CHEMICAL BIOLOGY
Two- and Three-Hybrid Systems
Jason E. Gestwicki, Department of Pathology and the Life Sciences Institute, University of Michigan, Ann Arbor, Michigan
Anuj Kumar, Department of Molecular, Cellular and Developmental Biology and the Life Sciences Institute, University of Michigan, Ann Arbor, Michigan
doi: 10.1002/9780470048672.wecb615
One goal of chemical biology is to document and understand the macromolecular interactions that take place in the cell. In this review, we discuss the classic yeast two-hybrid method (and its derivatives) and how this method continues to provide insight into the number of protein-protein partnerships in a cell. In turn, these platforms have been adapted to explore a variety of interactions, such as those between proteins and small molecules. These systems collectively are called three-hybrid assays, and they provide new opportunities for the discovery of potential binding pairs. However, two- and three-hybrid assays also have significant disadvantages, such as incomplete coverage and high rates of false positives. With these issues in mind, we discuss emerging ways of minimizing the impact of these limitations using microarrays and mass spectrometry. Finally, chemical probes related to the three-hybrid concept are going beyond observation and providing active, rational control over individual protein-protein contacts. In these systems, bifunctional compounds are used to homo- or heterodimerize target proteins reversibly, thus altering their colocalization. By purposefully controlling protein-protein contacts, these chemical dimerization methods have progressed beyond observation and towards synthetic manipulation of protein function.
Biological responses are shaped by macromolecular interactions among proteins, nucleic acids, and other organic molecules. One goal of chemical biology is to understand how, why, and when these contacts take place. Which proteins interact with each other? Where in the cell do these interactions take place? How do these complexes change in response to stimuli? By creating a list of the components that interact with each other under specific conditions, we hope to reveal the architecture, dynamics, and logic of cellular processes. In turn, by cataloging these interactions, we hope to learn how they are altered during disease and, ultimately, how imbalances might be corrected.
The first step toward this goal is to document the interactions. To this end, powerful tools have been developed to scan for macromolecular interactions. Arguably, the most significant and far-reaching of these methods is the yeast two-hybrid screen (1). Subsequent work has produced a host of thematic variations including the three-hybrid screen, a method first described explicitly in 1996 that employs small molecules or nucleic acids as “baits” (2). In this review, we discuss some strengths and limitations of the two- and three-hybrid techniques and suggest how they are being used to explore sophisticated questions in chemical biology. In addition, we review chemical dimerization technology, a three-hybrid-type method that allows purposeful manipulation of specific protein-protein interactions. Together, chemical dimerization and two- and three-hybrid systems are being used to define and control macromolecular interactions and, therefore, provide insight into the organization of cellular networks.
Two-Hybrid Screens
The classic yeast two-hybrid system
The architecture and organization of biological networks are maintained by a backbone of protein-protein interactions termed the “interact-ome.” Refining our understanding of this complex network is an important goal of chemical biology. The yeast two-hybrid approach was developed in the late 1980s (1) as a simple means of identifying physical interactions between binary protein pairs. As suggested by its name, the classic two-hybrid method uses two chimeric proteins expressed in the budding yeast Saccharomyces cerevisiae (Fig. 1a). One hybrid, referred to as the “bait,” consists of a target protein fused to the DNA-binding domain of a transcription factor, typically Gal4 or LexA (3). The second hybrid, referred to as the “prey,” consists of a putative interacting protein fused to the activation domain of this transcription factor. Physical interaction between bait and prey will bring the transcription factor DNA-binding domain and activation domain into close proximity of each other, thereby reconstituting a transcriptionally active chimera; transcriptional activity can be easily monitored through a convenient reporter system or selection scheme in yeast (e.g., lacZ) (1, 3).
The two-hybrid method is robust and lends itself well to automation; thus, high-throughput applications of the two-hybrid approach have become increasingly common over the last decade. These projects encompass two phases: an initial phase of reagent development and a subsequent phase of high- throughput, two-hybrid screening. Methods for recombination- based cloning (4) and increased experience with large-scale cloning projects have enabled the generation of many successful bait and prey collections in numerous organisms (5-11). Subsequent high-throughput screening procedures typically employ array and pooling strategies driven by simple robotics (Fig. 1b). In one approach, haploid yeast carrying bait and prey constructs, respectively, are mated to generate a diploid strain with both bait and prey for analysis of protein-protein interaction. This screen can be accomplished either by arraying the haploid strains before mating (generating an interaction “matrix”) or by using bait/prey libraries (6, 7). The efficiency of these methods is improved by pooling; small pools of baits and preys are tested first, and individual interactions are subsequently confirmed through analysis of a particular pair (12-14).
Although the two-hybrid approach has clearly proven to be a powerful tool for biological discovery, it is not without its caveats. For example, some genuine interactions may be missed in a two-hybrid screen because a given protein may not be expressed properly as a binding domain- and/or activation domain-fusion (15); also, certain interactions may be too weak to be detected by two-hybrid methods, although the minimum affinity threshold for detection has been difficult to determine. More significantly, the two-hybrid method is prone to identify false-positive interactions, and many possible sources of these artifacts exist (16). For example, a given protein pair may interact by yeast two-hybrid analysis but be precluded from interacting in vivo because of the distinct subcellular localizations of the two proteins (i.e., one protein may be nuclear, whereas the other may be cytoplasmic). In the two-hybrid method, both putative interacting proteins are forced to the nucleus by virtue of nuclear localization signals incorporated in the chimeras, which yields an artificial colocalization and correspondingly artificial potential for interaction. In general, this situation exemplifies a class of false-positive results by two-hybrid analysis, wherein the two hybrid-associated proteins possess the potential to interact in vivo but are restricted from doing so in their cellular context. Artifacts of the yeast two-hybrid assay itself are also evident, which results in observed transcriptional activity independent of protein-protein interaction. This additional class of false-positive results is exemplified by bait/prey proteins that are capable of activating reporter transcription in the absence of a second hybrid (15).
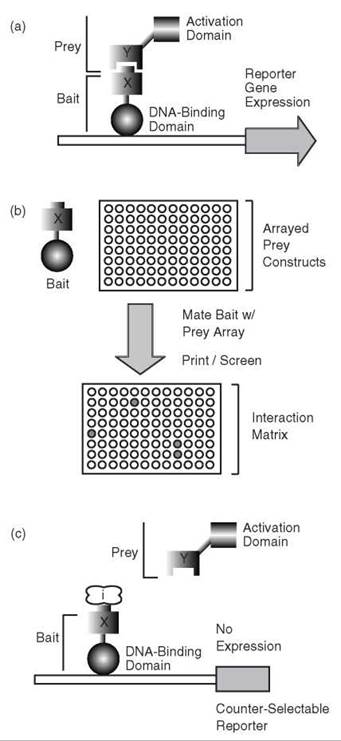
Figure 1. The classic yeast two-hybrid method and derivatives. (a) Schematic diagram of the yeast two-hybrid approach, describing an interaction between protein X and protein Y. Protein X is fused to a transcription factor DNA-binding domain (the ''bait'' construct), and protein Y is fused to a transcription factor activation domain (the ''prey'' construct). (b) High-throughput applications of the yeast two-hybrid method use mating of haploid strains carrying bait and prey, respectively. Hybrids can be mated in arrayed formats (as shown) or as libraries. (c) The reverse two-hybrid method uses a counter-selectable marker to indicate loss of protein interaction because of disruption by an inhibitor protein/small molecule (''/" illustrated in the diagram) or mutation(s) in proteins X and/or Y.
Considering the caveats indicated above, yeast two-hybrid methods are often coupled with secondary, high-confidence assays to define a truer interaction data set. Toward this end, Li et al. (8) undertook a large-scale yeast two-hybrid analysis of protein-protein interactions in Caenorhabditis elegans and used co-affinity purification assays to validate 143 putative interactions. Co-immunoprecipitation provides a high-confidence measure of protein interaction; however, the approach is labor intensive and presents a greater challenge for automation. Other related approaches have also been employed to verify protein interactions that are suggested by two-hybrid studies. For example, Tong et al. (17) integrated results from two-hybrid analysis with protein interaction data sets derived from phage display experiments to yield a subset of high-confidence interactions associated with SH3 domain-containing proteins in the budding yeast. Protein localization data have also proven useful in identifying biologically relevant interactions within large two-hybrid data sets because interacting proteins should localize within the same subcellular compartment (18). Collectively, these and other approaches (see the section titled “Combining Two- and Three-Hybrid Screens with Other Technologies”) offer significant promise toward the construction of high-confidence protein interaction maps on a genome-wide scale.
Reverse two-hybrid approaches
The classic two-hybrid method has been modified over time to yield a variety of derivative approaches that offer complementary data. For example, the “reverse” two-hybrid method was developed to identify drugs, mutations, and/or proteins capable of disrupting a known interaction (Fig. 1c). Briefly, the reverse two-hybrid approach employs a reporter gene that encodes a counter-selectable marker (e.g., the yeast gene URA3) (19). The URA3 gene product is required for growth on medium lacking uracil; however, this product is toxic to yeast cells grown on medium containing 5-fluoroorotic acid (5-FOA). By the reverse two-hybrid method, dissociation or inhibition of a two-hybrid interaction results in loss of URA3 expression, which permits cell growth in the presence of 5-FOA. Similar loss-of-interaction detection schemes have been devised using the Escherichia coli tet repressor (tetR) as a reporter (20). Expression of tetR represses expression from a second reporter containing an upstream tetR-binding site, and conversely, disruption of the interaction restores expression from this reporter. Although the affinity requirements and dynamic range of these methods have yet to be fully described, they have proven useful in proof-of-principle studies.
Membrane-associated two-hybrid systems
In the classic two-hybrid system, protein interaction is assayed in the nucleus, a suboptimal environment for the detection of many protein-protein interactions—particularly those involving membrane proteins. As an alternative to nuclear two-hybrid assays, several groups have developed membrane-associated two-hybrid systems that use signaling components, such as Ras (21). The membrane-associated Ras protein binds and hydrolyzes GTP, alternating between an active GTP-bound form and an inactive GDP-bound state. These states are modulated by GTPase-activating proteins that promote GTP hydrolysis and guanine nucleotide exchange factors (GEFs) that stimulate GDP release/GTP uptake. Aronheim et al. (22, 23) used Ras signaling as the basis for a two-hybrid system, substituting the human GEF hSos for the native yeast RAS GEF, Cdc25p, in yeast cells carrying a mutant allele of cdc25. Using a hybrid consisting of a target protein fused to hSos and a second putatively interacting protein localized to the cell membrane by a myristoylation tag, protein-protein interaction recruits hSos to the cell membrane, activating Ras, which, in turn, promotes cell survival and growth.
Isakoff and colleagues (24) presented an extension of this system for the identification of proteins binding 3-phosphoinositide second messengers, PtdIns(3,4)P2 and PtdIns(3,4,5)P3, in vivo. In this system, the putative PtdIns(3,4)P2/PtdIns(3,4,5)P3-binding protein (or specifically just its predicted pleckstrin homology, PH, domain) is fused to constitutively activated GTP-bound Ras. This hybrid protein is coexpressed with phos- phatidylinositol 3-kinase (PI3K), which generates the 3-phosphoinositides in a temperature-sensitive Ras mutant. Binding of the target protein’s PH domain to the 3-phosphoinositides draws the hybrid protein to the cell membrane where the activated Ras substitutes for the nonfunctional mutant, which enables cell growth. Medici and colleagues (25) developed an additional membrane-based two-hybrid assay with bait and prey constructs consisting of the yeast G protein a subunit Gpalp and the a-factor receptor Ste2p. By this approach, interaction between a Gpalp chimera and Ste2p hybrid drives a signaling cascade that permits cell growth in a gpa1 mutant background.
Imaging protein-protein interactions in live cells
In contrast to the classic yeast two-hybrid method and its close derivatives, chimeric protein-protein interactions may also be assessed in many organisms using readouts easily visualized in live cells. For example, Sanjiv Gambhir’s group developed a modified mammalian two-hybrid system using the Gal4p DNA-binding domain, herpes simplex virus VP16 activation domain, inducible promoters, and the firefly luciferase gene as a reporter (26). In a proof-of-principle study, the helix-loop-helix proteins Id and MyoD were shown to interact, which results in luciferase activity visualized by standard methods of bioluminescence imaging in cell culture and in 293 T cells implanted in mice (26).
Protein-protein interactions may also be detected in living cells using hybrids that consist of fluorescent proteins capable of undergoing fluorescence resonance energy transfer (FRET). FRET refers to an exchange of energy occurring between two fluorescent molecules, with fluors satisfying the following two criteria: 1) The emission spectrum of one fluorophore (the donor) must overlap with the excitation spectrum of the second fluorophore (the acceptor), and 2) the fluors must be within approximately 10 nm of each other (reviewed in Reference 27). If these requirements are met, the donor fluorophore (e.g., cyan fluorescent protein, CFP) can transfer the resonance energy of its excited state to the acceptor (e.g., yellow fluorescent protein, YFP), which causes it to fluoresce (Fig. 2a). The efficiency of this energy transfer depends on the inverse sixth power of the distance between donor and acceptor; therefore, the detection of FRET is a sensitive measure of the mutual proximity of two fluors. Thus, FRET can be used to detect protein-protein interactions with high confidence and is amenable to implementation on a fairly large scale, using YFP- and CFP-chimeras. Recently, Muller et al. (28) used FRET to identify core proteins of the yeast spindle pole body, thus highlighting the applicability of this approach toward defining a target subset of protein-protein interactions.
Along similar lines, bioluminescence resonance energy transfer (BRET) also presents a technology for the detection of protein-protein interactions based on energy transfer between two proteins in close spatial proximity. As presented by De and Gambhir (29), BRET can be implemented using the Renilla luciferase (Rluc) as a bioluminescent donor and mutant green fluorescent protein (GFP) as a fluorescent acceptor (Fig. 2b). Proteins predicted to interact are fused to luciferase and GFP; interaction between the Rluc and GFP chimeras results in excitation of GFP by resonance energy transfer from the reaction of Rluc with its substrate, deep blue coelenterazine. De and Gambhir used this approach to detect Id-MyoD and FKBP12FRAP interactions in both cultured cells and in deeper tissues of mice with implanted cells overexpressing the fusion constructs (29).
Although FRET- and BRET-based technologies offer many advantages associated with the ability to image protein interactions in live cells, both approaches are substantially limited by the spatial constraints of energy transfer. Many genuine protein interactions may be missed in FRET/BRET approaches because donor and acceptor are not brought in sufficiently close proximity for resonance energy transfer by target protein dimerization (27). The false-negative rate associated with these technologies is, therefore, presumed to be high. Conversely, however, these approaches are prone to a very low rate of false-positive results, and detected interactions can be considered genuine with few exceptions.

Figure 2. Imaging protein-protein interactions by FRET and BRET. (a) Diagram illustrating FRET between the donor CFP-fusion and the acceptor YFP-fusion. (b) Detection of protein-protein interaction between X and Y by BRET using the Renilla luciferase (Rluc) as a bioluminescent donor and GFP as a fluorescent acceptor. The substrate for Rluc is deep blue coelenterazine. Constructs not drawn to scale.
Since the mid-1990s, numerous studies have advanced clever alternatives to transcription factor-based two-hybrid methods through technologies that use various split-reporter systems (30). In particular, Johnson and Varshavsky (31) developed an interesting split-protein sensor of protein interactions using the small protein ubiquitin (Ub). Ubiquitin is a 76-amino acid protein best characterized for its role in protein degradation: Covalent attachment of Ub to a target protein marks that protein for degradation by the 26 S proteosome (32, 33). In eukaryotes, newly formed Ub fusions are cleaved by site-specific proteases after the last Ub residue at the Ub-target protein junction, provided that the Ub protein is properly folded. The Varshavsky group took advantage of this fact, splitting Ub into amino- and carboxy-terminal halves and fusing each to one of two putatively interacting membrane proteins (Fig. 3a). The amino-terminal Ub fragment was modified additionally by the introduction of a point mutation that decreased its affinity for the carboxy-terminal Ub fragment. This carboxy-terminal fragment was fused to a transcription factor, which provided a convenient readout for this system. During target protein interaction, the amino- and carboxy-terminal Ub fragments are brought into close proximity, which drives partial reassociation of Ub. This reconstituted Ub is recognized by its proteases, which results in cleavage and release of the transcription factor; protein interaction thus can be monitored by the activity of reporter genes (e.g., HIS3 and lacZ) recognized by the released transcription factor. This split-Ub system has been used successfully in several studies in organisms ranging from yeast to humans (34, 35) and, in fact, was used recently to isolate proteins interacting with human Bap31 and Erb3 (36). This method has also been applied to membrane proteins (37).

Figure 3. Protein fragment complementation strategies. (a) Schematic representation of the split-ubiquitin yeast two-hybrid system. The amino-terminal fragment of ubiquitin is designated ''Nub,'' and the carboxy-terminal fragment is designated ''Cub.'' Interaction of proteins X and Y yields properly folded ubiquitin, the recognition of which by ubiquitin-specific proteases results in cleavage and activation of a reporter (e.g., DHFR or a transcription factor). (b) Simple representation of the β-gal fragments used in β-gal complementation. The α peptide consists of β-gal amino acids 5-51, and the m fragment contains a deletion of residues 11-41. Fragments not drawn to scale. (c) Firefly luciferase complementation by protein-protein interaction results in detectable signal at 575-600 nm. (d) Bimolecular fluorescence complementation using split mutants of YFP, as applied in mammalian cells, results in fluorescence at 545 nm. Again, fragments are not drawn to scale.
The Blau group has presented an alternative protein fragment complementation approach for detecting protein-protein interactions using the well-studied reporter β-galactosidase (β-gal) (38). The lacZ gene is known to exhibit intracistronic complementation, wherein pairs of inactive β-gal deletion mutants are capable of complementing each other in trans, assembling to yield an active enzyme. To use β-gal fragments as the basis for a protein interaction system, Rossi et al. (38) selected β-gal mutants (∆α and ∆ω) with low affinity for each other and fused each mutant to one of two target proteins (Fig. 3b). Target protein interaction drives the formation of active β-gal, which provides a convenient and quantitative monitor of protein dimerization. Recently, Wehrman et al. (39) have modified this system, using a truncated low-affinity α-peptide that weakly complements the ω fragment, such that recognition between the two fragments in the absence of forced dimerization is insufficient to maintain a complemented enzyme. Accordingly, reversible interactions can be monitored by this method, and the activity of the enzyme is directly related to the local concentration of the enzyme fragments. This approach has been used successfully as an assay of nuclear import/export (39) and as a monitor of G-protein-coupled receptor internalization (40). As with other β-gal readout systems, this complementation approach allows for convenient signal visualization, which offers a method for the direct assessment of protein interaction in the context of native cell compartments and environments.
The Michnick group has also developed an elegant split- reporter system, based on reconstitution of dihydrofolate reductase (DHFR) (41). In this system, cell survival requires functional DHFR; thus, a successful protein-protein interaction can be identified readily by selection. This system has been applied to the identification of peptide sequences that bind the Ras-binding domain of Raf (42).
The firefly luciferase enzyme has also served as the basis of an informative protein fragment complementation approach for the detection of protein-protein interactions in cells and living animals (43). By this method, amino- and carboxy-terminal fragments of luciferase are fused to target proteins of interest, such that protein-protein interaction reconstitutes active luciferase (Fig. 3c). Luker et al. have used this approach to investigate STAT1 activity and yielded data suggesting that STAT1 proteins homodimerize in the absence of ligand-induced phosphorylation (43). In addition to the firefly enzyme, Renilla luciferase has also been used, for example, to explore protein kinase A interactions (44). These luciferase complementation imaging strategies share several advantages of the systems described above and, in particular, provide near real-time detection of protein-protein interactions in intact cells and living animals.
Tom Kerppola’s group has developed a distinct and powerful variation on split-protein detection strategies, by using fragments of a fluorescent protein as part of a technique termed bimolecular fluorescence complementation (BiFC) (45). In this method, nonfluorescent fragments of enhanced yellow fluorescent protein are fused to putatively interacting target proteins; protein interaction brings these fragments into close proximity, which results in detectable yellow fluorescence (Fig. 3d). For genuine in vivo interaction partners, the BiFC approach is typically quite successful, although the fluorescence intensity produced by target protein dimerization is routinely less than 10% of levels generated by intact yellow fluorescent protein (45). To date, BiFC has been used to identify interactions among many types of proteins in numerous cell types and organisms (reviewed in Reference 46) and provides a method not only to identify protein interactions but also to define the subcellular localization of protein complexes in living cells (47). The BiFC approach is amenable to many additional applications (47); however, it is not without its limitations. Specifically, the fluorescent protein fragments in a BiFC system associate irreversibly under some conditions and exhibit an intrinsic affinity for each other in the absence of target protein interaction, which limits both the sensitivity and scope of BiFC-based studies. Nonetheless, the approach holds significant potential as a generally applicable means of imaging protein interactions and complexes in single live cells, with future promise for even broader applications in a wide range of organisms.
Three-Hybrid Screens
The classic three-hybrid screen
Although the two-hybrid is an effective option for protein- protein interactions, this platform is not suitable for other types of macromolecular contacts, such as those involving small molecules or nucleic acids. The three-hybrid screen was developed specifically to address this need; for reviews see References 48-50. The third component in these systems is a small molecule or RNA that serves as the “bait” (Fig. 4). A general feature of three-hybrid “baits” is that they have two distinct, nonoverlapping domains; the anchoring end has a well-characterized binding partner, whereas the other side is used to query a collection of potential “preys.” For example, the first compound used in a three-hybrid was composed of a dexamethasone (Dex) anchor attached to FK506 (2). Dex has high affinity for the glucocorticoid receptor (GR), and, therefore, it is bound to the promoter via its tight association with a LexA-GR fusion (Fig. 4a). The query end of this divalent compound recruits a transcriptional activator that is fused to FK506-binding protein (FKBP). The reporter gene is typically a selection marker, such as LEU2, or a directly detectable signal, such as fluorescence or luminescence. Thus, this relatively straightforward adaptation of the two-hybrid platform has the potential to identify binding partners for a variety of macromolecules.

Figure 4. Architecture of three-hybrid screens. (a) The classic, small-molecule-based, three-hybrid platform is shown. The three components of this systems are: a DNA-binding protein fused to a known drug-binding domain (X), a bifunctional small molecule that binds X on one end and displays a query ''bait'' on the other, and a library of possible ''prey'' proteins (Y) expressed as fusions to transcriptional activation domains. Assembly of the ternary complex recruits the activation domain to the promoter and initiates reporter gene expression. A partial list of examples is shown in the inset boxes; because of space constraints, some relevant systems are absent. (b) The RNA-based, three-hybrid is assembled in a similar fashion except that the bifunctional ligand is an RNA molecule. The complex between the MS2 RNA and the MS2-coat protein is typically used as the anchoring interaction. (c) A key component of the three-hybrid system is the high-affinity interaction that anchors the bifunctional molecule to the promoter. The most common combinations of ligands and binding proteins are shown. Note that the site of attachment to the query compound is indicated by the box.
Several recent studies have examined the strengths and limitations of the three-hybrid. For example, this system has been successfully adapted for use in yeast (51) and bacteria (52). In addition, extensive analyses of the kinetic and affinity requirements have been reported (51, 53, 54). These studies have revealed that, although the dynamic range is small (~1 order of magnitude), the three-hybrid signal is dependent on the association strength of the interaction, with a minimal KD of ~50 nM (55). This in-depth work has helped refine our understanding of the key interactions, and, through these studies and others (48), the features of three-hybrid screens that influence the signal have begun to be revealed.
Target identification for small molecules
In forward chemical genetics, a system is treated with a collection of compounds and a candidate molecule that produces the desired phenotype identified (56-58). One main challenge in this field is that each new chemical probe requires follow-up studies to identify its cellular protein target (59). Accordingly, one primary use of the three-hybrid system is to find these targets. However, the challenge is that the molecule of interest must be covalently coupled to the anchoring compound without a loss of binding affinity (Fig. 4a). One of the most extensive synthetic studies was reported by the Verdine group, in which they generated a library of 320 tetrahydrooxazepines attached to an analog of FK506 on solid phase (60). This study validates that large collections of bifunctonal ligands are possible, but, in practice, only a few query molecules have been screened. For example, the Kley group used a three-hybrid approach, coupled with affinity chromatography and in vitro kinase assays, to identify CDK1, CDK2, and other kinases as the protein targets of the kinase inhibitors, purvalanol B, roscovitine, and indenopyrazole, IP-1 (51).
Reverse three-hybrid
The downside to the three-hybrid as a target validation tool is that a suitable attachment point must be found on the query molecule. Oftentimes, selection of an appropriate site requires structure-activity relationship (SAR) studies to avoid damaging the compound’s binding affinity. Moreover, each chemical entity requires individual synthetic effort because the properties of the linker can have important effects on the efficiency of the system (51, 54). One way to avoid the need for synthetic manipulation of the query molecule is to conduct a reverse three-hybrid screen. Similar to the reverse two-hybrid discussed above, a library of compounds is applied to a two- or three-hybrid reporter (Fig. 5a), and if the molecule interrupts the reconstituted complex, the reporter is deactivated. In one example, this approach was used to identify inhibitors of the helicase-primase interaction in Bacillus stearothermophilus (61). A related system involves expression of potential binding proteins in the presence of an intact three-hybrid contact (Fig. 5a) (62). These reverse three-hybrid systems do not require extensive synthetic manipulations and are ideal for those examples in which little SAR is available or no suitable attachment point is found.

Figure 5. Variations on the three-hybrid method. (a) Two common variations of the reverse three-hybrid are shown. In the first, potential inhibitors are applied to a two-hybrid system, and interruption of the protein-protein contact (X with Y) is expected to interrupt reporter gene levels. In the other variation, proteins are expressed in the presence of a functional three-hybrid system, and successful binding displaces the activation domain (Y). (b) Some three-hybrid systems have been assembled to rely on the activity of an enzyme for reporter gene activation; thus, these systems can be used to identify inhibitors or to identify enzymes with desired properties. (c) A membrane-associated three-hybrid is shown schematically. In this system, STAT3 is recruited to active Jak2 by association between the ''prey'' (Y) and the bifunctional three-hybrid probe.
Screens for enzymatic activity and enzyme inhibitors
The Cornish group has reported new variations of the three- hybrid, which all involve an enzyme as a necessary fourth component (Fig. 5b). In their first report, they developed a way to study cephalosporinase activity in yeast; they installed a cephalosporinase-sensitive site within a Dex-Mtx conjugate such that the enzymatic activity would cleave the bifunctional ligand and disrupt expression from a GR-DHFR transcriptional switch (63). Later variations of this platform have been used to evolve glycosynthase activity (64). Briefly, formation of a glycosidic link between detached sugar analogs reconstituted the bifunctional “bait” that subsequently activated the DHFR-GR-based LEU2 selection marker (Fig. 5b). Using this method, they evolved a glycosynthase with fivefold better enzymatic activity. Related methods have been used to study phosphorylation-dependent protein-protein contacts (65, 66), which demonstrates the versatility of this approach. These systems all take advantage of the signal-amplifying properties and high sensitivity of three-hybrids to monitor enzyme-dependent interactions in living systems.
RNA-RNA interactions and RNA-binding proteins
Another adaptation of the three-hybrid replaces the bifunctional small molecule with an RNA molecule [first described by SenGupta et al. in 1996 (67) and reviewed more thoroughly by Jaeger et al. (68)]. The bifunctional RNA is composed of two distinct domains; one part has a well-characterized affinity for an RNA-binding protein (typically MS2 coat protein or Hiv-1 RevM10), whereas the query RNA is used to pan for possible “preys’ ’ (Fig. 4b). This system has been used to verify aptamer interactions (69) and to identify RNA-RNA interactions (70); also it has been used successfully to characterize over 20 RNA-binding proteins (68). Additionally, this method has been quite successful in revealing the key interacting residues in known RNA-binding proteins, including p53, HIV Tat, FBF-1, and histone hairpin binding protein (HBP), by random or direct mutagenesis (68, 71-74). Like other three-hybrid systems, the kinetics and binding constants are important for the efficiency of this system (75). As evident by the more than 60 published reports of successful three-hybrid screens (68), this method has proven to be a powerful addition to the arsenal of molecular and chemical biologists.
Membrane-associated three-hybrid screens
As mentioned above, advantages are gained by screening in non-nuclear compartments, but adapting the two- and three- hybrid technologies for different reporter strategies can be challenging. The Kley group has developed a system that they call MASPIT as one solution (76). In this system, erythropoietin receptor is expressed as a fusion with DHFR in mammalian cells, and methotrexate-coupled bait molecules are used to recruit the prey (Fig. 5c). This membrane-proximal juxtaposition triggers Jak2-mediated phosphorylation of STAT3 and activation of a downstream reporter. This system was used to identify a family of ephrin receptor tyrosine kinases as targets of the inhibitor, PD173955 (76). Although this system is relatively complex, it represents an important advance because it allows screening in the cytosol and opens the possibility of finding contacts in their native subcellular environment.
Combining Two- and Three-Hybrid Screens with Other Technologies
Both two- and three-hybrid screens are limited by technical problems, such as high false-positive rates and incomplete coverage. This last problem is best illustrated by comparing the hits in otherwise identical screens; Parrish et al. compared the hits from three screens of 24,000 protein-protein interactions (~54% of the estimated total) in Drosophila and found remarkably little overlap (only 1 gene common to all three, and only 61 shared by at least 2 experiments) (77). Similar trends were seen in human two-hybrid systems, and, together, these findings strongly suggest that the coverage of possible interactions is incomplete (77). Insufficient data prevents similar comparisons in three-hybrid systems, but it is presumed that similar problems will be encountered. Minimizing the impact of these limitations might be best achieved by coupling two- and three-hybrid screens with other methods. Specifically, recent advances in mass spectrometry and microarrays are being used to investigate and confirm protein-protein interactions. High-throughput mass spectrometry is being used to both identify protein-protein contacts and to investigate these interactions on a proteome-wide scale (78-80). Thus, any interactions identified by two- or three-hybrid screens might be confirmed by this means. Finally, advances in microarray profiling offer promise to those screening for novel protein-protein or protein-ligand interactions. In these techniques, potential binding partners are labeled (e.g., with fluorescence or other reporter) and passed over patterned proteins, small molecules, or carbohydrates arrayed on a chip. After removing nonspecifically adsorbed material, the remaining “hits” represent interacting pairs. Technical advances over the past 10 years have positioned mass spectrometry and microarrays as viable tools for performing large-scale protein interaction studies (81-88). Thus, these methods provide potential synergy with two- and three-hybrid methods, and, most importantly, they can be used to verify suspected contacts independently.
Chemical Dimerization: Three-Hybrids for Conditional Regulation of Protein Function
Chemical inducers of dimerization
Although two- and three-hybrid screens are useful ways to identify new protein-protein and protein-drug contacts, the next set of questions require one to control these interactions rationally and to confirm their importance in a biological process. Toward this goal, the chemical dimerization systems developed in the mid-1990s by Crabtree and Schrieber are ideal tools (89-93). In these systems, a chemical inducer of dimerization (CID) is used to force the conditional homo- or hetero-dimerization of two proteins (for more in-depth reviews of these systems, see References 94-97). The target proteins are expressed as fusions with known drug binding domains such that addition of the bifunctional molecule causes their tight association (Fig. 6a). Although other pairs have been used (98-102), the most common systems are the homodimerization of FKBP by dimers of FK506 and the heterodimerization of FKBP with the FK506-rapamycin binding (FRB) domain by analogs of rapamycin. The shared feature of these CIDs is that they have two nonoverlapping domains; thus, they can bridge two proteins. Another common property of these systems is that they are reversible; thus, they offer the opportunity to regulate protein function conditionally. Whereas two- and three-hybrid screens are used to identify binding partners, CID systems are typically used to test whether protein colocalization is necessary and sufficient to instigate a biological response.
CIDs are used in many biological applications
Chemical dimerization has been remarkably successful at addressing previously inaccessible biological questions (Fig. 6a). For example, recent studies have explored the mechanisms of complex pathways, such as chromatid disassembly (103) and learning and memory (104). In addition, this method has proven quite adept in studies aimed at understanding signaling through the T-cell receptor (89), vasopressin receptor (105), FGF receptor (106-108), Akt (109), GTPases (110), and phosphoinositides (111, 112). Similarly, this procedure has been used to gain regulatory control over important cellular processes, such as apoptosis (113, 114), preRNA splicing (115), translation initiation (116), glycosylation (64, 117), pathogen recognition (118, 119), and secretion (120, 121). This list of applications is necessarily brief, because greater than 300 manuscripts have been published (across numerous scientific areas) using CID systems (http://www.ariad.com/wt/page/kits_references). This expansive literature illustrates the widespread acceptance of this method in the scientific community. In addition, this breadth makes a comprehensive overview of the area difficult, and the interested reader is encouraged to pursue several excellent reviews (49, 93-97, 122).
Inhibition of protein-protein interactions
Another recent use of CIDs is to inhibit protein-protein interactions. One reason that this class of interactions has been difficult to block is that they typically involve a large and complex surface (123). Therefore, typical drug-like molecules are often too small to block these contacts effectively. This problem is particularly evident in amyloid formation, in which high-affinity protein-protein interactions lead to formation of cytotoxic, oligomeric structures (124). To alleviate this problem, we used a bifunctional CID that binds tightly to amyloid on one end and to FKBP on the other (Fig. 6b) (125). Recruitment of FKBP to the amyloid interface blocked the interaction 40-fold better than “regular” inhibitors (125, 126). These studies established that CIDs could be used in unconventional ways to engage biological systems and achieve desired outcomes.
Conditional protein stability
The chemical dimerization platform has been adapted for several other new applications. One approach is to recruit the target protein to the proteolytic machinery using CIDs; forced engagement leads to proteosomal degradation and concurrent, conditional removal of its function (127, 128). In another approach, an intrinsically unstable FRB tag is added to the target, which causes degradation of the entire chimera (Fig. 6c) (129-131). More than 5kcal/mol of folding energy is rescued by CID-mediated recruitment of FKBP (130), and, thus, this method can be used in cultured cells, mice, and Xenopus laevis to regulate protein function conditionally (129, 130, 132). This method has proven useful in studies of developmental processes; in one recent example, unstable FRB fusions were used to learn that the kinase GSK3P is required for midline fusion during two brief times in mouse gestation (133). An improved version of this method has also been reported recently by the Wandless group; in these systems, stability of the tag can be controlled without the need for a recruited protein partner (134, 135). The Wandless group found that unstable FKBP proteins can be attached to a variety of targets and that a modified rapamycin analog, termed Shield-1, is sufficient to recover the stability of the chimeras. This simplified system has been applied to studying falcipain function in Plasmodium falciparum (136) and the phenotypes of essential genes in Toxoplasma gondii (137). The goal of these technologies is to regulate protein function at the posttranscriptional level. By targeting this stage of a protein’s lifetime, one avoids the lengthy transcriptional and translational process to generate more rapid conditional systems. In practice, these systems typically display changes in protein levels over 1 or more hours.

Figure 6. Chemical dimerization. (a) The general components of homo- and hetero-dimerization systems are shown schematically. The binding proteins (X and Y) expressed as chimeras with the targets and these fusions are brought into proximity by the bifunctional small molecule. A variety of molecules have been used as the chemical inducer of dimerization (CID; see text for details). (b) CID-based approach to inhibiting difficult protein-protein contacts; a readily available cellular protein, FKBP, is used to preclude amyloid formation sterically. (c) Inducible stabilization. These variations on the CID system involve the conditional stabilization or destabilization of one protein target. A representative system is shown; an unstable domain leads to degradation of the target, but recruitment of the binding partner recovers protein levels and function. (d) Conditional protein mislocalization is shown schematically, with the example of nuclear import used to illustrate the general components. Reconstitution of a target with an NLS favors temporary nuclear uptake, whereas removal of the small molecule reverses this process.
Conditional control over subcellular localization
Although some biological processes occur over hours, other questions may require more rapid regulation. Conditional protein mislocalization offers fast kinetics and, thus, may be suitable for these specific situations. In these systems, a CID is used to append a subcellular address signal reversibly to the protein of interest (Fig. 6d) (138, 139). Because a protein’s function is often linked to its position within the cell (e.g., a transcription factor cannot be active in the cytoplasm), this system offers rapid and selective control.
Conclusions and future directions
Chemical dimerization and three-hybrid systems are being applied to numerous different biological systems, in a variety of organisms, and in a growing number of technological variations. Likewise, the classic two-hybrid is being reimagined in new ways, such as fragment complementation. At the same time, improvements in parallel technologies, such as high-content microscopy screening, mass spectrometry, and microarray profiling, have offered new opportunities. Together, this suite of methods is being used to study and control macromolecular contacts. As chemical biologists continue to develop, refine, and reimagine two- and three-hybrid techniques, more insights will be made.
Acknowledgments
The authors thank Dennis Coles and members of our groups for comments on the manuscript.
References
1. Fields S, Song O. A novel genetic system to detect interactions. Nature 1989; 340:245-246.
2. Licitra EJ, Liu JO. A three-hybrid system for detecting small ligand-protein receptor interactions. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:12817-12821.
3. Chien CT, Bartel PL, Sternglanz R, Fields S. The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:9578-9582.
4. Walhout AJ, Boulton SJ, Vidal M. Yeast two-hybrid systems and protein interaction mapping projects for yeast and worm. Yeast 2000; 17:88-94.
5. Walhout AJ, Sordella R, Lu X, Hartley JL, Temple GF, Brasch MA, Thierry-Mieg N, Vidal M. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science 2000; 287:116-122.
6. Uetz P, Hughes RE. Systematic and large-scale two-hybrid screens. Curr. Opin. Microbiol. 2000; 3:303-308.
7. Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:4569-4574.
8. Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, et al. A map of the interactome network of the metazoan C. elegans. Science 2004; 303:540-543.
9. Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, et al. A protein interaction map of Drosophila melanogaster. Science 2003; 302:1727-1736.
10. Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005; 437:1173-1178.
11. Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005; 122:957-968.
12. Zhong J, Zhang H, Stanyon CA, Tromp G, Finley RL Jr. A strategy for constructing large protein interaction maps using the yeast two-hybrid system: regulated expression arrays and two-phase mating. Genome Res. 2003; 13:2691-2699.
13. Vermeirssen V, Deplancke B, Barrasa MI, Reece-Hoyes JS, Arda HE, Grove CA, Martinez NJ, Sequerra R, Doucette-Stamm L, Brent MR, Walhout AJ. Matrix and Steiner-triple-system smart pooling assays for high-performance transcription regulatory network mapping. Nat. Methods 2007; 4:659-664.
14. Jin F, Avramova L, Huang J, Hazbun T. A yeast two-hybrid smart-pool-array system for protein-interaction mapping. Nat. Methods 2007; 4:405-407.
15. Fields S. High-throughput two-hybrid analysis. The promise and the peril. FEBS J 2005; 272:5391-5399.
16. Serebriiskii IG, Golemis EA. Two-hybrid system and false positives. Approaches to detection and elimination. Methods Mol. Biol. 2001; 177:123-134.
17. Tong AH, Drees B, Nardelli G, Bader GD, Brannetti B, Castagnoli L, Evangelista M, Ferracuti S, Nelson B, Paoluzi S, Quondam M, Zucconi A, Hogue CW, Fields S, Boone C, Cesareni G. A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 2002; 295:321-324.
18. Wiwatwattana N, Kumar A. Organelle DB: a cross-species database of protein localization and function. Nucleic Acids Res. 2005; 33:D598-604.
19. Vidal M, Brachmann RK, Fattaey A, Harlow E, Boeke JD. Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:10315-10320.
20. Shih HM, Goldman PS, DeMaggio AJ, Hollenberg SM, Goodman RH, Hoekstra MF. A positive genetic selection for disrupting protein-protein interactions: identification of CREB mutations that prevent association with the coactivator CBP. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:13896-13901.
21. Aronheim A, Zandi E, Hennemann H, Elledge SJ, Karin M. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol. Cell Biol. 1997; 17:3094-3102.
22. Aronheim A. Improved efficiency sos recruitment system: expression of the mammalian GAP reduces isolation of Ras GTPase false positives. Nucleic Acids Res. 1997; 25:3373-3374.
23. Aronheim A, Engelberg D, Li N, al-Alawi N, Schlessinger J, Karin M. Membrane targeting of the nucleotide exchange factor Sos is sufficient for activating the Ras signaling pathway. Cell 1994; 78:949-961.
24. Isakoff SJ, Cardozo T, Andreev J, Li Z, Ferguson KM, Abagyan R, Lemmon MA, Aronheim A, Skolnik EY. Identification and analysis of PH domain-containing targets of phosphatidylinositol 3-kinase using a novel in vivo assay in yeast. EMBO J. 1998; 17:5374-5387.
25. Medici R, Bianchi E, Di Segni G, Tocchini-Valentini GP. Efficient signal transduction by a chimeric yeast-mammalian G protein alpha subunit Gpa1-Gsalpha covalently fused to the yeast receptor Ste2. EMBO J. 1997; 16:7241-7249.
26. Ray P, Pimenta H, Paulmurugan R, Berger F, Phelps ME, Iyer M, Gambhir SS. Noninvasive quantitative imaging of protein-protein interactions in living subjects. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:3105-3110.
27. Stryer L. Fluorescence energy transfer as a spectroscopic ruler. Ann. Rev. Biochem. 1978; 47:819-846.
28. Yoder TJ, McElwain MA, Francis SE, Bagley J, Muller EG, Pak B, O’Toole ET, Winey M, Davis TN. Analysis of a spindle pole body mutant reveals a defect in biorientation and illuminates spindle forces. Mol. Biol. Cell 2005; 16:141-152.
29. De A, Gambhir SS. Noninvasive imaging of protein-protein interactions from live cells and living subjects using bioluminescence resonance energy transfer. Faseb J 2005; 19:2017-2019.
30. Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat. Rev. Drug Discov. 2007; 6:569-582.
31. Johnsson N, Varshavsky A. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:10340-10344.
32. Varshavsky A. The N-end rule. Cell 1992; 69:725-735.
33. Rechsteiner M. Natural substrates of the ubiquitin proteolytic pathway. Cell 1991; 66:615-618.
34. Scheper W, Thaminy S, Kais S, Stagljar I, Romisch K. Coordination of N-glycosylation and protein translocation across the endoplasmic reticulum membrane by Sss1 protein. J. Biol. Chem. 2003; 278:37998-38003.
35. Cervantes S, Gonzalez-Duarte R, Marfany G. Homodimerization of presenilin N-terminal fragments is affected by mutations linked to Alzheimer’s disease. FEBS Lett. 2001; 505:81-86.
36. Thaminy S, Auerbach D, Arnoldo A, Stagljar I. Identification of novel ErbB3-interacting factors using the split-ubiquitin membrane yeast two-hybrid system. Genome Res. 2003; 13:1744-1753.
37. Stagljar I, Korostensky C, Johnsson N, te Heesen S. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:5187-5192.
38. Rossi F, Charlton CA, Blau HM. Monitoring protein-protein interactions in intact eukaryotic cells by beta-galactosidase complementation. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:8405-8410.
39. Wehrman TS, Casipit CL, Gewertz NM, Blau HM. Enzymatic detection of protein translocation. Nat. Methods 2005; 2:521-527.
40. Hammer MM, Wehrman TS, Blau HM. A novel enzyme complementation-based assay for monitoring G-protein-coupled receptor internalization. FASEB J. 2007; 21:3827-3834.
41. Remy I, Campbell-Valois FX, Michnick SW. Detection of protein-protein interactions using a simple survival protein- fragment complementation assay based on the enzyme dihydrofolate reductase. Nat. Protoc. 2007; 2:2120-2125.
42. Campbell-Valois FX, Michnick SW. Synthesis of degenerated libraries of the ras-binding domain of raf and rapid selection of fast-folding and stable clones with the dihydrofolate reductase protein fragment complementation assay. Methods Mol. Biol. 2007; 352:249-274.
43. Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:12288-12293.
44. Stefan E, Aquin S, Berger N, Landry CR, Nyfeler B, Bouvier M, Michnick SW. Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:16916-16921.
45. Hu CD, Chinenov Y, Kerppola TK. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 2002; 9:789-798.
46. Kerppola TK. Visualization of molecular interactions by fluorescence complementation. Nat. Rev. Mol. Cell. Biol. 2006; 7:449-456.
47. Hynes TR, Mervine SM, Yost EA, Sabo JL, Berlot CH. Live cell imaging of Gs and the beta2-adrenergic receptor demonstrates that both alphas and beta1gamma7 internalize upon stimulation and exhibit similar trafficking patterns that differ from that of the beta2-adrenergic receptor. J. Biol. Chem. 2004; 279:44101-44112.
48. Kley N. Chemical dimerizers and three-hybrid systems: scanning the proteome for targets of organic small molecules. Chem. Biol. 2004; 11:599-608.
49. Lin H, Cornish VW. In vivo protein-protein interaction assays: beyond proteins. Angew. Chem. Int. Ed. Engl. 2001; 40:871-875.
50. Lin H, Cornish VW. Screening and selection methods for large-scale analysis of protein function. Angew. Chem. Int. Ed. Engl. 2002; 41:4402-4425.
51. Becker F, Murthi K, Smith C, Come J, Costa-Roldan N, Kaufmann C, Hanke U, Degenhart C, Baumann S, Wallner W, Huber A, Dedier S, Dill S, Kinsman D, Hediger M, Bockovich N, Meier-Ewert S, Kluge AF, Kley N. A three-hybrid approach to scanning the proteome for targets of small molecule kinase inhibitors. Chem Biol 2004; 11:211-223.
52. Althoff EA, Cornish VW. A bacterial small-molecule three- hybrid system. Angew. Chem. Int. Ed. Engl. 2002; 41:2327-2330.
53. Baker K, Sengupta D, Salazar-Jimenez G, Cornish VW. An optimized dexamethasone-methotrexate yeast 3-hybrid system for high-throughput screening of small molecule-protein interactions. Anal. Biochem. 2003; 315:134-137.
54. Abida WM, Carter BT, Althoff EA, Lin H, Cornish VW. Receptor-dependence of the transcription read-out in a small- molecule three-hybrid system. ChemBioChem. 2002; 887-895.
55. de Felipe KS, Carter BT, Althoff EA, Cornish VW. Correlation between ligand-receptor affinity and the transcription readout in a yeast three-hybrid system. Biochemistry 2004; 43:10353-10363.
56. Lokey RS. Forward chemical genetics: progress and obstacles on the path to a new pharmacopoeia. Curr. Opin. Chem. Biol. 2003; 7:91-96.
57. Walsh DP, Chang YT. Chemical genetics. Chem. Rev. 2006; 106:2476-2530.
58. Strausberg RL, Schreiber SL. From knowing to controlling: a path from genomics to drugs using small molecule probes. Science 2003; 300:294-295.
59. Terstappen GC, Schlupen C, Raggiaschi R, Gaviraghi G. Target deconvolution strategies in drug discovery. Nat. Rev. Drug Discov. 2007; 6:891-903.
60. Koide K, Finkelstein JM, Ball Z, Verdine GL. A synthetic library of cell-permeable molecules. J. Am. Chem. Soc. 2001; 123:398-408.
61. Gardiner L, Coyle BJ, Chan WC, Soultanas P. Discovery of antagonist peptides against bacterial helicase-primase interaction in B. stearothermophilus by reverse yeast three-hybrid. Chem. Biol. 2005; 12:595-604.
62. Henthorn DC, Jaxa-Chamiec AA, Meldrum E. A GAL4-based yeast three-hybrid system for the identification of small molecule- target protein interactions. Biochem. Pharmacol. 2002; 63: 1619-1628.
63. Baker K, Bleczinski C, Lin H, Salazar-Jimenez G, Sengupta D, Krane S, Cornish VW. Chemical complementation: a reaction- independent genetic assay for enzyme catalysis. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:16537-16542.
64. Lin H, Tao H, Cornish VW. Directed evolution of a glycosynthase via chemical complementation. J. Am. Chem. Soc. 2004; 126:15051-15059.
65. Zang X, Loke P, Kim J, Wojnoonski K, Kusdra L, Allison JP. A genetic library screen for signaling proteins that interact with phosphorylated T cell costimulatory receptors. Genomics 2006; 88:841-845.
66. Shaywit AJ, Dove SL, Greenberg ME, Hochschild A. Analysis of phosphorylation-dependent protein-protein interactions using a bacterial two-hybrid system. Sci STKE 2002, PL11.
67. SenGupta DJ, Zhang B, Kraemer B, Pochart P, Fields S, Wickens M. (1996). A three-hybrid system to detect RNA-protein interactions in vivo. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:8496-8501.
68. Jaeger S, Eriani G, Martin F. Results and prospects of the yeast three-hybrid system. FEBS Lett. 2004; 556:7-12.
69. Konig J, Julius C, Baumann S, Homann M, Goringer HU, Feldbrugge M. Combining SELEX and the yeast three-hybrid system for in vivo selection and classification of RNA aptamers. RNA 2007; 13:614-622.
70. Piganeau N, Schauer UE, Schroeder R. A yeast RNA-hybrid system for the detection of RNA-RNA interactions in vivo. Rna 2006; 12:177-184.
71. Riley KJ, Cassiday LA, Kumar A, Maher LJ 3rd. Recognition of RNA by the p53 tumor suppressor protein in the yeast three-hybrid system. Rna 2006; 12:620-630.
72. Bieniasz PD, Grdina TA, Bogerd HP, Cullen BR. Analysis of the effect of natural sequence variation in Tat and in cyclin T on the formation and RNA binding properties of Tat-cyclin T complexes. J. Virol. 1999; 73:5777-5786.
73. Bernstein DS, Buter N, Stumpf C, Wickens M. Analyzing mRNA-protein complexes using a yeast three-hybrid system. Methods 2002; 26:123-141.
74. Martin F, Michel F, Zenklusen D, Muller B, Schumperli D. Positive and negative mutant selection in the human histone hairpin-binding protein using the yeast three-hybrid system. Nucleic Acids Res. 2000; 28:1594-1603.
75. Hook B, Bernstein D, Zhang B, Wickens M. RNA-protein interactions in the yeast three-hybrid system: affinity, sensitivity, and enhanced library screening. Rna 2005; 11:227-233.
76. Caligiuri M, Molz L, Liu Q, Kaplan F, Xu JP, Majeti JZ, Ramos-Kelsey R, Murthi K, Lievens S, Tavernier J, Kley N. MASPIT: three-hybrid trap for quantitative proteome fingerprinting of small molecule-protein interactions in mammalian cells. Chem. Biol. 2006; 13:711-722.
77. Parrish JR, Gulyas KD, Finley RL Jr. Yeast two-hybrid contributions to interactome mapping. Curr. Opin. Biotechnol. 2006; 17:387-93.
78. Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom-Cerajewski L, Robinson MD, O’Connor L, Li M, Taylor R, et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol. Syst. Biol. 2007; 3:89.
79. Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002; 415:141-147.
80. Ho Y, Grahler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 2002; 415:180-183.
81. Espejo A, Cote J, Bednarek A, Richard S, Bedford MT. A protein-domain microarray identifies novel protein-protein interactions. Biochem. J. 2002; 367:697-702.
82. Schweitzer B, Predki P, Snyder M. Microarrays to characterize protein interactions on a whole-proteome scale. Proteomics 2003; 3:2190-2199.
83. Zhu H, Snyder M. Protein arrays and microarrays. Curr. Opin. Chem. Biol. 2001; 5:40-45.
84. Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, Mitchell T, Miller P, Dean RA, Gerstein M, Snyder M. Global analysis of protein activities using proteome chips. Science 2001; 293:2101-2105.
85. MacBeath G, Schreiber SL. Printing proteins as microarrays for high-throughput function determination. Science 2000; 289:1760-1763.
86. Bradner JE, McPherson OM, Mazitschek R, Barnes-Seeman D, Shen JP, Dhaliwal J, Stevenson KE, Duffner JL, Park SB, Neuberg DS, Nghiem P, Schreiber SL, Koehler AN. A robust small-molecule microarray platform for screening cell lysates. Chem. Biol. 2006; 13:493-504.
87. Pilobello KT, Slawek DE, Mahal LK. A ratiometric lectin microarray approach to analysis of the dynamic mammalian glycome. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:11534-11539.
88. Ko KS, Jaipuri FA, Pohl NL. Fluorous-based carbohydrate microarrays. J Am Chem Soc 2005; 127:13162-13163.
89. Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Controlling signal transduction with synthetic ligands. Science 1993; 262:1019-1024.
90. Holsinger LJ, Spencer DM, Austin DJ, Schreiber SL, Crabtree GR. Signal transduction in T lymphocytes using a conditional allele of Sos. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:9810-9814.
91. Schreiber SL, Crabtree GR. Immunophilins, ligands, and the control of signal transduction. Harvey Lect. 1995; 91:99-114.
92. Belshaw PJ, Ho SN, Crabtree GR, Schreiber SL. Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:4604-4607.
93. Crabtree GR, Schreiber SL. Three-part inventions: intracellular signaling and induced proximity. Trends Biochem. Sci. 1996; 21:418-422.
94. Banaszynski LA, Wandless TJ. Conditional control of protein function. Chem. Biol. 2006; 13:11-21.
95. Clackson T. Dissecting the functions of proteins and pathways using chemically induced dimerization. Chem. Biol. Drug Des. 2006; 67:440-442.
96. Clackson T. Controlling Protein-Protein Interactions Using Chemical Inducers and Disrupters of Dimerization. 2007. Wiley CH Verlag GmbH & Co, New York.
97. Gestwicki JE, Marinec PS. Chemical control over protein-protein interactions: beyond inhibitors. Comb. Chem. High Throughput Screen. 2007; 10:667-675.
98. Farrar MA, Alberol I, Perlmutter RM. Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature 1996; 383:178-181.
99. Mizuguchi R, Hatakeyama M. Conditional activation of Janus kinase (JAK) confers factor independence upon interleukin-3- dependent cells. Essential role of Ras in JAK-triggered mitogenesis. J. Biol. Chem. 1998; 273:32297-32303.
100. Gendreizig S, Kindermann M, Johnsson K. Induced protein dimerization in vivo through covalent labeling. J. Am. Chem. Soc. 2003; 125:14970-14971.
101. Hussey SL, Muddana SS, Peterson BR. Synthesis of a beta-estradiol-biotin chimera that potently heterodimerizes estrogen receptor and streptavidin proteins in a yeast three-hybrid system. J. Am. Chem. Soc. 2003; 125:3692-3693.
102. Neddermann P, Gargioli C, Muraglia E, Sambucini S, Bonelli F, De Francesco R, Cortese R. A novel, inducible, eukaryotic gene expression system based on the quorum-sensing transcription factor TraR. EMBO Rep. 2003; 4:159-165.
103. Gruber S, Arumugam P, Katou Y, Kuglitsch D, Helmhart W, Shirahige K, Nasmyth K. Evidence that loading of cohesin onto chromosomes involves opening of its SMC hinge. Cell 2006; 127:523-537.
104. Karpova AY, Tervo DG, Gray NW, Svoboda K. Rapid and reversible chemical inactivation of synaptic transmission in genetically targeted neurons. Neuron 2005; 48:727-735.
105. Terrillon S, Bouvier M. Receptor activity-independent recruitment of betaarrestin2 reveals specific signalling modes. EMBO J 2004; 23:3950-3961.
106. Pownall ME, Welm BE, Freeman KW, Spencer DM, Rosen JM, Isaacs HV. An inducible system for the study of FGF signalling in early amphibian development. Dev. Biol. 2003;256:89-99.
107. Welm BE, Freeman KW, Chen M, Contreras A, Spencer DM, Rosen JM. Inducible dimerization of FGFR1: development of a mouse model to analyze progressive transformation of the mammary gland. J. Cell. Biol. 2002; 157:703-714.
108. Freeman KW, Gangula RD, Welm BE, Ozen M, Foster BA, Rosen JM, Ittmann M, Greenberg NM, Spencer DM. Conditional activation of fibroblast growth factor receptor (FGFR) 1, but not FGFR2, in prostate cancer cells leads to increased osteopontin induction, extracellular signal-regulated kinase activation, and in vivo proliferation. Cancer Res. 2003; 63:6237-6243.
109. Li B, Sun A, Youn H, Hong Y, Terranova PF, Thrasher JB, Xu P, Spencer D. Conditional Akt activation promotes androgen-independent progression of prostate cancer. Carcinogenesis 2007; 28:572-583.
110. Inoue T, Heo WD, Grimley JS, Wandless TJ, Meyer T. An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat. Methods 2005; 2:415-418.
111. Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, Meyer T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 2006; 314:1458-1461.
112. Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 2006; 314:1454-1457.
113. Belshaw PJ, Spencer DM, Crabtree GR, Schreiber SL. Controlling programmed cell death with a cyclophilin-cyclosporin-based chemical inducer of dimerization. Chem. Biol. 1996; 3:731-738.
114. Mallet VO, Mitchell C, Guidotti JE, Jaffray P, Fabre M, Spencer D, Arnoult D, Kahn A, Gilgenkrantz H. Conditional cell ablation by tight control of caspase-3 dimerization in transgenic mice. Nat. Biotechnol 2002; 20:1234-1239.
115. Graveley BR. Small molecule control of pre-mRNA splicing. Rna 2005; 11:355-358.
116. Schlatter S, Senn C, Fussenegger M. Modulation of translation-initiation in CHO-K1 cells by rapamycin-induced heterodimerization of engineered eIF4G fusion proteins. Biotechnol. Bioeng. 2003; 83:210-225.
117. Kohler JJ, Bertozzi CR. Regulating cell surface glycosylation by small molecule control of enzyme localization. Chem. Biol. 2003; 10:1303-1311.
118. Briesewitz R, Ray GT, Wandless TJ, Crabtree GR. Affinity modulation of small-molecule ligands by borrowing endogenous protein surfaces. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:1953-1958.
119. Braun PD, Barglow KT, Lin YM, Akompong T, Briesewitz R, Ray GT, Haldar K, Wandless TJ. A bifunctional molecule that displays context-dependent cellular activity. J. Am. Chem. Soc. 2003; 125:7575-7580.
120. Rivera VM, Wang X, Wardwell S, Courage NL, Volchuk A, Keenan T, Holt DA, Gilman M., Orci L, Cerasoli F Jr, Rothman JE, Clackson T. Regulation of protein secretion through controlled aggregation in the endoplasmic reticulum. Science 2000; 287:826-830.
121. Johnston J, Tazelaar J, Rivera VM, Clackson T, Gao GP, Wilson JM. Regulated expression of erythropoietin from an AAV vector safely improves the anemia of beta-thalassemia in a mouse model. Mol Ther 2003; 7:493-497.
122. Buskirk AR, Liu DR Creating small-molecule-dependent switches to modulate biological functions. Chem. Biol. 2005; 12:151-161.
123. Berg T. Modulation of protein-protein interactions with small organic molecules. Angew. Chem. Int. Ed. Engl. 2003; 42:2462-2481.
124. Selkoe DJ. Folding proteins in fatal ways. Nature 2003; 426:900-904.
125. Gestwicki JE, Crabtree GR, Graef IA. Harnessing chaperones to generate small-molecule inhibitors of amyloid beta aggregation. Science 2004; 306:865-869.
126. Bose M, Gestwicki JE, Devasthali V, Crabtree GR, Graef IA. ‘Nature-inspired’ drug-protein complexes as inhibitors of Abeta aggregation. Biochem. Soc. Trans. 2005; 33:543-547.
127. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U.S.A. 2001; 98, 8554-8559.
128. Janse DM, Crosas B, Finley D, Church GM. Localization to the proteasome is sufficient for degradation. J. Biol. Chem. 2004; 279:21415-21420.
129. Stankunas K, Bayle JH, Gestwicki JE, Lin YM, Wandless TJ, Crabtree GR. Conditional protein alleles using knockin mice and a chemical inducer of dimerization. Mol. Cell 2003; 12:1615-1624.
130. Stankunas K, Bayle JH, Havranek JJ, Wandless TJ, Baker D, Crabtree GR, Gestwicki JE. Rescue of Degradation-Prone Mutants of the FK506-Rapamycin Binding (FRB) Protein with Chemical Ligands. Chembiochem 2007; 8:1162-1169.
131. Edwards SR, Wandless TJ. The rapamycin-binding domain of the protein kinase mammalian target of rapamycin is a destabilizing domain. J. Biol. Chem. 2007; 282:13395-13401.
132. Liu KJ, Gestwicki JE, Crabtree GR. Bringing Small Molecule Regulation of Protein Activity to Developmental Systems. 2007. Taylor & Francis, Oxford, UK.
133. Liu KJ, Arron JR, Stankunas K, Crabtree GR, Longaker MT. Chemical rescue of cleft palate and midline defects in conditional GSK-3beta mice. Nature 2007; 446:79-82.
134. Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 2006; 126:995-1004.
135. Maynard-Smith LA, Chen LC, Banaszynski LA, Ooi AG, Wandless TJ. A directed approach for engineering conditional protein stability using biologically silent small molecules. J. Biol. Chem. 2007; 282:24866-24872.
136. Armstrong CM, Goldberg DE. An FKBP destabilization domain modulates protein levels in Plasmodium falciparum. Nat. Methods 2007; 4:1007-1009.
137. Herm-Gotz A, Agop-Nersesian C, Munter S, Grimley JS, Wandless TJ, Frischknecht F, Meissner M. Rapid control of protein level in the apicomplexan Toxoplasma gondii. Nat. Methods. 2007; 4:1003-1005
138. Klemm JD, Beals CR, Crabtree GR. Rapid targeting of nuclear proteins to the cytoplasm. Curr Biol 1997; 7:638-644.
139. Bayle JH, Grimley JS, Stankunas K, Gestwicki JE, Wandless TJ, Crabtree GR. Rapamycin analogs with differential binding specificity permit orthogonal control of protein activity. Chem. Biol. 2006; 13:99-107.
Further Reading
The Saccharomyces Genome Database (www.yeastgenome.org) details an extensive collection of two-hybrid screens and other large-scale protein interaction studies.
Ariad Pharmaceuticals maintains a useful and comprehensive database of CID-related references (http://www.ariad.com/wt/page/kits_references). They also have made ARGENT homo- and hetero-dimerizer systems available for research use.
See Also
Protein Interaction Networks, Chemical Tools to Elucidate
Protein-Protein Interactions
Array-Based Techniques for Proteins
Mass Spectrometry (MS) for Proteins and Protein Complexes
Forward Chemical Genetics