon P, Moras D, Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nature Struct. Biol. 1996; 3:87-94.
69. Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, Lambert MH, Moore JT, Pearce KH, Xu HE. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 2002; 110:93-105.
70. Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson J-A, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor.Nature 1997; 389:753-758.
71. Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, Katzenellenbogen JA, Agard DA, Greene GL. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat. Struct. Biol. 2002; 9:359-364.
72. Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998; 395:137-143.
73. Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science 2001; 292:2329-2333.
74. Williams SP, Sigler PB. Atomic structure of progesterone complexed with its receptor. Nature 1998; 393:392-396.
75. Sack JS, Kish KF, Wang C, Attar RM, Kiefer SE, An Y, Wu GY, Scheffler JE, Salvati ME, Krystek SRJ, Weinmann R, Einspahr HM. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:4904-4909.
76. Martinez L, Webb P, Polikarpov I, Skaf MS. Molecular dynamics simulations of ligand dissociation from thyroid hormone receptors: evidence of the likeliest escape pathway and its implications for the design of novel ligands. J. Med. Chem. 2006; 49:23-26.
77. Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F. Cyclic, proteasome- mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol. Cell. 2003; 11:695-707.
78. Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 2003; 115:751-763.
79. Huang J, Li X, Maguire CA, Hilf R, Bambara RA, Muyan M. Binding of estrogen receptor beta to estrogen response element in situ is independent of estradiol and impaired by its amino terminus. Mol. Endocrinol. 2005; 19:2696-2712.
80. Simons SS Jr. Factors influencing association of glucocorticoid receptor-steroid complexes with nuclei, chromatin, and DNA: interpretation of binding data. In Glucocorticoid Hormone Action. Baxter JD, Rousseau GG, eds. 1979.; Springer-Verlag, Berlin, pp. 161-187.
81. Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997; 18:306-360.
82. Pemberton LF, Paschal BM. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic 2005; 6:187-198.
83. Heneghan AF, Connaghan-Jones KD, Miura MT, Bain DL. Cooperative DNA binding by the B-isoform of human progesterone receptor: thermodynamic analysis reveals strongly favorable and unfavorable contributions to assembly. Biochemistry 2006; 45:3285-3296.
84. Cranz S, Berger C, Baici A, Jelesarov I, Bosshard HR. Monomeric and dimeric bZIP transcription factor GCN4 bind at the same rate to their target DNA site. Biochemistry 2004; 43:718-727.
85. von Hippel PH. Completing the view of transcriptional regulation. Science. 2004; 305:350-352.
86. Lieberman BA, Bona BJ, Edwards DP, Nordeen SK. The constitution of a progesterone response element. Mol. Endocrinol. 1993; 7:515-527.
87. Hall JM, McDonnell DP. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 1999; 140:5566-5578.
88. Trapp T, Rupprecht R, Castren M, Reul JMHM, Holsboer F. Heterodimerization between mineralocorticoid and glucocorticoid receptor: a new principle of glucocorticoid action in the CNS. Neuron 1994; 13:1457-1462.
89. Nordeen SK, Suh BJ, Kuhnel B, Hutchinson CA. Structural determinants of a glucocorticoid receptor recognition element. Mol. Endocrinol. 1990; 4:1866-1873.
90. Song L-N, Huse B, Rusconi S, Simons SS Jr. Transactivation specificity of glucocorticoi vs. progesterone receptors: Role of functionally different interactions of transcription factors with amino- and carboxyl-terminal receptor domains. J. Biol. Chem. 2001; 276:24806-24816; Szapary D, Song L-N, He Y, Simons SS, Jr. Comparison of modulatory actions of GME, GMEB-2, Ubc9, and STAMP with PR vs. GR, submitted.
91. Nelson CC, Hendy SC, Shukin RJ, Cheng H, Bruchovsky N, Koop BF, Rennie PS. Determinants of DNA sequence specificity of the androgen, progesterone, and glucocorticoid receptors: evidence of differential steroid receptor response elements. Mol. Endocrinol. 1999; 13:2090-2107.
92. Roemer SC, Donham DC, Sherman L, Pon VH, Edwards DP, Churchill ME. Structure of the progesterone receptor-deoxyribonucleic acid complex: novel interactions required for binding to half-site response elements. Mol. Endocrinol. 2006; 20:3042- 3052.
93. Jantzen H-M, Strahle U, Gloss B, Stewart F, Schmid W, Boshart M, Miksicek R, Schutz G. Cooperativity of glucocorticoid response elements located far upstream of the tyrosine aminotransferase gene. Cell 1987; 49:29-38.
94. Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005; 122:33-43.
95. Hao H, Rhodes R, Ingbar DH, Wendt CH. Dexamethasone responsive element in the rat Na, K-ATPase beta1 gene coding region. Biochim. Biophys. Acta 2003; 1630:55-63.
96. Beato M, Chalepakis G, Schauer M, Slater EP. DNA regulatory elements for steroid hormones. J. Steroid Biochem. 1989; 32:737-748.
97. Webster JC, Cidlowski JA. Mechanisms of glucocorticoid-receptor-mediated repression of gene expression. Trends Endocrinol. Metab. 1999; 10:396-402.
98. Jakacka M, Ito M, Weiss J, Chien PY, Gehm BD, Jameson JL. Estrogen receptor binding to DNA is not required for its activity through the nonclassical AP1 pathway. J. Biol. Chem. 2001; 276:13615-13621.
99. Diamond MI, Miner JN, Yoshinaga SK, Yamamoto KR. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science 1990; 249:1266- 1272.
100. McKay LI, Cidlowski JA. Cross-talk between nuclear factor- kappaB and the steroid hormone receptors: mechanisms of mutual antagonism. Mol. Endo. 1998; 12:45-56.
101. Conroy AT, Sharma M, Holtz AE, Wu C, Sun Z, Weigel RJ. A novel zinc finger transcription factor with two isoforms that are differentially repressed by estrogen receptor-alpha. J. Biol. Chem. 2002; 277:9326-9334.
102. Schwabe JWR, Chapman L, Finch JT, Rhodes D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell 1993; 75:567-578.
103. Archer TK, Lee H-L, Cordingley MG, Mymryk JS, Fragoso G, Berard D, Hager GL. Differential steroid hormone induction of transcription from the mouse mammary tumor virus promoter. Mol. Endocrinol. 1994; 8:568-576.
104. Smith CL, Hager GL. Transcriptional regulation of mammalian genes in vivo. A tale of two templates. J. Biol. Chem. 1997; 272:27493-27496.
105. Peterson CL, Laniel MA. Histones and histone modifications. Curr. Biol. 2004; 14:R546-R551.
106. Kinyamu HK, Archer TK. Modifying chromatin to permit steroid hormone receptor-dependent transcription. Biochim. Biophys. Acta. 2004; 1677:30-45.
107. Chen J, Kinyamu HK, Archer TK. Changes in attitude, changes in latitude: nuclear receptors remodeling chromatin to regulate transcription. Mol. Endocrinol. 2006; 20:1-13.
108. Nagaich AK, Walker DA, Wolford R, Hager GL. Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol. Cell. 2004; 14:163-174.
109. Pugh BF, Tjian R. Mechanism of transcriptional activation by Sp 1: evidence for coactivators. Cell 1990; 61:1187-1197.
110. Kumar R, Lee JC, Bolen DW, Thompson EB. The conformation of the glucocorticoid receptor AF1/tau1 domain induced by osmolyte binds co-regulatory proteins. J. Biol. Chem. 2001; 276:18146-18152.
111. Reid J, Kelly SM, Watt K, Price NC, McEwan IJ. Conformational analysis of the androgen receptor amino-terminal domain involved in transactivation. Influence of structure-stabilizing solutes and protein-protein interactions. J. Biol. Chem. 2002; 277:20079-20086.
112. Copik AJ, Webb MS, Miller AL, Wang Y, Kumar R, Thompson EB. Activation function 1 of glucocorticoid receptor binds TATA-binding protein in vitro and in vivo. Mol. Endocrinol. 2006; 20:1218-1230.
113. McKenna NJ, O’Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002; 108:465-474.
114. Wang Q, Blackford JA Jr, Song L-N, Huang Y, Simons SS Jr. Equilibrium interactions of corepressors and coactivators modulate the properties of agonist and antagonist complexes of glucocorticoid receptors. Mol. Endocrinol. 2004; 18:1376-1395.
115. Wu Y, Kawate H, Ohnaka K, Nawata H, Takayanagi R. Nuclear compartmentalization of N-CoR and its interactions with steroid receptors. Mol. Cell. Biol. 2006; 26:6633-6655.
116. Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998; 12:3343-3356.
117. Hu X, Li Y, Lazar MA. Determinants of CoRNR-dependent repression complex assembly on nuclear hormone receptors. Mol. Cell. Biol. 2001; 21:1747-1758.
118. Szapary D, Huang Y, Simons SS Jr. Opposing effects of corepressor and coactivators in determining the dose-response curve of agonists, and residual agonist activity of antagonists, for glucocorticoid receptor regulated gene expression. Mol. Endocrinol. 1999; 13:2108-2121.
119. Simons SS Jr. How much is enough? Modulation of dose-response curve for steroid receptor-regulated gene expression by changing concentrations of transcription factor. Curr. Top. Med. Chem. 2006; 6:271-285.
120. Jung SY, Malovannaya A, Wei J, O’Malley BW, Qin J. Proteomic analysis of steady-state nuclear hormone receptor coactivator complexes. Mol. Endocrinol. 2005; 19:2451-2465.
121. Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal- dependent programs of transcriptional response. Genes Dev. 2006; 20:1405-1428.
122. Cho S, Kagan BL, Blackford JA Jr, Szapary D, Simons SS Jr. Glucocorticoid receptor ligand binding domain is sufficient for the modulation of both the dose-response curve of receptor-agonist complexes and the partial agonist activity of receptor-antisteroid complexes by glucocorticoid receptors, coactivator TIF2, and Ubc9. Mol. Endo. 2005; 19:290-311.
123. He Y, Simons SS Jr. STAMP: a novel predicted factor assisting TIF2 actions in glucocorticoid receptor-mediated induction and repression. Mol. Cell. Biol. 2007; 27:1467-1485.
124. van Tilborg MAA, Lefstin JA, Kruiskamp M, Teuben J, Boelens R, Yamamoto KR, Kaptein R. Mutations in the glucocorticoid receptor DNA-binding domain mimic an allosteric effect of DNA. J. Mol. Biol. 2000; 301:947-958.
125. Wood JR, Likhite VS, Loven MA, Nardulli AM. Allosteric modulation of estrogen receptor conformation by different estrogen response elements. Mol. Endocrinol. 2001; 15:1114-1126.
126. Onate SA, Tsai SY, Tsai M-J, O’Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 1995; 270:1354-1357.
127. Ko MSH, Nakauchi H, Takahashi N. The dose dependence of glucocorticoid-inducible gene expression results from changes in the number of transcriptionally active templates. EMBO J. 1990; 9:2835-2842.
128. Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 2005; 6:451-464.
129. Voss TC, John S, Hager GL. Single-cell analysis of glucocorticoid receptor action reveals that stochastic post-chromatin association mechanisms regulate ligand-specific transcription. Mol Endocrinol. 2006; 20:2641-2655.
130. Nagy L, Schwabe JW. Mechanism of the nuclear receptor molecular switch. Trends Biochem. Sci. 2004; 29:317-324.
131. Rousseau GG, Baxter JD. Glucocorticoid receptors. In Glucocorticoid Hormone Action. Baxter JD, Rousseau GG, eds. 1979. Springer-Verlag, Berlin, pp. 49-77.
132. Mercier L, Miller PA, Simons SS Jr. Antiglucocorticoid steroids have increased agonist activity in those hepatoma cell lines that are more sensitive to glucocorticoids. J. Steroid Biochem. 1986; 25:11-20.
133. Westley BR, Holzel F, May FEB. Effects of oestrogen and the antioestrogens, tamoxifen and LY117018, on four oestrogen-regulated RNAs in the EFM-19 breast cancer cell line. J. Steroid Biochem. 1989; 32:365-372.
134. Simons SS Jr, Oshima H, Szapary D. Higher levels of control: modulation of steroid hormone-regulated gene transcription. Mol. Endocrinol. 1992; 6:995-1002.
135. Simons SS Jr. The importance of being varied in steroid receptor transactivation. Trends Pharmacol. Sci. 2003; 24:253-259.
136. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella, R, Rosenfeld, MG, Sawyers, CL. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004; 10:33-39.
137. Kian Tee M, Rogatsky I, Tzagarakis-Foster C, Cvoro A, An J, Christy RJ, Yamamoto KR, Leitman DC. Estradiol and selective estrogen receptor modulators differentially regulate target genes with estrogen receptors alpha and beta. Mol. Biol. Cell. 2004; 15:1262-1272.
138. MacGregor JI, Jordan VC. Basic guide to the mechanisms of antiestrogen action. Pharmacol. Rev. 1998; 50:151-196.
139. Zajchowski DA, Kauser K, Zhu D, Webster L, Aberle S, White FAr, Liu HL, Humm R, MacRobbie J, Ponte P, Hegele-Hartung C, Knauthe R, Fritzemeier KH, Vergona R, Rubanyi GM. Identification of selective estrogen receptor modulators by their gene expression fingerprints. J. Biol. Chem. 2000; 275:15885-15894.
140. Cole TJ. Glucocorticoid action and the development of selective glucocorticoid receptor ligands. Biotechnol. Annu. Rev. 2006; 12:269-300.
141. Yang X, Downes M, Yu RT, Bookout AL, He W, Straume M, Mangelsdorf DJ, Evans RM. Nuclear receptor expression links the circadian clock to metabolism. Cell 2006; 126:801-810.
142. Reichardt HM, Umland T, Bauer A, Kretz O, Schutz G. Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol. Cell. Biol. 2000; 20:9009-9017.
143. Stavreva DA, Muller WG, Hager GL, Smith CL, McNally JG. Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol. Cell. Biol. 2004; 24:2682-2697.
144. Muratani M, Tansey WP. How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell. Biol. 2003; 4:192-201.
145. Lee D, Ezhkova E, Li B, Pattenden SG, Tansey WP, Workman JL. The proteasome regulatory particle alters the SAGA coactivator to enhance its interactions with transcriptional activators. Cell 2005; 123:423-436.
Further Reading
Beato M. Gene regulation by steroid hormones. Cell 1989; 56:335-344.
Freedman LP. Anatomy of the steroid receptor zinc finger region. Endo. Rev. 1992; 13:129-145.
Glass CK, Rose DW, Rosenfeld MG. Nuclear receptor coactivators. Curr. Opin. Cell Biol. 1997; 9:222-232.
Leonhardt SA, Edwards DP. Mechanism of action of progesterone antagonists. Exp. Biol. Med. 2002; 227:969-980.
Moriarty K, Kim KH, Bender JR. Estrogen receptor-mediated rapid signaling. Endocrinology 2006; 147:5557-5563.
Nuclear Receptor Signaling Atlas. A web sited aimed at developing a comprehensive understanding of the structure, function, and role in disease of nuclear receptors. Available at: http://www.nursa.org/index.cfm.
Robyr D, Wolffe AP, Wahli W. Nuclear hormone receptor coregulators in action: diversity for shared tasks. Mol. Endocrinol. 2000; 14:329-347.
Tasker JG, Di S, Malcher-Lopes R. Rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology 2006; 147:5549-5556.
Xu J, Li Q. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol. Endocrinol. 2003; 17:1681-1692.
See Also
Steroids, Synthesis of
Nuclear Receptors, Chemistry of
Chromatin Remodeling
Transcriptional Control
Transcription, Activators and Repressors of