CHEMICAL BIOLOGY
Xenobiotic Metabolism
Costas loannides, Molecular Toxicology Croup, School of Biomedical and Molecular Sciences, University of Surrey, Guildford, Surrey, United Kingdom
doi: 10.1002/9780470048672.wecb628
Humans are exposed continuously and unavoidably to a myriad of potentially toxic chemicals that are inherently lipophilic and, consequently, very difficult to excrete. To effect their elimination, the human body has developed appropriate enzyme systems that can transform metabolically these chemicals to hydrophilic, readily excretable, metabolites. This biotransformation process occurs in two distinct phases, Phase I and Phase II, and involves several enzyme systems, the most important being the cytochromes P450. The expression of these enzyme systems is regulated genetically but can be modulated also other factors, such as exposure to chemicals that can either increase or impair activity. Paradoxically, the same xenobiotic-metabolizing enzyme systems also can convert biologically inactive chemicals to highly reactive intermediates that interact with vital cellular macromolecules and elicit various forms of toxicity. Thus, xenobiotic metabolism does not always lead to deactivation but can result also in metabolic activation with deleterious consequences.
It is unlikely that the human body could survive and thrive in the chemical environment it lives in if it were not endowed with effective means to protect itself from the adverse effects of the myriad of chemicals to which it is continuously and unavoidably exposed. Although scientists focus on anthropogenic chemicals, humans are exposed to even more naturally occurring chemicals, mostly phytochemicals that, like their man-made counterparts, have the potential to induce toxicity. Both anthropogenic and natural chemicals are referred to as xenobiotics (Gr. foreign to life). Humans are exposed to huge numbers of xenobiotics, in the order of 5000 to 10,000 per day, most of which emanate from the diet. Of the chemicals that humans ingest, 99.9% are natural, largely of plant origin (1).
Biologic Background
Most chemicals to which humans are exposed cannot be exploited by the body either to generate energy or to use as structural blocks to build new tissue; they cannot be used as essential cofactors for enzyme reactions nor as chemical messengers. During the last three decades, however, it has become evident that some dietary phytochemicals possess biologic activity and have the potential to afford protection against major degenerative diseases of high morbidity, such as cancer and cardiovascular disease; this protection may explain the epidemiological findings that populations that consume diets with high vegetable and fruit content are less susceptible to these fatal diseases. Because most of xenobiotics offers no benefit to the human body, its immediate response is to prevent exposure and/or eliminate them. For this objective to be achieved, the body has developed as a first line of defense several transporter systems, such as the P-glycoprotein, which prevent the absorption of chemicals through the gastrointestinal tract by facilitating their efflux from the enterocytes into the intestinal lumen (2). For chemicals that at least partly overcome this obstacle and reach the blood circulation, its immediate response is to excrete them, primarily through the kidney and biliary routes. Chemicals that reach the systemic blood circulation through which they are distributed to the body tissues must traverse lipoid membranes, so they are inherently lipophilic. Because the body finds it difficult to eliminate such lipophilic chemicals, to prevent them from accumulating, it has developed as a defense mechanism several enzyme systems that are adept at metabolically converting them to hydrophilic products, facilitating in this way their elimination. The metabolic conversion of a lipophilic compound to a hydrophilic metabolite is known as biotransformation. Moreover, in most cases, such metabolism abolishes biologic activity because the generated metabolites, unlike the parent compound, cannot reach their site of action and/or fail to interact with the appropriate receptor. However, less frequently, metabolism of an inert chemical can produce a biologically active metabolite(s). In clinical therapy, chemicals may be administered as prodrugs, where the pharmacologically active entity is a metabolite (Table 1). Very occasionally, a change in pharmacological activity may occur following biotransformation; for example, iproniazid, an antidepressant, is metabolized to isoniazid, an antitubercular drug. Thus, metabolism has a profound effect on the biologic activity of a chemical. It is unlikely that the human body could withstand the constant onslaught of chemicals and survive in such a hostile chemical environment if such effective defense mechanisms were not operative.
Table 1. Examples of prodrugs
|
Parent drug |
Pharmacological activity |
Active metabolite |
|
Prontosil |
Antibacterial |
Sulphanilamide |
|
Levodopa |
Antiparkinson |
Dopamine |
|
Sulindac |
Anti-inflammatory |
Sulindac sulphide |
|
Cyclophosphamide |
Anticancer |
4-Hydroxycyclophosphamide |
|
Terfenadine |
Antihistaminic |
Fexofenadine |
|
Codeine |
Analgesic |
Morphine |
|
Sulfasalazine |
Anti-inflammatory |
5-Aminosalicylic acid |
|
Tamoxifen |
Anticancer |
Endoxifen |
|
Enalapril |
Antihypertensive |
Enalaprilat |
|
Glyceryt trinitrate |
Antianginal |
Nitric oxide |
The metabolic conversion of a lipophilic compound to a hydrophilic metabolite(s) proceeds through two distinct phases, namely Phase I and Phase II, each being catalyzed by different enzyme systems, localized largely in the endoplasmic reticulum and cytosol. Although the xenobiotic-metabolizing systems are found in almost every tissue in the body, the liver has the highest concentration, and for this reason it is the center of metabolism. The location of the drug-metabolizing enzymes in the liver is logical because most foreign compounds to which humans are exposed, including drugs, are ingested orally and, when they are absorbed through the gastrointestinal tract, they enter the portal blood supply that takes them to the liver before being distributed into the rest of the body. During absorption, a compound may be metabolized by intestinal, as well as microbial, enzymes (vide infra).
During Phase I metabolism, also referred to as functionalization, the xenobiotic acquires a functional group such as -OH, -COOH, and -NH2; alternatively, such functional groups may be unmasked, for example, an alkoxy (-C-OR) group can be dealkylated to unmask a functional group (-C-OH). For example, 7-hydroxycoumarin can be formed either by the hydroxylation of coumarin (insertion of an oxygen atom) or by the deethylation of 7-ethoxycoumarin (unmasking) (Fig. 1). Phase I metabolism involves oxidation, reduction, and hydrolysis reactions and is catalyzed by various enzyme systems (Table 2). The predominant reactions are oxidations, and the most important system catalyzing these is the cytochromes P450 (vide infra). Reductions are catalyzed by reductases, which are not very active in mammalian cells but are very active in gut bacteria that, consequently, may contribute extensively to the metabolism of orally administered drugs and other xenobiotics; the most common reductions are azo- and nitroreductions. The enzymes catalyzing hydrolysis reactions are esterases and amidases that hydrolyze esters and amides, respectively (Table 2).

Figure 1. Phase I and Phase II metabolic pathways. PAPS, adenosine 3'-phosphate 5'-phosphosulphate; UDPGA, uridine diphosphate glucuronic acid.
Phase II metabolism
During Phase II metabolism, also referred to as conjugation, the metabolites generated during Phase I metabolism combine with endogenous substrates, such as sulphate, glucuronic acid, glutathione, and amino acids, to form highly hydrophilic metabolites that are excreted with ease in the urine and feces. As an example, 7-hydroxycoumarin, formed from Phase I metabolism, forms a sulphate and a glucuronide conjugate (Fig. 1). Sulphation is catalyzed by the sulphotransferases and glucuronidation by the glucuronosyl transferases that attach glucuronic acid to the substrate. Chemicals, such as the drug paracetamol (acetaminophen) that already possesses a functional group, may bypass Phase I metabolism and be metabolized predominantly through conjugation (Fig. 2). Conjugation with glutathione represents an effective cellular defense mechanism that neutralizes toxic chemicals that otherwise would cause toxicity. Another Phase II pathway, usually minor, is conjugation with amino acids, the most common being glycine. Finally, methylation and acetylation are unusual pathways in that the generated metabolites are less polar than the parent compound; in this case, metabolism hinders rather than facilitates excretion and additional metabolism is required for eventual elimination.

Figure 2. Metabolism of paracetamol (acetaminophen).
Enzyme Systems that Catalyze Phase I Metabolism
Although several enzymes contribute to the Phase I metabolism of xenobiotics (Table 2), by far the most prominent are the cytochromes P450, so called because in the reduced state they form a complex with carbon monoxide that is characterized by an absorption maximum at 450 nm.
Table 2. Enzyme systems catalyzing Phase I and II xenobiotic metabolism
|
Phase I |
Phase II |
||
|
Enzyme system |
Principal cellular localization |
Enzyme system |
Principal cellular localization |
|
Cytochromes P450 Flavin monooxygenases Amine oxidases Molybdenum hydroxylases Prostaglandin synthases* Lipoxygenases Alcohol/aldehyde dehydrogenases Esterases/amidases |
Endoplasmic Endoplasmic reticulum Mitochondria Cytosol Endoplasmic reticulum Cytosol Cytosol Cytosol |
Glucuronosyl transferases Sulphotransferases Glutathione S-transferases Acetyl transferases Methyl transferases Epoxide hydrolases Amino acid conjugases |
Endoplasmic reticulum Cytosol Cytosol Cytosol Cytosol Endoplasmic reticulum Mitochondria |
*also referred to as cyclooxygenases
A widespread enzyme system in nature, cytochromes P450 are found in both prokaryotic and eukaryotic cells and, with the exception of striated muscle and red blood cells, are encountered in every tissue but predominate in the liver, which, as a result, is the principal site of xenobiotic transformations. However, cytochrome P450 enzymes also are active in portals of xenobiotic entry, such as the lungs, the gastrointestinal tract, and the nasal mucosa. Although the cytochrome P450 enzyme system can function as a reductase under anaerobic conditions, its main role is to facilitate the oxidation of a myriad of structurally diverse xenobiotics. It is a haem-containing, membrane-bound enzyme that requires molecular oxygen and NADPH and inserts an atom of oxygen to the xenobiotic (X) while the other is reduced to water.
![]()
The flavoprotein cytochrome P450 reductase channels the electrons from NADPH to the cytochromes P450, which function as a terminal oxidase (Fig. 3).
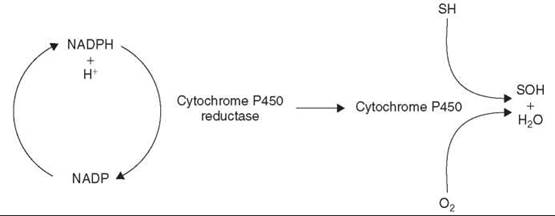
Figure 3. Cytochrome P450 electron transport system. S, Substrate.
A principal attribute of the cytochrome P450 system is the unprecedented broad substrate specificity it displays, which explains its pivotal role in xenobiotic metabolism. It catalyzes efficiently the metabolism of thousands of structurally diverse chemicals with markedly different molecular shape and size. It achieves this very broad substrate specificity by existing as a “superfamily” of enzymes. Each family is subdivided further into subfamilies that may contain one or more enzymes. Enzymes that share a structural similarity of at least 40% belong to the same family, which is indicated by Arabic numbers, whereas if the structural similarity exceeds 55%, then they are classified within the same subfamily, which is denoted by capital letters; finally, enzymes belonging to the same subfamily are denoted by Arabic numbers. For example, the CYP1 family comprises two subfamilies, namely CYP1A and CYP1B; the former consists of two enzymes, CYP1A1 and CYP1A2, whereas within the latter only a single enzyme has been identified so far, CYP1B1. The cytochrome P450 families active in xenobiotic metabolism are CYP1, CYP2, and CYP3. Body distribution of individual cytochrome P450 enzymes may be tissue-dependent; CYP1A2 is expressed exclusively in the liver, whereas CYP1B1 is expressed poorly in the liver but predominates in tissues such as the adrenal glands and the lung (3).
Although this article is concerned with their role in xenobiotic metabolism, it should be emphasized that cytochromes P450 play a critical role in the metabolism, both biosynthesis and catabolism, of endogenous substrates, including hormones such as steroids and melatonin, eicosanoids such as prostaglandins, fatty acids such as lauric acid, and vitamins such as vitamin D. Cytochrome P450 enzymes may be engaged almost exclusively in the metabolism of endogenous substrates, with negligible catalytic activity toward xenobiotics. The cytochrome P450 enzymes responsible for the metabolism of endogenous compounds are characterized by narrow substrate specificity, frequently entailing a single, or very few, structurally related substrates, and do not contribute to xenobiotic metabolism. The fact that some xenobiotic-metabolizing cytochrome P450 isoforms are expressed polymorphically (vide infra) and that individuals may lack totally a certain isoform without any detriment to health indicates that the role of these cytochromes P450 in endogenous metabolism is far from life threatening.
Cytochromes P450 are believed to have evolved from a common ancestor some three thousand million years ago. The function of the earliest forms is thought to have been in the metabolism of essential endogenous chemicals, such as steroid hormones, and then to have evolved to enzymes capable of metabolizing foreign compounds. As a result, their initial narrow substrate specificity toward steroids was lost and, to cope with the new, increasingly chemical environment, they developed into broad-specificity enzymes that could metabolize the diverse chemicals to which they now were exposed. It has been proposed that what forced the evolution of these proteins was the necessity to develop defense mechanisms to protect against plant toxins present in the food chain that were produced to discourage predators (5).
Enzyme Systems that Catalyze Phase II Metabolism
Conjugation reactions of xenobiotics or their metabolites with endogenous substrates produce highly hydrophilic metabolites; however, to a lesser extent functional groups may be also methylated or acetylated to produce less hydrophilic compounds.
Glucuronide conjugation
Glucuronide conjugation is a most frequently used routes of conjugation by xenobiotics where glucuronic acid in its active form, uridine diphosphate glucuronic acid (UDPGA), is added to the molecule (Fig. 1). Some drugs like oxazepam and morphine are catalyzed predominantly by glucuronidation because they do not require prior Phase I metabolism to generate a functional accepting group. The most readily conjugated functional groups are phenols and alcohols, which yield ester glucuronides, and carboxylic acids, which form ether glucuronides.
Sulphate conjugation
Conjugation with sulphate, catalyzed by cytosolic sulphotransferases, is also a major route of Phase II metabolism, where inorganic sulphate, made available in the activated form of 3'-phosphoadenosine-5'-phosphosulphate (PAPS), is added to the molecule (Fig. 1). This is the most important pathway in the metabolism of phenols and is a very efficient conjugating system as long as inorganic sulphate is available.
Glutathione conjugation
Glutathione conjugation is an important pathway of metabolism that allows the cell to defend itself from chemical insult. It uses the nucleophilic tripeptide glutathione, which possesses a nucleophilic sulphur atom, to detoxify chemically reactive metabolites, preventing them from interacting with critical cellular macromolecules with adverse consequences and, thus, preserves cellular integrity. This reaction is catalyzed by the ubiquitously distributed glutathione S-transferases, which transfer a molecule of reduced glutathione to the toxic chemical that results in its neutralization. Glutathione conjugates additionally are processed further metabolically in the body are excreted and usually as N-acetylcysteine conjugates (mercapturates).
Amino acid conjugation
In this minor route of xenobiotic metabolism, the carboxylic group of organic acids may conjugate with amino acids, glycine being the most common. The carboxyl group of the xenobiotic forms a peptide bond with the a-amino group of the amino acid. Initially, in an ATP-dependent reaction, the carboxylic group reacts with CoA to form an acyl-CoA thioester derivative, which then interacts with the amino acid to form the conjugate.
Hydration
Hydration involves the addition of water to epoxides to form dihydrodiols and is catalyzed by microsomal epoxide hydrolase that displays, broad substrate specificity. As epoxides are generally toxic entities, this is a very important route for their detoxification.
Methylation
Hydroxyl, as well as amino and thiol groups, may be metabolized through methylation, the methyl donor being S-adenosyl methionine, the product of the interaction of ATP with methionine. Usually, it is a minor metabolic route in xenobiotic metabolism, but it plays a major role in the metabolism of endogenous substrates such as noradrenaline (norepinephrine). Methylation is catalyzed by methyltransferases located in the mitochondria.
Acetylation
Acetylation is an important metabolic route for aromatic and heterocyclic amines, hydrazines and sulphonamides. An amide bond is formed between the amino group of the chemical and the acetate. This reaction is catalyzed by acetylases, the acetyl group being donated by acetyl CoA.
The First-Pass Effect
If an orally taken drug is not absorbed, then there will be no pharmacological effect and the drug will be excreted in the feces. However, it is conceivable that a drug is very well absorbed and yet fails to manifest the expected pharmacological effect. Following oral administration, drugs are absorbed through the intestine into the portal circulation that takes them to the liver and then to the systemic circulation. A drug may undergo metabolism either in the intestine and/or through its first passage through the liver to such an extent that very little remains available for distribution to the other tissues, including the site of action, and, consequently, biologic activity is either not manifested or attenuated. In the intestine, metabolism may be catalyzed not only by intestinal enzymes, such as cytochromes P450 and Phase II conjugation enzymes, but also by microorganisms. This phenomenon is known as the first-pass effect or presystemic metabolism. Thus, poor response to drug treatment after oral intake may be due to the fact that, despite complete absorption, only a small fraction of the drug reaches systemic circulation intact because of extensive metabolism in the intestine or liver.
First-pass metabolism, therefore, will decrease the pharmacological effect of the drug, and if the metabolism is extensive, the pharmacological effect may be abolished completely as a result, drugs that are subject to first-pass metabolism are administered through alternative routes or at higher doses to compensate for the loss during presystemic metabolism. Glyceryl trinitrate, a drug used in the treatment of heart angina, when taken orally undergoes > 99% of first-pass metabolism being denitrated in the liver, and, as a result, it is never administered through this route but is taken sublingually to bypass the intestine. The patient places a tablet under the tongue, and because of the good network of blood vessels, the drug is absorbed very rapidly and the patient benefits from it within a few minutes.
Factors that Influence Xenobiotic Metabolism
Several factors can modulate the activity of xenobiotic-metabo- lizing enzymes, such as age, the nature of diet, and the presence of disease, but the most important appear to be genetic makeup and concurrent or prior exposure to chemicals (enzyme induction and enzyme inhibition).
Polymorphic expression of xenobiotic-metabolizing enzyme systems: clinical implications
The presence of xenobiotic-metabolizing enzymes and this level of expression are governed by our genes. Xenobiotic- metabolizing cytochromes P450 may be polymorphically expressed, and if this is not appreciated and the necessary steps taken to adjust drug dosage accordingly, it may have a dramatic impact in clinical therapeutics (5, 6). It is recognized now that xenobiotic-metabolizing enzymes, both in Phase I and Phase II, are polymorphically expressed, resulting in inter individual differences in metabolic capacity, so that drug dose regimens are unlikely to be optimal for all patients. For a certain drug, some individuals are poor metabolizers, whereas others are extensive metabolizers. Furthermore, polymorphic expression affects not only drug efficacy but also the appearance of adverse effects. The etiology of adverse effects experienced by some patients exposed to the therapeutic doses of drugs, especially of drugs with a narrow therapeutic index, i.e., drugs whose plasma concentrations must be maintained within a narrow range to achieve the desired pharmacological effect, may be attributed to their individual enzyme profile (7).
One of the first polymorphisms to be identified involved acetylation (N-acetyltrasferase) and the antitubercular drug isoniazid. Several people are slow acetylators, the proportion being race related and varying from about 10% in the Japanese to 70% in Caucasians. Acetylation of isoniazid results in loss of pharmacological activity because acetylisoniazid is devoid of antitubercular activity, but it provokes toxicity that occurs more frequently in slow acetylators. Since acetylation is not catalyzed efficiently, isoniazid blood levels are high and the drug interacts with pyridoxal 5-phosphate, the active form of vitamin B6, resulting in its depletion; this vitamin deficiency causes neuropathy that leads to seizures. This situation can be avoided by the concurrent administration of the vitamin to patients treated with isoniazid. Genetic polymorphism in other conjugating systems that result in metabolic deficiencies also can lead to predisposition to the toxicity of chemicals that rely heavily on these enzymes for their deactivation. Gilbert’s syndrome is a condition where the patient experiences intermittent jaundice because of reduced capacity in eliminating bilirubin through glucuronide conjugation. The prodrug irinotecan, used in the treatment of advanced colorectal cancer, provokes severe gastrointestinal toxicity in these patients because of suppressed glucuronidation (8, 9). The active metabolite (SN-38) that is generated metabolically cannot be detoxicated by glucuronidation and consequently accumulates.
Another early example of polymorphism involves the drug succinylcholine (suxamethonium), a muscle relaxant used primarily during surgery. Its action lasts only a few minutes because it is very efficiently metabolized by cholinesterases present in the liver and plasma. A few people, about 1 in 3000 who genetically lack this enzyme, develop sustained apnoea as a result of paralysis because its effect is prolonged from 30 minutes to hours.
Genetic polymorphism in cytochromes P450 is believed to be responsible for many adverse effects associated with drug intake. The first cytochrome P450 protein to be recognized as being polymorphically expressed was CYP2D6, an enzyme that catalyzes many current psychoactive drugs, such as tricyclic antidepressants; subsequent studies have revealed that polymorphism may involve also the CYP2 C9 and CYP2 C19 enzymes. Persons lacking an active gene, i.e., a gene that can generate a functional protein, when exposed to drugs relying on this enzyme for their metabolic deactivation will show exaggerated adverse effects as a consequence of accumulation, and ideally should be prescribed lower doses. One such drug is the antihypertensive debrisoquine, which in normal individuals undergoes extensive CYP2D6-mediated 4-hydroxylation that leads to loss of activity. About 10% of European Caucasians and 1% of Japanese have been identified as poor metabolizers because of the lack of a functional CYP2D6. Subjects with poor metabolism inherited two copies of a gene that encodes either an enzyme with low activity or one with no activity. When exposed to dose regimens of debrisoquine developed for normal metabolizers, as a consequence of diminished metabolism, the drug accumulates on repeated administration and adverse effects commensurate with overdose are experienced, such as sustained drop in blood pressure. A number of individuals are classified as intermediate metabolizers, having one copy of the inactive gene; they display CYP2D6 activity but at low level. As a result of CYP2D6 polymorphic expression, the dose required to produce the same plasma levels may differ by as much as 20-fold among individuals. Similarly, the β-blocking agent timolol is metabolized largely by CYP2D6; when exposed to this drug, poor metabolizers experience a prolonged, more intense pharmacological effect (10). If one is dealing with a prodrug, however, where metabolism leads to the production of the pharmacologically active form, a less intense or a complete loss of the pharmacological effect may occur in poor metabolizers. The analgesic effect of codeine is associated with morphine, which is formed because of CYP2D6-mediated metabolism. In poor metabolizers, the drug displays poor efficacy, and the lack of analgesia is due to the active metabolite is not being produced; indeed, morphine plasma levels are extremely low (11, 12).
Polymorphism also can enhance the metabolism of a drug in persons carrying multiple copies of a cytochrome P450 gene, for example, as a result of gene amplification and duplication, so that a drug may be deactivated more rapidly through metabolism, leading to a less intense or totally absent pharmacological effect because therapeutic plasma levels are not achieved. Such individuals are known as ultrarapid metabolizers and require far higher doses than normal to achieve therapeutic response. However, in the case of intake of a prodrug, the dose should be decreased to prevent an exaggerated effect and toxicity. Ultrarapid metabolizers show symptoms of codeine intoxication, such as severe abdominal pain, when they take a normal therapeutic dose (13).
Cytochrome P450 regulation by chemicals and clinical consequences
The levels of cytochrome P450 are not only genetically determined but may be modulated also by factors such as age, nutritional status, and the presence of disease. The most important factor, however, is previous exposure to xenobiotics, either natural or synthetic, that either can induce, largely as a result of enhanced enzyme synthesis, or impair cytochrome P450 activity; both can lead to serious consequences during multiple drug intake. Such drug interactions involve in particular, CYP3A4, as this enzyme is the most active cytochrome P450 in drug metabolism and, moreover, is the dominant form in the liver and intestine, the principal site and the first site of metabolism following oral drug intake, respectively.
Cyclosporin is an immunosuppressant drug taken chronically by organ transplant patients. It has a narrow therapeutic index, and plasma levels have to be maintained within a narrow range to ensure efficacy and to avoid the appearance of serious adverse effects that can be life threatening. The principal catalyst of its metabolism is CYP3A4, and even small changes in the activity of this enzyme can have major impact on its efficacy. The antifungal drug ketoconazole is a potent inhibitor of CYP3A4, and, as a result, it increases plasma levels of cyclosporin leading to toxicity when coadministered with it (14). Similarly, serious interactions can occur with prodrugs that necessitate CYP3A4-metabolism to generate the pharmacologically active form. Terfenadine undergoes nearly complete first-pass effect in the liver; it is metabolized by CYP3A4 to generate a metabolite, fexofenadine, which possesses antihistaminic activity. Normally, terfenadine is never seen in the blood of patients because of the extensive first-pass metabolism. When, however, it is taken together with ketoconazole, its metabolism is impaired and terfenadine can escape into the systemic circulation causing cardiotoxicity (14). To avoid such complications, the active metabolite fexofenadine has replaced now the original drug.
In the 1990s, it became apparent that clinically important metabolic interactions could occur also between drugs and food components. It was realized that concomitant intake of grapefruit juice with drugs that are subject to presystemic metabolism mediated by CYP3A4 led to higher plasma drug levels and possible toxicity (15). Subsequently, it was recognized that a constituent of grapefruit, the furanocoumarin bergamottin, selectively inhibited intestinal CYP3A4 so that a larger fraction of the dose survived the presystemic metabolism leading to higher, toxic levels in the plasma (16). The furanocoumarin is metabolized by cytochrome P450 to a metabolite, which subsequently interacts covalently with the cytochrome and leads to its destruction. Activity is restored only when a new enzyme is synthesized; this type of inhibition is referred to as mechanism-based inhibition.
CYP3A4 activity, however, may be induced also by prior exposure to chemicals so that metabolism of drugs reliant on this enzyme will be accelerated, leading to lower plasma levels and an attenuated pharmacological response. Patients with transplanted organs who were stabilized successfully on cyclosporin displayed signs of rejection after self-medicating with St John’s Wort, an herbal remedy taken to treat mild depression (17). It appears that a component(s) of this remedy, such as hyperforin, increases CYP3A4 activity, which leads to enhanced cyclosporin metabolism and results in suboptimal plasma levels of the drug. Since the norethindrone and ethinyl oestradiol components of the contraceptive pill also are deactivated by CYP3A4, intake of St. John’s Wort has been associated also with intracyclic bleeding episodes because of contraceptive failure (18, 19).
Since cytochromes P450 also are important contributors to the metabolism of endogenous substrates, drugs potentially can cause adverse effects by interfering with the metabolism of critical endogenous substrates. Cytochrome P450 is responsible for the conversion of vitamin D3 to its biologically active form. Vitamin D3 undergoes hydroxylation at the 25-position in the liver (CYP27A1); in the kidney, 25-hydroxyvitamin D3 is further hydroxylated at the 1α-position (CYP27B1) to form 1α,25-dihydroxyvitamin D3, the active form, which through interaction with the vitamin D nuclear receptor exerts its biologic activity, maintaining calcium homeostasis (Fig. 4). However, 1α,25-dihydroxyvitamin D3 is deactivated by cytochrome P450-mediated hydroxylations at the 24- and 23-positions, originally shown to be catalyzed by CYP24A1. More recent work established that CYP3A4 is the dominant cytochrome P450 enzyme responsible for the deactivation reactions (20). These observations provide a likely explanation for the established fact that epileptics chronically prescribed antiepileptic drugs, such as diphenylhydantoin and phenobarbitone, to control seizures develop various bone conditions, such as osteomalacia and osteoporosis. These drugs, being CYP3A4 inducers, lead to a state of vitamin D deficiency and impairment of calcium homeostasis, so vitamin supplementation is necessary to compensate for the loss.
Modulation of xenobiotic-metabolizing enzymes, not surprisingly, has been exploited in clinical therapy. The principal elimination process of bilirubin, an endogenous toxic product of haemoglobin degradation, is through glucuronide conjugation to produce the highly polar and readily excretable bilirubin diglucuronide, the reaction being catalyzed by glucuronosyl transferase. In conditions of hyperbilirubinaemia, such as Crigler-Najjar syndrome type II, elimination of bilirubin may be accelerated by stimulating the activity of this enzyme by using drugs like phenobarbitone. Moreover, to lower the dose of the expensive drug cyclosporin, several studies have addressed the possibility of coadministering it with the CYP3A4-inhibiting drug ketoconazole (21).
Although hepatic enzymes are the most frequently induced, extrahepatic induction has been also documented, but in these cases the route of administration of the inducing agent may be critical. Pulmonary cytochrome P450 activity is more likely to be increased when exposure to the inducing agent occurs by inhalation; smokers, for example, display higher pulmonary cytochrome P450 activity (CYP1) compared with nonsmokers, which is ascribed to the inhalation of polycyclic aromatic hydrocarbons.
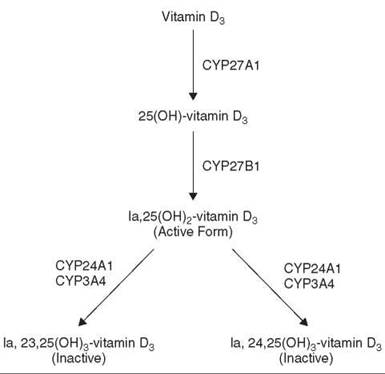
Figure 4. Metabolism of vitamin D3.
A striking paradox that was first recognized in the 1970s was that the xenobiotic-metabolizing enzyme systems, long regarded as being exclusively involved in the deactivation and elimination of chemicals, such as the cytochromes P450, could in fact assume the reverse role, i.e., they could convert innocuous, chemically inert xenobiotics to highly reactive and toxic metabolites with deleterious consequences to the body. This process is referred to frequently as “metabolic activation” or “bioactivation.”
Although the chemicals that undergo activation are structurally diverse, the basic mechanism of activation appears to be quite similar. Activation involves primarily oxygenation, although reduction is also important with some chemicals (Fig. 5). The reactive intermediates that are formed are electrophiles, having electron-deficient atoms, and so are chemically very highly reactive. Since these reactive species are generated intracellularly, they can interact readily and irreversibly with vital cellular macromolecules, such as DNA, RNA, and proteins, to provoke various types of toxicity; thus, in this case, metabolism confers to the chemical adverse biologic activity. Alternatively, these reactive intermediates may interact with tissue oxygen, giving rise to toxicity indirectly through redox cycling, by acting as radical generators. These free radicals are capable of inducing cellular damage similar to that resulting from the covalent binding of electrophiles to cellular constituents. Simultaneously, a chemical will be subject to metabolism through pathways that lead to deactivation so that the extent of toxicity will be dependent on the balance of activation and deactivation. An extensively studied example of such drug bioactivation involves the drug paracetamol (acetaminophen), which is primarily metabolized through conjugation with sulphate and glucuronic acid leading to deactivation. However, to a very small extent, it undergoes cytochrome P450-dependent oxidation to form a reactive quinoneimine that has the potential to cause hepatotoxicity following covalent interaction with proteins (Fig. 2). The low levels of the quinoneimine are, however, effectively neutralized by conjugation with glutathione. As a result, paracetamol is a very safe drug at therapeutic doses, despite the formation of a cytotoxic metabolite, and only becomes unsafe when this fine balance of activation/deactivation is disturbed. Some groups of people, such as the chronic alcoholics, are vulnerable to the toxicity of paracetamol. Alcohol is the prototype inducer of CYP2E1, one of the cytochrome P450 enzymes that catalyzes the oxidation of paracetamol to the quinoneimine. Consequently, alcoholics, because of the higher CYP2E1 activity, form more of the toxic intermediate of paracetamol and, therefore, are sensitive to the hepatotoxicity of this drug. Thus, in this case, toxicity ensues because of increased activation that leads to the enhanced production of the toxic metabolite.

Figure 5. Bioactivation of xenobiotics.
Most chemical carcinogens also rely on bioactivation to genotoxic metabolites that readily interact with DNA and set into motion the processes that eventually lead to tumorigenesis. The metabolic pathways once again are catalyzed by both Phase I and Phase II enzymes, and the chemical is concurrently subject to deactivation pathways. Although cytochromes P450 frequently catalyze the first step in the bioactivation of most chemicals, other enzyme systems, such as the sulphotransferases and acetylases, also are essential in the generation of the ultimate toxic species, the entity that interacts with the cellular macromolecules. For example, the heterocyclic amine 2-amino-3-methylimidazo-(4,5-f)quinoline (IQ), a carcinogen formed during the grilling/frying of meat and fish, requires bioactivation to express its carcinogenicity. The activation pathway involves CYP1A2-catalyzed N-hydroxylation, followed by esterification of the hydroxylamine with sulphate and acetate to generate the sulphatoxy and acetoxy esters, respectively, that break down spontaneously to yield the nitrenium ion, the presumed ultimate carcinogen (Fig. 6). Ring hydroxylation at the 5-position and direct conjugation of the parent compound with glucuronide or sulphate are strictly deactivation pathways; the ring-hydroxylated metabolite of IQ is excreted eventually in conjugated form.

Figure 6. Metabolic activation and deactivation of IQ. IQ, 2-amino-3-methylimidazo-(4,5-f)quinoline.
Conclusions
The human body is equipped with an array of enzyme systems that enable it to transform the myriad of chemicals to which it is inevitably exposed to metabolites that are readily and efficiently eliminated. In this way, the residence time of chemicals in the body is minimized and their accumulation prevented. These enzyme systems, in turn, are regulated genetically but also are modulated by environmental factors such as exposure to chemicals. The initial view, however, that these enzyme systems function simply in the elimination of xenobiotics through metabolism is anachronistic in the face of mounting evidence that these enzyme systems also can convert innocuous chemicals to toxic products that are detrimental to the welfare of the human body. Numerous examples are documented where metabolism of a chemical, especially by cytochromes P450, results in toxicity. Any factor that modulates the enzymes involved in the metabolism of a certain chemical will influence also its biologic activity, including toxicity. Clearly, toxicity is not merely a consequence of the intrinsic molecular structure of the chemical but is determined by the nature of the enzymes active in the body at the time of exposure. A chemical is subject to several metabolic pathways, the majority of which will bring about its deactivation and facilitate its excretion. However, some routes of metabolism will transform the chemical to a metabolite capable of inducing toxicity and carcinogenicity. Obviously, the amount of reactive intermediate produced, and hence incidence and degree of toxicity, will be largely dependent on the competing pathways of activation and deactivation, and whatever factor influences this delicate balance of activation/deactivation of a chemical will impact also on its biologic activity and safety.
References
1. Ames BN, Gold LS. The prevention of cancer. Drug Met. Rev. 1998; 30:201-223.
2. Van Tellingen O. The importance of drug-transporting P-glycoproteins in toxicology. Toxicol. Lett. 2001; 120:31-41.
3. Bhattacharyya KK, Brake PB, Eltom SE, Otto SA, Jeffcoate CR. Identification of a rat adrenal cytochrome P450 active in polycyclic-hydrocarbon metabolism as rat CYP1B1 demonstration of a unique tissue-specific pattern on hormonal and aryl-hydrocarbon receptor-linked regulation. J. Biol. Chem. 1995; 270:11595-11602.
4. Gonzalez FJ, Nebert DW. Evolution of the P450 gene superfamily animal plant ‘warfare’, molecular drive and human differences in drug oxidation. Trends Genet. 1990; 6:182-186.
5. Ingelman-Sundberg M, Oscarson M, McLellan RA. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol. Sci. 1999; 20:342-349.
6. Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005; 5:6-13.
7. Phillips KA, Veenstra DL, Oren E, Lee JK, Sadee W. Potential role of pharmacogenomics in reducing adverse drug reactions. JAMA 2001; 286:2270-2279.
8. Wasserman E, Myara A, Lokiec F, Goldwasser F, Trivin F, Mahjoubi M, Misset JL, Cvitkovic E. Severe CPT-11 toxicity in patients with Gilbert’s syndrome: two case reports. Annals Oncol. 1997; 8:1049-1051.
9. Innocenti F, Iyer L, Ratain MJ. Pharmacogenetics of anticancer agents: Lessons form amonafide and irinotecan. Drug Met. Disp. 2001; 29:596-600.
10. Lewis RV, Lennard MS, Jackson PR, Tucker GT, Ramsay LE, Woods HF. Timolol and atenolol: relationships between oxidation phenotype, pharmacokinetics and pharmacodynamics. Br. J. Clin. Pharmacol. 1985; 19:329-333.
11. Sindrup SH, Brosen K, Bjerring P, Arendt-Nielsen L, Larsen U, Angelo HR, Gram LF. Codeine increases pain thresholds to copper vapor laser stimuli in extensive but not poor metabolizers of sparteine. Clin. Pharmacol. Ther. 1991; 49:686-693.
12. Poulsen L, Brosen K, Arendt-Nielsen L, Gram LF, Elbaek K, Sindrup SH. Codeine and morphine in extensive and poor metabolizers of sparteine: pharmacokinetics, analgesic effects and side effects. Eur. J. Clin. Pharmacol. 1996; 51:289-295.
13. Gasche Y, Daali Y, Fathi M, Chiappe A, Cottini S, Dayer P, Desmeules J. Codeine intoxication associated with ultrarapid CYP2D6 metabolism. N. Engl. J. Med. 2004; 351:2827-2831.
14. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism—Clinical relevance. Clin. Pharmacokin. 2000; 38:111-180.
15. Mertens-Talcott SU, Zadezensky I, De Castro WV, Derendorf H, Butterweck V. Grapefruit-drug interactions: can interactions with drugs be avoided? J. Clin. Pharmacol. 2006; 46:1690-1416.
16. Goosen TC, Cillie D, Bailey DG, Yu CW, He K, Hollenberg PF, Woster PM, Cohen L, Williams JA, Rheeders M, Dijikstra HP. Bergamottin contribution to the grapefruit juice-felodipine interaction and disposition in humans. Clin. Pharmacol. Ther. 2004; 76:607-617.
17. Ioannides C. Pharmacokinetic interactions between herbal remedies and medicinal drugs. Xenobiotica 2002; 32:451-478.
18. Hall SD, Wang ZQ, Huang SM, Hamman MA, Vasavada N, Adigun AQ, Hilligoss JK, Miller M, Gorski JC. The interaction between St John’s wort and an oral contraceptive. Clin. Pharmacol. Ther. 2003; 74:525-535.
19. Murphy PA,Kern SE, Stanczyk FZ, Westhoff CL. Interaction of St. John’s Wort with oral contraceptives: effects on the pharmacokinetics of norethindrone and ethinyl estradiol, ovarian activity and breakthrough bleeding. Contraception 2005; 71:402-408.
20. Xu Y,Hashizume T, Shuhart MC, Davis CL, Nelsos WL, Sakaki T, Kalhorn TF, Watkins PB, Schuetz EG, Thummel KE. Intestinal and hepatic CYP3A4 catalyze hydroxylation of 1 alpha,25-dihydroxyvitamin D-3: Implications for drug-induced osteomalacia. Molec. Pharmacol. 2006; 69:56-65.
21. Gerntholtz T,Pascoe MD, Botha JF, Halkett J, Kahn D. The use of a cyclosporin-ketoconazole combination: making renal transplantation affordable in developing countries. Eur. J. Clin. Pharmacol. 2004; 60:143-148.
Further Reading
Gardiner SJ, Begg EJ. Pharmacogenetics, drug-metabolizing enzymes, and clinical practice. Pharmacol. Rev. 2006; 58:521-590.
Ioannides C, ed. Cytochromes P450: Metabolic and Toxicological Aspects. 1996. CRC Press, Boca Raton, FL.
Ioannides C, ed. Enzyme Systems that Metabolise Drugs and other Xenobiotics. 2002. John Wiley & Sons, Chichester.
Ioannides C, Lewis DFV. Cytochromes P450 in the bioactivation of chemicals. Curr. Topics Medic. Chem. 2004; 4:1767-1788.
Park BK,Kitteringham NR, Maggs JL, Pirmohamed M, Williams DP. The role of metabolic activation in drug-induced toxicity. Annu. Rev. Pharmacol. Toxicol. 2005; 45:177-202.
Tang C, Lin JH, Lu AYH. Metabolism-based drug-drug interactions: what determines individual variability in cytochrome P450 induction? Drug Met. Disp. 2005; 33:603-613.
Xu G, Li CY-T, Kong A-NT. Induction of Phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 2005; 28:249-268.
Zhou S, Chan SY, Goh BC, Chan E, Duan W, Huag M, McLeod HL. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacokin. 2005; 44:279-304.
See Also
ADMET Properties of Drugs
Cytochrome P450 Monooxygenases, Chemistry of
Drug Metabolizing Enzymes, Chemistry of
Enzyme Inhibition, Chemistry and Mechanisms of
Polymorphisms, Detection of