CHEMICAL BIOLOGY
Cellular Membranes, Dynamics of
Anne K. Kenworthy, Vanderbilt University School of Medicine, Nashville, Tennessee
doi: 10.1002/9780470048672.wecb064
Cell membranes are two-dimensional fluids that exhibit a wide range of dynamic behaviors. Recent technical advances have enabled unprecedented views of membrane dynamics in living cells. In this technical review, we provide a brief overview of three well-studied examples of membrane dynamics: lateral diffusion of proteins and lipids in the plane of the membrane, vesicular trafficking between intracellular compartments, and exchange of proteins on and off membranes. We then discuss experimental approaches to monitor membrane protein and lipid dynamics, and we place a special emphasis on the use of genetically encoded fluorescent probes and live cell-imaging techniques.
The concept that membranes are fluid, dynamic structures is now over 35 years old (1, 2). In this review, we describe three of the best-studied examples of cell membrane dynamics—lateral diffusion of proteins and lipids within the plane of the bilayer, membrane trafficking between intracellular compartments, and cycling of proteins on and off membranes—along with the recent technical advances that have enabled researchers to visualize these motions directly within living cells. In particular, we describe how the use of green fluorescent protein (GFP) from Aequorea Victoria and other, more recently developed labeling technologies can be used to mark molecules to study inside cells. We also summarize common fluorescence microscopy techniques for live cell imaging, including conventional techniques such as wide-field and confocal microscopy, and more specialized techniques such as total internal reflection fluorescence (TIRF) microscopy. Finally, we cover advanced methods used to study cell membrane dynamics, including single particle tracking (SPT), fluorescence recovery after photobleaching (FRAP), photoactivation, fluorescence correlation spectroscopy (FCS) and image correlation spectroscopy (ICS).
Types of Cell Membrane Dynamics
Lateral diffusion
Membranes are two-dimensional fluids whose protein and lipid components continuously exchange positions because of Brownian motion, a process commonly referred to as lateral diffusion. Lateral diffusion enables proteins and lipids to explore their environment, which encourages interactions between molecules. Thus, the speed of lateral diffusion is one of the limiting factors that regulate intermolecular interactions, and, consequently, cellular function. Diffusional mobility of molecules can be estimated based on their size and the viscosity of the lipid bilayer and surrounding aqueous phase. As a result, deviations from this behavior can provide important insights into the environment that proteins and lipid encounter in biologic membranes.
How rapidly diffusion occurs is characterized by the diffusion coefficient D, a parameter that provides a measure of the mean of the squared displacement x of a molecule per unit time t. For diffusion in two dimensions such as a membrane, this is given by
Interestingly, the diffusional behavior of membrane proteins measured experimentally by FRAP, FCS, or single particle tracking in cells is more complex than predicted by this model. This technique is described best for the case of cell surface proteins, as assessed by FRAP. Such measurements indicate that diffusion is typically much slower than one would expect based on membrane viscosity. In cell membranes, typical values of D for transmembrane proteins are approximately 0.05 μm2/s or less, which is much slower than observed in artificial membranes composed of purified lipids. In addition, a significant fraction of proteins is often immobile over the timescale of diffusion experiments (4, 5). Furthermore, diffusional mobilities vary among proteins, and sometimes they differ for the same protein expressed in different cell lines (4, 5). Deviations from pure diffusion are more readily apparent when the trajectories of single molecules are analyzed (Fig. 1). Individual molecules exhibit a range of diffusive behaviors, characterized as immobilization, transient confinement, free diffusion, and in some cases directed motion (6).
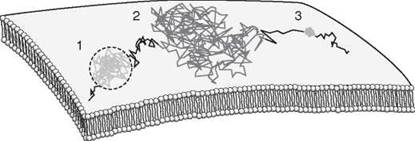
Figure 1. Modes of diffusion of individual membrane proteins as revealed by single-molecule tracking techniques. The hypothetical trajectory of an individual plasma membrane protein as traced by single-particle tracking techniques is shown. An individual protein can switch between several different modes of over time, which include confined diffusion (region 1), free diffusion (region 2), and immobilization (region 3).
Features of cell membranes that contribute to this complex diffusional behavior include the presence of membrane domains, interactions of proteins with the cytoskeleton and extracellular matrix, and molecular crowding because of the high protein concentrations found in cellular membranes (4, 5). The diffusional mobility of proteins and lipids can also be modulated actively. For example, crosslinking of certain cell surface molecules causes dramatic changes in their mobility, which reflects interactions with other cellular components. A well-studied example of such a protein is FcsR1, the high affinity IgE receptor, which undergoes a substantial loss of mobility on the formation of small aggregates of IgE-FcsR1 (7). Using an advanced technique known as FCS cross correlation analysis, it is now possible to detect transient interactions of membrane proteins that occur during cell signaling by virtue of their codiffusion (8).
Until recently, experiments that probe protein diffusion relied on fluorescent antibody-based probes, and thus were limited to plasma membrane proteins with extracellular epitopes. In contrast, the mechanisms that regulate diffusion of intracellular membrane proteins remained unexplored because of their inaccessibility to labeling. With the development of genetically encoded fluorescent probes, such studies have become tractable because proteins targeted to a particular organelle can be fluorescently labeled directly with GFP. In addition, improved technology has now made it possible to monitor the movement of multiple types of proteins or lipids, tagged with different markers simultaneously. Some examples of the types of questions it is now possible to address include:
How is protein diffusional mobility regulated in intracellular compartments?
Some of the first investigations of diffusion in organelle membranes such as the Golgi complex and endoplasmic reticulum suggest that unlike the plasma membrane, protein diffusion in intracellular membranes approaches theoretic limits (9). These findings suggest that intracellular membranes contain considerably fewer barriers to free diffusion than the cell surface. Diffusion studies can also be used to test whether protein immobilization is responsible for retaining proteins within a specific organelle, the effect of unfolding on protein mobility, and how various perturbations influence both the luminal and membrane environment experienced by proteins (10).
Is the mobility of proteins influenced by how they are attached to membranes?
Lateral diffusion is predicted to be relatively insensitive to the size of the transmembrane domain of the diffusing species. This prediction suggests that membrane anchorage should not strongly influence diffusion. However, membrane anchorage would be expected to influence the types of interactions that occur between a given protein and the complex environment of the cell. For example, the diffusion of peripheral membrane proteins localized to the inner leaflet of the plasma membrane by lipid anchors has been shown to be much faster than transmembrane proteins and nearly as fast as that of lipid probes in some instances (11). This finding suggests that because of their lipid anchors, these molecules do not experience the same barriers to diffusion as transmembrane proteins. It also likely reflects the ability of some proteins to undergo rapidly reversible binding to membranes (12, 13).
Do cholesterol-enriched lipid domains organize proteins into functional complexes?
Interest in the role of membrane domains in regulating protein and lipid diffusion has recently been revitalized by the lipid raft model, which proposes that cell membranes are divided into cholesterol and sphingolipid-enriched and -depleted microdomains. As a result, much effort has been made recently to relate lipid and protein diffusion in cells and model membranes (14) and to determine the effect of cholesterol levels on protein and lipid diffusion by both FRAP and FCS (11, 15). The role of cholesterol-dependent domains in regulating events such as T-cell signaling has also been investigated by examining closely the mechanisms of diffusional trapping of proteins at the single molecule level by TIRF microscopy; unexpectedly, protein-protein interactions seem to play a more important role than cholesterol-dependent domains in this process (16).
Membrane trafficking
Although integral membrane proteins and lipids are laterally mobile within the plane of a given cell membrane, they cannot exchange between different membrane compartments unaided because of the high energetic barriers to exposing their hydrophobic portions to water. The movement of transmembrane proteins between cellular compartments is accomplished by the formation of closed bilayer structures known as transport vesicles that bud off from one compartment and fuse with another. The term “membrane trafficking” refers to the movement of these vesicular structures between organelles and/or the plasma membrane, and includes the processes of endocytosis and exocytosis.
Membrane trafficking involves many highly regulated events, which include cargo selection, recruitment of coat and accessory proteins, vesicle budding, intra-organelle transport, targeting to and fusion with the target membrane, and recycling of cellular machinery for another cycle of transport (17). Recent work has now begun to focus on the temporal and spatial regulation of these events in living cells (18-20). Such approaches are especially powerful when coupled with cell biologic manipulations such as drug treatments, the introduction of mutant proteins into cells, or knockdown of protein expression to probe the cellular mechanisms that underlie membrane trafficking (20). Examples of the types of questions being addressed are listed in the following sections.
What are the kinetics of intracellular transport through the secretory pathway?
The transmembrane protein VSVG is a classic tool used to study protein transport through the secretory pathway. Some early studies of vesicular transport in living cells visualized the movement of a GFP-tagged version of VSVG by time-lapse confocal microscopy (21, 22). From these data, it was possible to derive a kinetic model of the movement of this well-studied protein between intracellular compartments, while at the same time providing information about the nature of the vesicular structures involved in this process (21, 22).
What pathways do pathogens use to enter cells?
Certain viruses and bacterial toxins are endocytosed by a specialized pathway that involves invaginations of the plasma membrane known as caveolae. Recent studies have traced out this caveolar endocytic pathway by jointly tagging the cellular machinery with GFP and labeling viruses or bacterial toxins with fluorescent dyes, which enables their dual visualization in real time (23, 24). Such work has revealed several unanticipated properties of this pathway, such as the ability of viruses to induce formation of actin comets that propel virus-containing vesicles (23) and the stable, immobile nature of the caveolar coat that encases these vesicles (24).
How do cargo molecules and trafficking machinery progress through the endocytic pathway?
Movement of cargo through the endocytic pathway could occur by vesicular transport between stable compartments. Alternatively, it could involve progressive maturation of endosomal membrane. Recent work has tested these models by using fast, live cell imaging in combination with tools to quantify dynamic changes in the levels of the small GTPases Rab5 and Rab7 on endosomal structures during transport of fluorescently labeled cargo (25). These data suggest a model in which conversion in Rab proteins is a mechanism by which cargo progresses between early and late endosomes. Other studies have focused on how specific cargo molecules that enter the cell via identical pathways, such as cholera toxin and SV40, ultimately are sorted to various intracellular destinations. Sorting requires the pH-dependent release of cholera toxin from caveolar domains in endosomes; SV40 remains immobilized under these conditions (24).
Cycling of proteins on and off membranes
Although integral membrane proteins must rely on membrane trafficking as their sole mechanism for traversing the cell, many peripheral membrane proteins can move on and off membranes in a reversible manner by shifting between a membrane-bound and a soluble state. This reversible binding is critical for the function of coat proteins involved in the formation of transport vesicles (clathrin, COPI, and COPII), proteins that play roles in organelle identity and membrane trafficking (Rabs and Arf proteins), and a wide variety of signaling proteins (Ras, Raf, and protein kinase Cy) (26-28). Biochemically, such exchange can be detected by fractionating cells into membrane and soluble fractions, and quantifying the amount of protein found in each. Using live cell imaging approaches, it is possible to begin to monitor these events in real time to address questions such as the following.
On what cellular membranes does exchange occur?
One major advantage to studying membrane/cytosol exchange of proteins in living cells is that it is possible to compare the recruitment of proteins with different organelles simultaneously. For example, such studies have been instrumental in showing that Ras activation, as reported by the membrane recruitment of the FP-tagged Ras binding domain of Raf from the cytosol, occurs on Golgi membranes as well as the cell surface, and that the kinetics of recruitment are different for the two compartments (29).
How rapidly do proteins cycle on and off membranes?
Compared with membrane trafficking, which occurs with characteristic rates of 3% per minute for the case of VSVG movement from the endoplasmic reticulum to the Golgi complex (22), the cycling of proteins on and off membranes can occur over very rapidly. In our own work (30), we have observed in photobleaching studies of GFP fusion proteins that a mutant form of HRas that lacks both palmitoylation sites is able to constitutively cycle on and off membranes of the Golgi complex with halftimes of ~5 s (Fig. 2).

Figure 2. Visualization of reversible protein binding to the Golgi complex. (a) COS-7 cells that express a GFP-tagged HRas palmitoylation mutant were imaged over time before and after photobleaching fluorescent molecules localized to the Golgi complex (circle). Fluorescence recovers rapidly and completely within 1 min. Bar, 10 pm. (b) Kinetics of recovery for the GFP-HRas palmitolyation mutant after photobleaching the entire Golgi complex. Data are from a representative experiment similar to that shown in panel A. The rapid recovery kinetics are highly suggestive of reversible membrane binding. (Reproduced from Reference 30. Copyright 2005 Rockefeller University Press.)
Is exchange constitutive or regulated?
Membrane/cytosol exchange is sometimes constitutive, but more often it occurs in a tightly regulated manner. Many proteins are recruited to membranes in response to the transient generation of protein or lipid binding sites on the membrane, which are recognized by modular protein interacting or lipid interacting domains contained within the recruited protein. Alternatively, membrane binding can be regulated by loss of membrane binding sites or perturbation of binding motifs within proteins. For example, recent evidence suggests that KRas, which contains a cluster of basic amino acids that facilitates its binding to the plasma membrane, can be released via phosphorylation of residues in the polybasic domain. This release in turn allows for the regulated redistribution of KRas to mitochondria, where it promotes apoptosis (31).
Chemical Tools and Techniques to Study Cell Membrane Dynamics
Probes
Fluorescent probes of membrane dynamics fall into two general classes: exogenous and genetically encoded. Exogenous probes, which include small organic dyes and quantum dots, usually are targeted to the protein of interest via immunolabeling approaches. They are used commonly to label cell surface proteins, although they can be introduced into cells by microinjection or by permeabilizing cells. Genetic approaches make use of intrinsically fluorescent proteins (FP) or genetic tags that generate binding sites for small dyes. Here, molecular biology tools are used to engineer DNA constructs that contain fusions of the protein of interest and the genetic tag. The DNA is then introduced into cells by transfection or transduction (Fig. 3).

Figure 3. Size of typical probes used in studies of membrane dynamics. Relative sizes of (a) Immunoglobulin G, (b) green fluorescent protein, (c) the Discoma red fluorescent protein (DsRed) tetramer, and (d) biarsenical tetracysteine. (Reprinted with permission from Reference 61.)
Small organic dyes
Small organic dyes are available with a wide range of spectral properties encompassing the entire visible range, including the near UV and far red (32). Prior to the advent of FPs, small organic dyes were the workhorses for fluorescent labeling of plasma membrane proteins, either by using dye-labeled antibodies (Fig. 3a) or by labeling proteins directly that bind to plasma membrane proteins or lipids such as bacterial toxins or growth factors. Indeed, most early work that defines the diffusional mobility of proteins on the plasma membrane relied on immunodetection of cell surface proteins with antibody fragments labeled covalently with fluorescent dyes (5). The use of small organic dyes to label proteins fluorescently remains a valuable tool to study the endocytic itineraries of ligands, viruses, and bacterial toxins (24). However, dye-labeled proteins have historically been much less useful in studies of intracellular proteins in living cells, because this task requires their introduction by either microinjection or permeabilization of the plasma membrane. The susceptibility of many fluorescent dyes to photobleaching also limits their use in live cell imaging applications.
Probes of lipid dynamics
Synthetic lipid analogs such as Dil and BODIPY-labeled lipids are classic reporters of lipid dynamics (32, 33). Fluorescent glycolipid analogs have also been used for many years, especially in studies of endocytosis and lipid trafficking (34). However, it is important to note the limitations of these probes. Many make use of short-chain analogs to enable their delivery into cells. In addition, some fluorophores used to label these lipids are used to replace a fatty acid chain. As a result, these lipids may not exhibit behavior similar to that of their native counterparts. To address these limitations, polyene-lipids, which have a structure similar to that of natural lipids, have been developed recently as an alternative to classic fluorescent lipid analogs (35). Fluorescent cholesterol analogs have joined the growing list of fluorescent lipid probes (36). Of these, dihydroergosterol is most likely to be a useful structural analog of cholesterol.
An alternative approach to the use of fluorescent lipid analogs to study lipid dynamics is the use of fluorescent reporter proteins that bind specific lipids. Such experiments are also of interest from the biologic standpoint of protein function. For example, plekstrin homolog (PH)-domain-containing proteins bind phosphoinositide lipids (26, 27), whereas cholera toxin binds ganglioside GM1 (20). It should be noted however that protein reporters that interact with a specific lipid might not be “neutral” from a biologic point of view, as they may compete for binding with endogenous molecules.
GFP and its derivatives
Many recent advances in the study of cell membrane dynamics (and indeed in much of cell biology) have been driven by the discovery of green fluorescent protein from Aequorea Victoria (37). Importantly, expression of GFP as part of a fusion protein results in visible green fluorescence, and despite its relatively large size (27kDa), GFP tagging often does not interfere with normal protein targeting or function. Since the initial discovery and cloning of GFP, mutational analysis has been performed to increase its brightness, photostability, and speed of folding as well as to generate a range of spectral variants. Of particular importance to the study of membrane proteins was the discovery of a point mutation that prevents the weak tendency of GFP to dimerize (38). Similar proteins were isolated from reef corals and sea anemones, which expanded the spectrum of available colors. Photoactivatable fluorescent proteins undergo a substantial change in their spectral properties (switching either to a dark state or to another color) in response to irradiation with light of a particular wavelength, intensity, and duration. This effect can be either reversible or irreversible depending on the particular protein (39). The recently discovered “KEIMA” protein (40) is a very promising fluorescent protein that provides an easy way to analyze dynamics of cellular membranes by simultaneously following two fluorescent-tagged subsets of proteins. Additional properties of the fluorescent proteins are described in Reference 41.
Despite the many attractive features of fluorescent fusion proteins, they are not without their limitations. First, GFP is relatively large (27 kDa) and thus has the potential to disrupt the structure, function, and/or localization of the protein to which it is linked (Fig. 3b). Second, the expression of fusion proteins typically relies on transfection, which can be problematic in some primary cell lines and can lead to overexpression artifacts. Third, certain fluorescent proteins form obligate oligomers (Fig. 3c). Finally, spectral overlap limits the number of different fluorescent proteins that can be used in the same experiment, although this problem is diminishing as new variants are developed.
Chemical labeling of fusion proteins
Recently, methods have been developed to label proteins site-specifically with small molecules for live cell imaging studies as an alternative to the use of FP fusion proteins (42-44). The strategy of these approaches relies on genetically incorporating a “receptor” domain that can serve as a specific binding site for a small molecule to the protein of interest. After expressing the fusion protein in cells, it can then be labeled using cell-permeant small molecule probes. This general scheme offers several major advantages over FP fusion proteins, which include 1) the potential to label proteins with relatively small, and thus in principle, minimally perturbing tags; 2) the possibility of taking advantage of small molecule probes with a wide range of chemical properties; and 3) the ability to control the time at which the proteins of interest are tagged, which allows for temporal regulation of labeling.
The tetracysteine-biarsenical system is one of the first examples of such technology (45). Here, a 12-residue sequence that includes four cysteines is incorporated into the protein of interest to enable binding of membrane permeant biarsenic dyes (FlAsH and ReAsH) (42). In addition to the small size of the tetracysteine motif (Fig. 3d), advantages of this approach include the possibility to perform correlative electron microscopy analysis and pulse-chase labeling (42). Furthermore, the fluorescence intensity of the bisarsenic dyes increases substantially on binding, which decreases background fluorescence. However, the biarsenical dyes exhibit nonspecific binding to cysteine-rich proteins and require a reducing environment for labeling. Compared with the FP, this and other methods that combine genetic tags with small molecules are still in their infancy, but with additional iterations of refinement, they are likely to become useful in the future.
Quantum dots
Quantum dots are fluorescent semiconductor nanocrystals that have been incorporated recently into the toolbox of fluorescent labeling techniques (42). When coated appropriately, quantum dots can be conjugated with streptavidin or antibodies for protein labeling applications in cells (46). Quantum dots are exceptionally bright and photostable, -have a broad absorbance spectrum, and can be tuned to emit at specific wavelengths depending on their size. Because of their brightness and photostability, they can be very useful for detecting low abundance proteins and are attractive probes for long-term single particle tracking studies. However, their applications in live cells are hampered somewhat by their large size (~10 to 30 nm) and “blinking” behavior. For example, the size of quantum dots has limited their use primarily to studies of extracellularly localized plasma membrane proteins because their introduction into cells requires membrane permeabilization. The potential for multivalent binding of quantum dots is also a potential concern in single-molecule tracking studies.
Fluorescence microscopy techniques for live cell imaging
Many dynamic processes, such as membrane trafficking and regulated cycling of proteins on and off membranes, can be visualized by capturing sequential images of living cells by fluorescence microscopy. The forms of fluorescence microscopy used most commonly for live cell imaging are wide field and confocal microscopy. In wide field microscopy, fluorescence is typically excited with an arc lamp and emission is collected using a CCD camera. Such systems can be configured readily for imaging live cells over time. An advantage of this approach is its relative simplicity compared with other imaging modalities. However, it collects light emitted from the entire depth of the specimen including out-of-plane fluorescence.
In confocal microscopy, out of plane fluorescence is eliminated by the incorporation of a pinhole in the light path, which enables the collection of three-dimensionally resolved images. This task is accomplished by using a single pinhole for the case of laser scanning confocal microscopy or with a series of rotating pinholes in spinning disk confocal microscopy. Because the intensity and position of the laser used to excite samples can be modulated rapidly in many laser scanning confocal microscopes, it is possible to use them for specialized applications such as photobleaching and photoactivation (see below). Spinning disk confocal microscopes are not well suited for photobleaching or photoactivation applications, but they have a greater image acquisition rate than laser scanning confocals.
In addition to confocal microscopy, a technique known as multiphoton microscopy can also be used to generate three-dimensionally resolved fluorescence images. In this case, excitation is limited to those fluorophores that are present within the small region where the laser is focused and thus are of sufficiently high power to enable a single fluorophore to absorb two or more photons simultaneously (47). Other advantages of multiphoton microscopy include reduced photobleaching outside of the focal plane as well as increased penetration into samples because of the use of longer wavelength excitation, which makes this a technique of choice for tissue imaging in vivo.
In some instances, it is of interest to focus exclusively on events that occur at the plasma membrane. Here, a technique known as TIRF microscopy is particularly valuable (48). TIRF uses an evanescent wave generated by a process referred to as total internal reflection to excite the sample. Total internal reflection occurs when light traveling from a medium of high refractive index arrives at an interface with a medium of lower refractive index above a so-called critical angle. Under these conditions, the light is reflected back into the high refractive index material, and an evanescent field is generated in the lower refractive index medium. The field decays exponentially away from the interface, with a typical depth between 50 to 150 nm. Because experimentally, this interface represents the surface at which cells attach to a coverslip, it is possible to selectively excite and visualize only those fluorescent molecules found at this surface. TIRF can also be combined with the advanced methods described in more detail below, which include single molecule imaging (16), FCS (49), and FRAP (50). Thus, TIRF offers an attractive imaging modality for probing plasma membrane dynamics using a variety of techniques. For a more in depth discussion of the pros and cons of each of these approaches in live cell imaging studies, we refer the reader to several recent reviews (51, 52).
Advanced techniques used to study membrane dynamics
FRAP
FRAP (also known as fluorescence photobleaching recovery or FPR) has been used for many years to study lateral diffusion of plasma membrane proteins as characterized by their diffusion coefficient and mobile fraction (5, 53). In these experiments, a population of fluorescence molecules is bleached irreversibly by exciting with an intense, focused laser spot. Recovery of fluorescence in the bleached region is then monitored over time to determine to what extent and how rapidly the bleached molecules are replaced by unbleached molecules from other regions within the cell (Fig. 4a). Two fundamental assumptions that underlie these experiments are that the bleaching event does not damage the labeled protein or surrounding region of the cell and that the bleaching is irreversible, in other words, that recovery occurs because of diffusional exchange and not by recovery of fluorescence of an individual fluorophore. FRAP curves are typically fit by equations for free diffusion plus an immobile fraction (53). A variation on this approach is to measure FRAP as a function of spot size, which is a technique that is sensitive to the cycling of proteins on and off membranes (13).
Until the development of the fluorescent proteins, FRAP measurements were confined to measurements of protein or lipid diffusion in the plasma membrane using fluorescently labeled Fabs or fluorescent lipid analogs that could be added to cells exogenously. For such experiments, little need existed for spatially resolved measurements because the fluorescence signal was localized to the plasma membrane. Thus, most classic FRAP studies made use of a spot photobleaching apparatus that consisted of an epifluorescence microscope, computer-controlled shutter, laser used both for bleaching and for low-level excitation of the sample, and detector to collect fluorescence emission coupled with electronics to record the measured fluorescence intensity (54). In recent years, commercially available confocal microscopes have incorporated FRAP protocols, which allows for imaging-based FRAP measurements of intracellular fluorescent proteins. These two developments have brought FRAP into the mainstream of cell biologic techniques, especially those related to questions of membrane dynamics.
Examples of useful applications of confocal FRAP include selective photobleaching and fluorescence loss in photobleaching (FLIP) (18, 55). In contrast with conventional spot photobleaching FRAP, in confocal FRAP it possible to visualize both the bleach region and the surrounding area of the cell. In addition, confocal FRAP techniques typically use large and/or complex regions of interest and may incorporate repetitive bleaching protocols instead of a single bleaching event. For example, in selective photobleaching experiments, an entire subcellular compartment such as the Golgi complex is photobleached to examine the mechanism and kinetics of recovery from elsewhere in the cell (Fig. 4d-e). Using this approach, it is possible to monitor the kinetics of coat protein cycling on and off membranes (28) as well as to assess the kinetics of vesicular and nonvesicular transport in the secretory and endocytic pathways (18) (Fig. 2). In FLIP, a single region of interest is bleached repetitively, which allows for recovery of fluorescent material to occur in between each repetition. This technique causes a gradual depletion of fluorescence material in regions of the cell that are in communication with the bleach region (Fig. 4c). Thus, the rate and the extent of loss of fluorescent material from the area outside of the bleach region depends on the degree of connectivity between compartments (18).

Figure 4. Principles of photoactivation and photobleaching experiments. For purposes of illustration, regions of bright fluorescence are shaded gray, and areas that contain little or no fluorescence are shown in white. (a) FRAP. Here, molecules in a region of interest (box) are photobleached, and their exchange with fluorescent molecules from the surrounding region is monitored over time. (b) Photoactivation. Similar to FRAP in principle except here the molecules in the region of interest (box) are converted to a different state. The redistribution of photoactivated molecules can then be monitored selectively. (c) FLIP. Repeated photobleaching of a region of interest (square) is performed, while monitoring the loss of fluorescence from other regions of the cell. (d, e) Selective photobleaching. Fluorescent molecules in an individual compartment (as illustrated here for the Golgi complex, circled) are photobleached. The sample is then monitored over time to determine whether fluorescence can recover (d) or not recover (e) from other regions of the cell. (Adapted from Reference 62 with permission from Elsevier.)
Photoactivation
Although simple time-lapse imaging is often sufficient to monitor vesicular trafficking and transient membrane binding events, for other experiments it is advantageous to “mark” a particular group of fluorescent molecules and watch their redistribution over time. The generation of photoactivatable fluorescent proteins (39) has now made it possible to “highlight” a population of molecules for optical tracking experiments (55). As a result, movement of the photoactivated molecules can be visualized directly in a “pulse-chase” experiment (Fig. 4b). Many of the same concepts described above for FRAP experiments can also be applied to photoactivation.
Single particle tracking
Single particle tracking is a technique that visualizes directly the movements of individual molecules, small groups of molecules, or even viruses by either fluorescence microscopy or light microscopy (54, 56). For such experiments, individual proteins can be expressed as GFP fusion proteins, labeled with fluorescent antibodies, or labeled with quantum dots. Alternatively, they can be immunolabeled with probes that can be detected by light microscopy such as 30-40 nm gold particles or latex beads. In the limit of sparse labeling, the trajectory of individual tagged molecules can then be tracked with high temporal and spatial accuracy using highly sensitive cameras (54). Although the resolution of fluorescence microscopy is ~250nm, the centroid of a single molecule can be determined with an accuracy of ~10nm (54). Typical rates of image acquisition are ~30 frames/sec, although much faster acquisition rates can be obtained with specialized cameras. The resulting trajectories can be plotted directly to show the movements of each molecule, or can be analyzed even more to determine the mean squared displacement as a function of time (Fig. 1). For example, during free diffusion, the molecule moves randomly, which results in a characteristic linear relationship between mean squared displacement and time. A hallmark of confined diffusion is that the mean squared displacement of the particle is at first linear, then plateaus, which reflects the limited movement of the molecule within a confined region of the membrane. A given molecule may shift between several modes of motion during the observation period. Thus, the percentage of molecules that undergo each type of motion and/or the fraction of time they exhibit a particular behavior is often reported.
An obvious advantage of single molecule tracking is that it allows for highly detailed analyses of the movements of individual molecules that are obscured in population-based measurements such as FRAP and photoactivation experiments. However, SPT experiments are not trivial to perform and are subject to several potential artifacts. The first challenge is to demonstrate that single molecules are being studied. Crosslinking can occur in studies that use antibody-labeled gold beads or quantum dots as probes, and lead to changes in mobility. In addition, large probes can potentially interact with the extracellular matrix. Photo- bleaching of organic dyes or FP-fusions can be rapid, which limits visualization times. However, photobleaching can also be used to confirm that single molecules are being visualized, because they will undergo a single-step photobleach. Finally, because of the intrinsic variability in trajectories, careful analysis is required to distinguish between motions that arise from free diffusion and confined diffusion.
FCS and ICS
FCS is not an imaging technique per se, although it is often performed using a microscope-based system. Instead, FCS measures the movement of individual fluorescent molecules through a defined observation volume, recorded as fluctuations in fluorescence over time (57). Such measurements require a sensitive photodetector, a dilute sample (~nM) and a sampling volume with femptoliter dimensions. The observation volume can be generated using a laser focused to a diffraction-limited spot with a confocal pinhole placed in front of the detector, multiphoton illumination, or TIRF excitation in conjunctions with a pinhole. A notable strength of FCS is its sensitivity to fluorescence fluctuations over a wide range of timescales, from fast kinetics corresponding to photophysical properties of fluorescent proteins to the diffusion of proteins in cell membranes (58). In addition, FCS measurements can be collected within user-defined regions in an individual cell or artificial membrane vesicle, which allows for comparison of protein or lipid dynamics in distinct membrane environments (15). FCS can also be used to measure kinetic rate constants, for example, the association and dissociation of fluorescently labeled molecules from the plasma membrane (49).
The fluorescence fluctuations measured by FCS can be analyzed in several ways. The most common technique, autocorrelation analysis, provides information about characteristic diffusion time of fluorescent molecules through the observation volume. It also reports on the average number of molecules present in the observation volume, and thus the concentration of fluorescent moleculesn (14, 49, 56, 57). Other types of FCS analysis can be used to analyze molecular brightness and the oligomeric state of the fluorescent molecule. Finally, cross-correlation FCS monitors fluctuations jointly from molecules labeled with two or more different fluorophores. This technique provides a powerful approach to assay for intermolecular interactions, because molecules that are bound either directly or indirectly to one another should diffuse as a single unit (8, 59).
Whereas FCS measures fluorescence fluctuations over time, a related technique, ICS, measures fluorescence fluctuations over space, in particular from images collected using a laser-scanning microscope (56). ICS analysis of pixels within a single image provides information about protein clustering and density. A variation of ICS known as image cross-correlation spectroscopy evaluates the interactions of molecules labeled with different fluorescent probes. ICS can also be performed on stacks of images collected as a function of time. This spatial-temporal version of ICS can be used to monitor slow protein dynamics, and even be used to generate vector maps of directed protein movements in living cells (60).
Acknowledgments
I apologize to those whose original research could not be cited because of space limitations. I thank Maria Kiskowski, Lynne Lapierre, Erik Snapp, and Michael Edidin for their comments on the manuscript and Erik Snapp for providing Figure 2.
Supported by R01GM-073846.
References
1. Frye LD, Edidin M. The rapid intermixing of cell surface antigens after formation of mouse-human heterokaryons. J. Cell. Sci. 1970; 7:319-335.
2. Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science 1972; 175:720-731.
3. Saffman PG, Delbruck M. Brownian motion in biological membranes. Proc. Natl. Acad. Sci. USA 1975; 72:3111-3113.
4. Saxton MP. Lateral diffusion of lipids and proteins. Curr. Topics in Membranes 1999; 48:229-282.
5. Edidin M. Fluorescence photobleaching and recovery, FPR, in the analysis of membrane structure and dynamics. In: Mobility and Proximity in Biological Membranes. Damjanovich S, Edidin M, Szollosi J, Tron L, Eds.1994. CRC Press, Boca Raton, FL.
6. Jacobson K, Sheets ED, Simson R. Revisiting the fluid mosaic model of membranes. Science 1995;268:1441-1442.
7. Holowka D, Baird B. Antigen-mediated IGE receptor aggregation and signaling: a window on cell surface structure and dynamics. Annu. Rev. Biophys. Biomol. Struct. 1996; 25:79-112.
8. Larson DR, Gosse JA, Holowka DA, Baird BA, Webb WW. Temporally resolved interactions between antigen-stimulated IgE receptors and Lyn kinase on living cells. J. Cell. Biol. 2005; 171:527-536.
9. Cole NB, Smith CL, Sciaky N, Terasaki M, Edidin M, Lippincott- Schwartz J. Diffusional mobility of Golgi proteins in membranes of living cells. Science 1996; 273:797-801.
10. Nehls S, Snapp EL, Cole NB, Zaal KJ, Kenworthy AK, Roberts TH, Ellenberg J, Presley JF, Siggia E, Lippincott-Schwartz, J. Dynamics and retention of misfolded proteins in native ER membranes. Nat. Cell. Biol. 2000; 2:288-295.
11. Kenworthy AK. Fleeting glimpses of lipid rafts: how biophysics is being used to track them. J. Investig. Med. 2005; 53:312-317.
12. Yokoe H, Meyer T. Spatial dynamics of GFP-tagged proteins investigated by local fluorescence enhancement. Nat. Biotechnol. 1996; 14:1252-1256.
13. Henis YI, Rotblat B, Kloog Y. FRAP beam-size analysis to measure palmitoylation-dependent membrane association dynamics and microdomain partitioning of Ras proteins. Methods 2006; 40:183-190.
14. Bacia K, Scherfeld D, Kahya N, Schwille P. Fluorescence correlation spectroscopy relates rafts in model and native membranes. Biophys. J. 2004; 87:1034-1043.
15. Korlach J, Schwille P, Webb WW, Feigenson GW. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. 1999; 96:8461-8466.
16. Douglass AD, Vale RD. Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell 2005; 121:937-950.
17. Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell 2004; 116:153-166.
18. Lippincott-Schwartz J, Snapp E, Kenworthy AK. Studying protein dynamics in living cells. Nature Reviews: Mol. Cell Biol. 2001; 2:444-456.
19. Lippincott-Schwartz J, Roberts TH, and Hirschberg K. Secretory protein trafficking and organelle dynamics in living cells. Annu. Rev. Cell. Dev. Biol. 2000; 16:557-589.
20. Watson P, Jones AT, Stephens DJ. Intracellular trafficking pathways and drug delivery: fluorescence imaging of living and fixed cells. Adv. Drug Deliv. Rev. 2005; 57:43-61.
21. Presley JH, Cole NB, Schroer TA, Hirschberg K, Zaal KJM, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature 1997; 389:81-85.
22. Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, Lippincott-Schwartz J. Kinetic analysis of secretory protein traffic and characterization of Golgi to plasma membrane transport intermediates in living cells. J. Cell. Biol. 1998; 143:1485-1503.
23. Pelkmans L, Puntener D, Helenius A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 2002; 296:535-539.
24. Pelkmans L, Burli T, Zerial M, Helenius A. Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell 2004; 118:767-780.
25. Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005; 122:735-749.
26. Teruel MN, Meyer T. Translocation and reversible localization of signaling proteins: a dynamic future for signal transduction. Cell 2000; 103:181-184.
27. Behnia R, Munro S. Organelle identity and the signposts for membrane traffic. Nature 2005; 438:597-604.
28. Presley JF, Ward TH, Pfeifer AC, Siggia ED, Phair RD, Lippincott- Schwartz J. Dissection of COPI and Arf1 dynamics in vivo and role in Golgi membrane transport. Nature 2002; 417:187-193.
29. Chiu VK, Bivona T, Hach A, Sajous JB, Stilleti J, Wiener H, Johnson RL, Cox AD, Philips MR. Ras signalling on the endoplasmic reticulum and the Golgi. Nature Cell. Biol. 2002; 4:343-350.
30. Goodwin JS, Drake KR, Rogers C, Wright L, Lippincott-Schwartz J, Philips MR, Kenworthy AK. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell. Biol. 2005; 170:261-272.
31. Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, Miura J, Wiener HH, Wright L, Saba SG, Yim D, Fein A, Perez de Castro I, Li C, Thompson CB, Cox AD, Philips MR. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell. 2006; 21:481-493.
32. Haughland RP. The Handbook—A Guide to Fluorescent Probes and Labeling Technologies. 2005.
33. Maier O, Oberle V, Hoekstra D. Fluorescent lipid probes: some properties and applications (a review). Chem. Phys. Lipids 2002; 116:3-18.
34. Marks DL, Singh RD, Choudhury A, Wheatley CL, Pagano RE. Use of fluorescent sphingolipid analogs to study lipid transport along the endocytic pathway. Methods 2005; 36:186-195.
35. Kuerschner L, Ejsing CS, Ekroos K, Shevchenko A, Anderson KI, Thiele C. Polyene-lipids: a new tool to image lipids. Nat. Methods 2005; 2:39-45.
36. Wustner D. Fluorescent sterols as tools in membrane biophysics and cell biology. Chem. Phys. Lipids 2007; 146:1-25.
37. Schmid JA, Neumeier H. Evolutions in science triggered by green fluorescent protein (GFP). Chembiochem. 2005; 6:1149-1156.
38. Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat. Methods 2005; 2:905-909.
39. Lukyanov KA, Chudakov DM, Lukyanov S, Verkhusha VV. Innovation: Photoactivatable fluorescent proteins. Nat. Rev. Mol. Cell. Biol. 2005; 6:885-891.
40. Kogure T, Karasawa S, Araki T, Saito K, Kinjo M, Miyawaki A. A fluorescent variant of a protein from the stony coral Montipora facilitates dual-color single-laser fluorescence cross-correlation spectroscopy. Nat. Biotechnol. 2006; 24:577-581.
41. Olenych SG, Claxton NS, Ottenberg GK, Davidson MW. The fluorescent protein color palette. In: Current Protocols in Cell Biology. Bonifacino JS, Dasso M, Harford JB, Lippincott-Schwartz J, Yamada KM, eds. 2006. John Wiley & Sons, Inc. New York.
42. Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science 2006; 312:217-224.
43. Chen I, Howarth M, Lin W, Ting AY. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2005; 2:99-104.
44. Gronemeyer T, Godin G, Johnsson K. Adding value to fusion proteins through covalent labelling. Curr. Opin, Biotechnol. 2005; 16:453-458.
45. Griffin, BA, Adams, SR, and Tsien, RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science 1998; 281:269-272.
46. Pinaud F, Michalet X, Bentolila LA, Tsay JM, Doose S, Li JJ, Iyer G, Weiss S. Advances in fluorescence imaging with quantum dot bio-probes. Biomaterials 2006; 27:1679-1687.
47. Piston DW. Imaging living cells and tissues by two-photon excitation microscopy. Trends Cell. Biol. 1999; 9:66-69.
48. Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol. 2003; 361:1-33.
49. Thompson NL, Steele BL. Total internal reflection with fluorescence correlation spectroscopy. Nat. Protoc. 2007; 2:878-890.
50. Sund SE, Axelrod D. Actin dynamics at the living cell submembrane imaged by total internal reflection fluorescence photobleach- ing. Biophys. J. 2000; 79:1655-1669.
51. Stephens DJ, Allan VJ. Light microscopy techniques for live cell imaging. Science 2003; 300:82-86.
52. Jaiswal JK, Simon SM. Imaging single events at the cell membrane. Nat. Chem. Biol. 2007; 3:92-98.
53. Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 1976; 16:1055-1069.
54. Chen Y, Lagerholm BC, Yang B, Jacobson K. Methods to measure the lateral diffusion of membrane lipids and proteins. Methods 2006; 39:147-153.
55. Lippincott-Schwartz J, Altan-Bonnet N, Patterson G. Photobleach- ing and photoactivation: following protein dynamics in living cells. Nat. Cell Biol. 2003; 7-14.
56. Bates IR, Wiseman PW, Hanrahan JW. Investigating membrane protein dynamics in living cells. Biochem. Cell. Biol. 2006; 84:825-831.
57. Elson EL. Quick tour of fluorescence correlation spectroscopy from its inception. J. Biomed. Opt. 2004; 9:857-864.
58. Kohl T, Schwille P. Fluorescence correlation spectroscopy with autofluorescent proteins. Adv. Biochem. Eng. Biotechnol. 2005; 95:107-142.
59. Haustein E, Schwille P. Fluorescence correlation spectroscopy: novel variations of an established technique. Annu. Rev. Biophys. Biomol. Struct. 2007; 36:151-169.
60. Hebert B, Costantino S, Wiseman PW. Spatiotemporal image correlation spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophys. J. 2005; 88:3601-3614.
61. Snapp E Design and use of fluorescent fusion protiens in biology. In: Current Protocols in Cell Biology. Bonifacino J, et al., eds. 2005. John Wiley and Sons, Inc., New York.
62. Goodwin JS, Kenworthy AK. Photobleaching approaches to investigate diffusional mobility and trafficking of Ras in living cells. Methods 2005; 37:154-164.
Further Reading
Edidin M. Lipids on the frontier: a century of cell-membrane bilayers.
Nature Revi. Mol. Cell. Biol. 4:414-418.
Gennis RB. Biomembranes. 1989. Springer-Verlag, New York.
Jans DA. The Mobile Receptor Hypothesis. 1997. Chapman & Hall, New York.
Lodish H, Berk A, Matsudaira P, Kaiser CA, Kreiger M, Scott MP, Zipursky SL, Darnell J. Molecular Cell Biology. 2004. WH Freemand and Company, New York.
Waters JC. Live-cell fluorescence imaging. Methods Cell.Biol. 2007; 81:115-140.
See Also
Imaging Techniques: Overview of Applications in Chemical Biology
Lipid Bilayers, Properties of
Membrane Assembly in Living Systems
Membranes, Fluidity of
Membrane Trafficking