CHEMICAL BIOLOGY
Lead Optimization in Drug Discovery
Craig W. Lindsley, Departments of Pharmacology and Chemistry, Vanderbilt Institute of Chemical Biology, Vanderbilt University Medical Center, Nashville, Tennessee
David Weaver and Thomas M. Bridges, Department of Pharmacology, Vanderbilt Institute of Chemical Biology, Vanderbilt University Medical Center, Nashville, Tennessee
J. Phillip Kennedy, Department of Chemistry, Vanderbilt University, Nashville, Tennessee
doi: 10.1002/9780470048672.wecb634
Lead optimization in drug discovery has changed significantly over the past five years and no longer is fragmented into separate hit-to-lead and lead optimization phases. Chemical lead optimization from high-throughput screening (HTS) to clinical candidate identification is now one seamless process that draws on new technologies for accelerated synthesis, purification, and screening of directed, iterative compound libraries. Advances in high-throughput screening technologies allow detection of new allosteric modes of target modulation, which provides new chemotypes and target opportunities. With the incorporation of drug metabolism and pharmacokinetics (DMPK) inputs early in the lead optimization workflow, molecules are not optimized solely for target potency and selectivity. Moreover, ''closed-loop'' workflows are in place such that synthesis and primary screening operate on a 1 -week turnaround for up to 48 compounds/week with DMPK data cycling every other week to guide compound design, which provides expedited timelines for the development of proof-of-concept compounds and clinical candidates with limited human resources.
The competitive drug discovery environment, whether in industry or academia, requires constant innovation and refinement as a prerequisite for success. A combination of market, patient, and regulatory concerns requires that new chemical entities act on truly novel, and therefore not clinically validated, molecular targets. With the high attrition rates and limited human resources, drug discovery efforts must focus on a large and diverse collection of molecular targets and judiciously employ enabling technologies and new paradigms to develop simultaneously multiple early stage programs. Importantly, the goal at the outset of a nascent program is to provide rapid target validation in vivo with a novel small molecule or to deliver a quick kill for the program so that resources can be reassigned. Coupled with these concerns is the need to establish intellectual property to support broad generic patent claims early in the development process, as chemical space is shrinking at an alarming rate (1).
Historically, the scope, mission, and technology platforms of lead optimization groups varied considerably across the drug discovery industry, which led to highly variable success rates (2-5). Some organizations had defined “hand-off” criteria and fragmented lead optimization into a hit-to-lead phase and a chemical lead optimization phase. Hit-to-lead focused on optimizing screening hits, usually by library synthesis (solution phase and/or solid phase), for target potency with minimal concern for selectivity, ancillary pharmacology, and pharmacokinetics (PK). Leads that met certain potency criteria and displayed robust structure-activity relationships (SAR) then would be “handed-off” to a second group for the lead optimization phase, wherein more classic medicinal chemistry [single compound synthesis and intense drug metabolism and pharmacokinetics (DMPK) profiling] would occur (2-5).
In the past five years, lead optimization in drug discovery has changed significantly and no longer needs to be fragmented into separate hit-to-lead and lead optimization phases (2-5). Major advances have been made in HTS technologies, which have enabled detection of novel modes of target modulation. Once limited to detection of classic agonists and antagonists by HTS, kinetic imaging plate readers, such as Flourescence Destection Screening System (FDSS) and Flourescence Imaging Plate Reader (FLIPR), allow for the HTS identification of positive and negative allosteric modulators of both known and novel targets, which offers new chemotypes as well as improved selectivity and safety profiles (6, 7). Chemical lead optimization, from evaluation of screening hits to clinical candidate identification, now can be a seamless process that draws on new technologies for accelerated synthesis, purification, and screening. Directed, iterative compound libraries now are employed throughout the lead optimization continuum with single compound synthesis restricted to an “as needed” basis. With the incorporation of DMPK inputs at the initiation of a lead optimization program, molecules are not optimized solely for target potency and selectivity but also for the optimization of protein binding and pharmacokinetics and for diminishing CYP inhibition (1-11). Moreover, “closed-loop” work flows are in place such that chemical synthesis and primary screening data operate on a 1-week turnaround for hundreds of compounds/week, with DMPK data cycling every other week to guide compound design and to provide expedited timelines for the development of proof-of-concept compounds to validate/kill novel molecular targets and to deliver clinical candidates with limited human resources. To avoid the negative stigma of combinatorial chemistry, both industrial and academic laboratories, this new paradigm for lead optimization is coined “technology-enabled synthesis” or “TES”; however, a more accurate moniker would be “technology-enhanced medicinal chemistry” (12-21).
Advances in High-Throughput Screening Technologies
The first step toward a successful lead optimization campaign begins in a state-of-the-art screening facility, which is ideally based on a philosophy that values the ability to automate complex biologic assays to allow screening of difficult-to-screen targets and to detect novel mechanisms of target modulation. Historical HTS paradigms valued the use of automation only to increase throughput; however, the focus now is to execute faithfully complex tasks with high precision. Modern HTS facilities employ automated screening systems composed of state-of-the-art liquid handling, plate readers, incubators, and other instruments to support a wide variety of cell-free and cell-based assays that range from enzyme assays on purified proteins to phenotypic screens on model organisms like C. elegans and zebrafish embryos (6, 7, 21-24). Advances in analysis software allow for information-rich assay forms, primarily in cell-based or organism-based environments, with read modes based on either parallel acquisition of kinetic data that use instruments like the Hamamatsu FDSS kinetic imaging plate readers (21, 25) or on object-based screening that uses high spatial resolution devices like automated microscopes or the BlueShift Isocyte (21, 26). Both of these read modes yield complex, information-rich data sets. The analysis and storage of such data can be challenging; however, the success of a lead optimization campaign is linked directly to the ability to acquire, synthesize, store, and present compounds as well as to the ability to collect/analyze data from the biologic systems for which we hope to discover proof-of-concept compounds and clinical candidates (6, 7, 21-26).
A triplicate screen to identify classic and allosteric modes of target modulation
Miniaturization of assays that employ kinetic imaging plate readers allow for the development of robust high-throughput calcium mobilization-based assays that detect the activation/ inhibition of molecular targets through both classic and allosteric modes of target modulation. For instance, we can measure receptor-induced intracellular release of calcium by using an imaging-based plate reader that makes simultaneous measurements of calcium levels in each well of a 384-well plate. In a novel triplicate-screening paradigm (Fig. 1), either vehicle or a test compound was added to cells expressing a G Protein-coupled receptor (GPCR) that has been loaded with fluorescent dye, Fluo-4. After a 2.5-minute incubation period, a submaximally effective (EC20) concentration of orthosteric agonist was added, followed by a nearly maximal (EC80) concentration added 1 minute later. In this manner, we can screen for and identify classic agonists/antagonists, allosteric potentiators, and antagonists simultaneously, which maximizes the efficiency of each screen and delivers a diverse collection of hits for chemists to optimize. This paradigm affords the medicinal chemists with options, both in terms of a modulatory mechanism for their therapeutic target and in terms of a chemotype, for the lead optimization campaign in a manner previously unavailable (27).
Of course, technology has not advanced only for the screening of GPCRs but also for kinases and ion channels. Kinase screens now employ both low and high concentrations of ATP to identify both ATP-competitive and allosteric inhibitors. Numerous technology platforms recently have appeared for ion channels targets, such as highly automated Ion-Works and Q-patch, which avoid the need for burdensome and slow single patch-clamp experiments (21-27).

Figure 1. Triplicate screen to identify allosteric modes of target modulation. (a) Vehicle with an EC20 and EC80 of agonist, (b) waveform profile of an agonist; (c) waveform profile of an antagonist (flowup necessary to distinguish orthosteric versus allosteric antagonist), and (d) waveform profile of a potentiator, aka, positive allosteric modulator. A single screen generates an entire spectrum of hits.
Solution-Phase Parallel Synthesis for Lead Optimization
The chemical technologies and platforms for chemical lead optimization have undergone a major paradigm shift in the past 10 years. In the 1990s, hit-to-lead efforts were driven by combinatorial chemistry and characterized by large (1000-10,000 member) solid-phase libraries that required months to synthesize and characterize (28). Often, by the time the library was ready for screening, the SAR of the program and/or lead series had moved on, and the value of the library was minimal (28, 29). As a result, most pharmaceutical companies disbanded their combinatorial chemistry groups, and lead optimization relied primarily on single compound synthesis or small collections (less than 12) of compounds. Driven to make the lead optimization process more efficient, the concept of solution-phase parallel synthesis began to gain favor, and technologies rapidly began to develop to create this new approach (30-34). In the last five years, major advances were made in the availability of polymer-supported reagents and scavengers and in the advent of precision-controlled, single-mode microwave synthesizers for organic synthesis. Along with the development of robust mass-directed, preparative HPLC purification, platforms have revolutionized and accelerated lead optimization (20, 30-36).
Solution-phase parallel synthesis (SPPS)
Key to the success of SPPS was the development of “scavenging reagents.” Scavenging (quenching) reagents are highly effective tools for the rapid purification and isolation of the desired product(s) from a solution-phase reaction by forming either covalent or ionic bonds with excess reactants and/or reaction by-products. In general terms, scavenging can be considered a “phase switching” technique wherein a chemo-selective reaction is employed to switch the phase of one product relative to another by virtue of a “tag” attached to the scavenging reagent (30-34). Three major classes of scavenging reagents are categorized by the nature of the phase tag: solid-phase polymers, ionizable functional groups, and fluoroalkyl chains (30-34, 37). In a typical scenario, an excess of reactant B is combined with A to provide product P along with B and other reaction by-products X in a homogeneous solution-phase reaction. Then, B and X are chemo-selectively removed from solution in a subsequent “scavenging” step with a scavenging reagent 1 linked to a phase tag. After separation of the resulting phases, the product, P, is obtained in high purity by simply evaporating the solvent (Fig. 2).
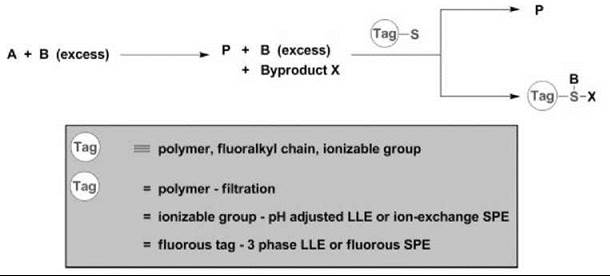
Figure 2. SPPS and ''phase switching''
The most commonly used tags are solid-phase polymers, and hence, a wealth of literature centers on the applications of polymer-supported scavenger reagents to transfer a captive species from the organic liquid phase to the solid phase for removal by filtration. Indeed, this approach has gained widespread acceptance because of the commercial availability of a diverse array of electrophilic and nucleophilic polymer-supported scavenging reagents along with an abundance of polymer-supported reagents. Moreover, because of site isolation, “cocktails” of polymer-supported reagents and scavengers can be used simultaneously (30-34).
Another commonly used tagging strategy involves linking a scavenger to an ionizable functional group, such as a COOH (pKa < 5) or an NR2 (pKa > 10). In this instance, the captured species can be phase transferred selectively by either pH-adjusted liquid/liquid extraction or by solid-phase extraction (SPE) on an ion-exchange cartridge that leaves the desired product either in the organic liquid phase or in the SPE cartridge eluent (34). SPE is a very attractive method for purification because a crude reaction simply is applied to a disposable silica plug and grafted with either a sulfonic acid (SCX—strong cation exchange) or a tertiary amine (SAX—strong anion exchange), and neutral molecules are eluted off with methanol, whereas ionizable functional groups are retained on the SPE cartridge. Unfortunately, this strategy impacts the diversity of a library by limiting the presence of ionizable groups to either neutral or orthogonally charged library members (34).
Relying on the affinity that fluoroalkyl chains have for each other and the phobia that they exhibit toward both organic molecules/solvents and aqueous solvents, researchers began examining fluorous tags as a means of phase switching (35). Initially, efforts centered on “heavy” fluorous tags (60% or more fluorine content by molecular weight; for example, 18 or more difluoromethylene, CF2, groups) that used liquid/liquid phase separation to isolate fluorous-tagged molecules from untagged organics. Typically, a three-phase liquid/liquid extraction, which requires an organic layer, an aqueous layer, and a fluorous layer (a perfluorohexane such as FC-72), delivers pure material. More recently, fluorous solid-phase extraction (FSPE) that employs fluorous silica gel (reverse-phase silica gel with a fluorocarbon bonded phase) has been developed to separate effectively both “heavy” fluorous-tagged molecules as well as “light” fluorous-tagged molecules (4 to 10 CF2 groups) from untagged organics. The FSPE columns, referred to as FluoroFlash (Fluorous Technologies, Pittsburgh, PA) columns, retain the fluorous-tagged material when eluted with a fluorophobic solvent, such as 80/20 MeOH/H2O, which allows the untagged organic molecule to elute rapidly from the column. Homogeneous reaction kinetics, generally with respect to charged and neutral functional groups and a variety of efficient phase-separation options, have spurred a dramatic increase in the development of fluorous scavenging reagents and protocols (37).
Microwave-assisted organic synthesis (MAOS)
MAOS, fueled by the development of precision-controlled, single-mode microwave reactors, has a profound impact on organic and parallel synthesis. Reaction times typically are cut by orders of magnitude, and it is usual to observe a diminution in side product formation. Moreover, MAOS reactions tend to be general in scope and lend themselves to the synthesis of libraries to develop SAR rapidly. These advantages, easily appreciated when considering established routes with successful reactions, are even more valuable when working out robust conditions for a synthesis. Exploratory reactions can be conducted in minutes to hours instead of days, and speculative, higher-risk ideas can be pursued with minimal time investment. Indeed, MAOS allows any chemistry to be pursued in parallel and allows chemistries that historically were avoided for library synthesis (mulitcomponent reactions, organometallics, transition-metal catalyzed couplings, etc.) to be completed successfully in minutes (20, 38, 39).
Beyond the speed advantage, two additional merits of MAOS and modern reactors should be highlighted: precision and reaction scope. As has been noted in these pages and elsewhere, the benefits of MAOS have been studied in multimode “kitchen microwaves” for decades; what prevented acceptance in the wider community was irreproducibility because of a lack of pressure and temperature control (Fig. 3). In addition, kitchen microwaves employ multimode resonators that lead to a heterogeneous field and local “hot” spots (Fig. 3a); despite this disadvantage, early work demonstrated the use of MAOS. Modern systems (Fig. 3b) provide a homogeneous field and precise control of temperature and pressure, and they bare little resemblance to kitchen microwaves. Importantly, MAOS relies on dipolar oscillations and ionic conduction, for example, molecular friction, to generate heat and to afford uniform heating of the sample. In contrast, conventional thermal heating relies on heat transfer from the walls of a reaction vessel and affords nonuniform heating of the sample (Fig. 3c). This uniform heating and rapid time to set temperature delivers reproducible results with fewer side products and, as a result, higher chemical yields (20).
Also, MAOS technology significantly has impacted library design and synthesis. For instance, when presented with a small heterocycle as a hit from an HTS, MAOS technology allows one not only to synthesize and evaluate substitutions on the parent heteocyclic scaffold rapidly but also to synthesize and evaluate multiple heterocyclic templates with diverse substituents in parallel (Fig. 4). Therefore, a single library will contain multiple heterocyclic cores with varying degrees of basicity and topology, while broadening the generic scope for a composition of matter patent (38).

Figure 3. Microwave-assisted organic synthesis. (a) A domestic kitchen microwave with local ''hot spots.'' (b) A single-mode microwave reactor for organic synthesis. (c) Comparison of surface temperature between microwave and conventional heating.
Mass-directed preparative HPLC
Despite the purity obtainable by SPPS scavenging (typically >90%) and the high purities obtained by MAOS (also typically >90%), modern lead optimization programs require >95% purity of all compounds that contribute to the development of SAR and that advance into DMPK assays. For years, many labs employed UV-directed preparative HPLC and often multichannel units to increase throughput. Although this approach worked, purification of a single sample might lead to 30-40 fractions per sample, which then required analysis by analytical Liquid Chromatography/Mass Spectrometer (LCMS) to identify which fractions contained the desired product (34, 40). In 2000, several vendors launched preparative LCMS units that offered mass-directed fractionation. Now, purification of a single crude sample afforded only one or two pure fractions—a significant advance. Additional modifications for library purification included DMSO slugs to bracket sample injections or “at-column dilution” to provide robust chromatography and prevent in-line sample precipitation before the column. These modified systems were capable of purifying, in a single pass, 60 to 80 compounds per day with purity levels exceeding 98%; however, the systems required an expert chromatographer to develop custom gradients for each sample in a library (34, 40). Recently, several vendors launched an analytical-to-preparative LCMS software package that addressed the need for a dedicated, expert chromatographer to operate each prep LCMS instrument. With analytical-to-preparative software, a file containing the compound ID and exact mass for each sample in a library to be purified is uploaded into the preparative LCMS system, which then electronically accesses the analytical LCMS data and extracts the retention time of the mass of interest from the crude sample chromatograms. The preparative LCMS system analytical-to-preparative software then calculates a customized gradient for each sample in a library and therefore reduces the need for an experienced chromatographer to achieve excellent first-pass purification results. This feature also allows the instrument to run overnight unattended and additionally increases operational efficiency.

Figure 4. MAOS to access rapidly diverse heterocyclic scaffolds form a common intermediate.
Postpurification sample handling and compound characterization
Modern parallel synthesis laboratories, for example, high- throughput medicinal chemistry laboratories, have borrowed a page from the automotive industry and have developed highly efficient assembly lines for postpurification sample handling and compound characterization. Automated weighing systems with bar code readers scan and record weights on unique bar-coded vials into which pure compounds from the preparative LCMS systems are transferred for concentration in a sample evaporator. After the dry-down step, the bar-coded vials with pure, solid sample are transferred to a liquid handling robot. This instrument scans each bar code, weighs the vial, and determines the net weight of the pure product. This data file is merged with a registration file that contains the molecular weight of the compound, and the system software then calculates the volume of DMSO required to dilute the samples to a preset concentration for screening. The system then dilutes the samples, transfers the DMSO stock solution to a 96-well plate, and generates an electronic plate map file for submission to the primary screen, in vitro drug metabolism assays, pharmacokinetic cassettes, and for flow cell NMR (vide infra). With this highly automated workflow, a single scientist can oversee the postpurification sample handling of thousands of samples per week (1-5).
Lack of complete compound characterization has been a major shortcoming of combinatorial chemistry and early high-throughput medicinal chemistry laboratories that led to poor adoption by traditional medicinal chemists. Once again, technology has advanced such that every member of a compound library is fully characterized to the same standard as a single compound prepared by a traditional medicinal chemist. After purification by preparative LCMS, a final analytical LCMS is generated for each sample at two wavelengths (214 nM and 254 nM) and evaporative light scattering detection (ELSD). The LCMS vials then are delivered to a quadrupole time of flight (QTOF) mass spectrometer system with a 100-position autosampler for accurate mass measurement (high-resolution mass specification) determinations. For each sample, 1H NMR spectra are obtained. Initially, NMR tubes were prepared for each sample, and chemists took advantage of NMRs with autosamplers. More recently, flow-cell NMR and solvent suppression software allow for quality NMR spectra to be obtained from DMSO stock solutions in 96-well plate (41). Importantly, this ensures high-purity samples, prepared in directed libraries, to drive lead optimization programs and generate quality SAR at the same level as compounds prepared by singleton synthesis.
Expedited Drug Metabolism and Pharmacokinetics
Lead optimization involves more than just optimizing for target potency. New technology allows early lead optimization campaigns to address and consider multiple parameters and inputs for each round of iterative library synthesis. These inputs allow for the rapid development of potent compounds with drug-like profiles, as opposed to just potent compounds. These data also provide “quick kills” to individual leads or series and allow the lead optimization effort to re-direct resources toward more productive leads (1-5, 8-20).
High-throughput in vitro drug metabolism assays
Significant effort has been applied to the miniaturization and DMSO compatibility of in vitro drug metabolism assays. Now, a 48-member library in a 96-well plate of DMSO stock solution can be evaluated rapidly in cytochrome P450 inhibition assays (CYP3A4, 2D6, 2C9), protein binding assays (rat, dog, and human), logP, hERG binding, and other standard assays (8-11, 42, 43). Previously, chemists were forced to choose which compounds to evaluate in these assays, and often only the most potent analogs would be selected; the most potent analogs were not necessarily the ones with the most promise as clinical candidates. Being able to acquire these data for an entire library provides opportunities to pursue leads within a series with the most balanced potency and DMPK profiles. This ability is of critical importance in lead optimization to ensure that drug-like leads are being pursued and additionally refined. Similarly, cassette dosing of compounds in liver microsomes and hepatocytes enables evaluation of an entire library in short order (8-11, 42, 43). This timely evaluation is especially valuable in the lead optimization of a backup clinical candidate program, wherein the clinical candidate is the positive control and new compounds (typically five to six per cassette) are viewed qualitatively as more or less stable than the first clinical candidate. Once an in vitro /in vivo correlation can be established for a given series, these rapid cassette experiments can drive a lead optimization program and require only intermittent in vivo experiments (8-20, 42, 43).
Pharmacokinetic cassettes
Resource and practical constraints prohibit acquiring single rat and dog pharmacokinetics (PK) (i.v. and p.o.) for every member of a library; however, the compound with the best PK may not be the most potent analog in a library, and knowing this data is crucial for lead optimization. In fact, chemical lead optimization programs have been guided solely by optimization of PK. With the success of in vitro cassette paradigms for microsomal and hepatocyte stability, the concept recently was extended to in vivo PK in rats and dogs (Fig. 5) so that an entire library could be evaluated in vivo employing a limited number of animals (8-11). However, some caveats exist. Combinatorial oral dosing to determine oral bioavailability (%F) in cassette format generally proved not very reproducible nor in agreement with single PK experiments. In contrast, intraveneous cassette dosing in both rats and dogs proved highly reproducible and within the error of a single PK experiment and is a valuable tool to determine qualitative rates of clearance between five to six new compounds and an internal control of known clearance (8-11). PK cassettes employ an overall low dose of test compounds to minimize potential drug-drug interactions. These rapid cassette experiments prioritize which compounds from a library then should be studied in single i.v./p.o. single animal PK studies. As shown in Fig. 5, compound 2 has qualitatively lower intrinsic plasma clearance rate than an internal control compound with known bid (twice daily predicted dosing) PK (Cl = 12 mL/min/kg), whereas the other four compounds in the cassette have higher clearance and need not be studied more.

Figure 5. Pharmacokinetic evaluation of libraries: cassette dosing to evaluate clearance rates relative to a bid control compound.
Expedited, "Closed-Loop" Work Flow for Lead Optimization
Combining all above-mentioned technologies and paradigms for synthesis, screening and DMPK evaluation affords an aggressive, expedited process for chemical lead optimization (1, 12-21). This protocol allows one to two synthetic chemists to support a chemical lead optimization effort with accelerated timelines that deliver proof-of-concept compounds within 6 months and clinical candidates within 12 months of the initiation of a lead optimization campaign.
Library design
Independently, the technologies and strategies described herein provide improvements for chemical lead optimization; however, when they become closely aligned with screening and DMPK resources in a “closed-loop” paradigm, the impact on drug discovery is exponential (Fig. 6). Starting from an HTS hit, considerable attention is devoted first to library design, without question the most important component of a successful lead optimization effort. Library design changes over the course of a lead optimization campaign. The initial design strategy is to explode SAR around a screening hit and to be as diverse as possible with respect to monomer input and analog synthesis to rapidly identify productive changes for additional optimization. In addition, this component of lead optimization is conducted often in parallel, wherein a single chemist simultaneously will synthesize diversity libraries around four to six hits to identify expediently the best leads for additional optimization. After this initial diversity-oriented explosion, library design must become more focused to impact drug discovery goals: Random libraries do not accelerate programs. It is important to approach directed library design from a medicinal chemistry perspective and to assemble the library as a collection of single compounds designed to address a particular issue. For example, the design of a 24-member library should involve careful thought regarding what four single compounds would be synthesized first to increase potency, improve PK, and so forth. Then, for each of the first four analogs synthesized, designers should consider what the next four analogs should be if the first changes were productive or nonproductive. This exercise in library design generates quality data that drive a lead optimization program toward proof-of-concept compounds and clinical candidates very quickly (1, 12-21).
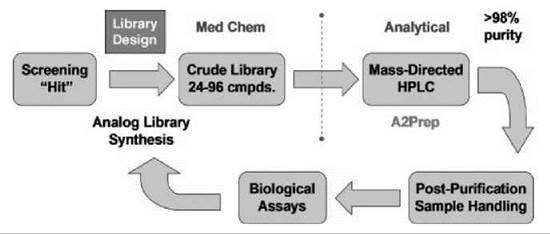
Figure 6. Expedited ''closed-loop" lead optimization paradigm.
Division of labor
Another key feature of the “closed-loop” approach to lead optimization involves division of labor and the transfer of samples from medicinal chemists to the analytical chemists. In this paradigm, the medicinal chemists design and synthesize the compound libraries (24 to 96 compounds) and obtain analytical LCMS reports for each member of the library. At this point, the medicinal chemists transfer the crude samples to the analytical chemists who purify the libraries by mass-directed preparative HPLC to >98% by using analytical-to-preparative software and perform all postpurification sample handling and coordinate submission of samples, in a 96-well plate format, to the biologists and DMPK personnel for screening (vide supra) (12-20). If resources allow, this division of labor affords opportunities for the medicinal chemists to focus on library design, develop and optimize new chemistries, and pursue multiple lead series in parallel (1, 12-21).
Data turnaround
The success of this paradigm hinges on rapid screening and dissemination of data to the medicinal chemists so that the next iteration of library synthesis can be initiated. To facilitate this, the delivery of compounds is coordinated with the biologists and assays are run the same day the compound libraries are delivered. Biologic data then is returned within 24 hours of receipt of the libraries. This allows lead optimization to operate on a 1 week turnaround between the initiation of chemical synthesis and the generation of primary assay data. Secondary and/or selectivity data typically trail primary data by 1 to 2 days. As these data trigger the need for DMPK information, DMPK data typically follow 1 week after the initial assay data is obtained. Overall, this expedited process parallels traditional singleton medicinal chemistry work flows but generates data on hundreds of compounds in the time it used to take to evaluate just a few compounds. Moreover, this protocol allows one to two synthetic chemists to support a chemical lead optimization effort with accelerated timelines delivering proof-of-concept compounds within 6 months and clinical candidates within 12 months of the initiation of a lead optimization campaign. It is important to note that this lead optimization paradigm requires collaboration, close and frequent communication with biology and DMPK colleagues, sophisticated databases to store the volumes of data generated, and a major investment in technology (1, 12-20).
References
1. Glaser V. Optimizing strategies for hit-to-lead process. Gen. Engin. Biotech. News. 2005; 25:31-37.
2. Gillespie P, Goodnow RA Jr. The hit -to- lead process in drug discovery. Ann. Rep. Med. Chem. 2004; 39:293-304.
3. Keseru GM, Makara GM. Hit discovery and hit -to- lead approaches. Drug Discov. Today 2006; 11:741-748.
4. Bleicher KH, Boehm H-J, Mueller K, Alanine AI. A guide to drug discovery: hit and lead generation: beyond high-throughput screening. Nat. Rev. Drug Disc. 2003; 2:369-378.
5. Meador V, Jordan W, Zimmermann J. Increasing throughput in lead optimization in vivo toxicity screens. Curr. Opin. Drug Discov. Devel. 2002; 5:72-78.
6. Wood M, Smart D. Real time receptor function in vitro: micro-physiometry and the fluorometric imaging plate reader (FLIPR). In: Receptors: Structure and Function. 2nd edition. Stanford C, Horton R, eds. 2001. Oxford University Press, Oxford. pp. 175-191.
7. Wood MD, Jerman J, Smart D. The FLIPR: a major advance in the study of neurotransmitter receptors. Rec. Res. Dev. Neurochem. 2000; 3:135-142.
8. Huang R, Qian M, Chen S, Lodenquai P, Zeng H, Wu J-T. Effective strategies for the development of specific, sensitive and rapid multiple-component assays for cassette dosing pharmacokinetic screening. Int. J. Mass Spect. 2004; 238:131-137.
9. Halladay JS, Wong S, Jaffer SM, Sinhababu AK, Khojasteh-Bakht SC. Metabolic stability screen for drug discovery using cassette analysis and column switching. Drug Metabol. Lett. 2007; 1:67-72.
10. Bu H-Z, Magis L, Knuth K, Teitelbaum P. High-throughput cytochrome P450 (CYP) inhibition screening via a cassette probe-dosing strategy. VI. Simultaneous evaluation of inhibition potential of drugs on human hepatic isozymes CYP2A6, 3A4, 2C9, 2D6 and 2E1. Rapid Commun. Mass Spect. 2001; 15:741-748.
11. Cai Z, Sinhababu AK, Harrelson S. Simultaneous quantitative cassette analysis of drugs and detection of their metabolites by high performance liquid chromatography/ion trap mass spectrometry. Rapid Commun. Mass Spect. 2000; 14:1637-1643.
12. Lindsley CW, Wisnoski DD, Leister WH, O’Brien JA, Lemiare W, Williams DL Jr, Burno M, Sur C, Kinney GG, Petti- bone DJ, Miller PR, Smith S, Duggan ME, Hartman GD, Conn PJ, Huff JR. Discovery of positive allosteric modulators for the metabotropic glutamate receptor subtype 5 from a series of N-(1,3-Diphenyl-1H-pyrazol-5-yl) benzamides that potentiate receptor function in vivo’ J. Med. Chem. 2004; 47:5825.
13. Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME. Allosteric Akt (PKB) kinase inhibitors. Discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem Lett. 2005; 15:761-765.
14. Wolkenberg SW, Lindsley CW. MAOS: a ‘diversity engine’ for parallel synthesis. Discov Devel 2004; 4:1-5.
15. Zhao Z, O’Brien JA, Lemiare W, Williams DL Jr, Jacobson MA, Sur C, Pettibone DJ, Tiller PR, Smith S, Hartman GD, Lindsley CW. Synthesis and SAR of GlyTl inhibitors derived from a series of N-((4-(morpholine-4-carbonyl)-l-(propylsulfonyl)piperidin-yl)methyl) benzamides’ Bioorg. Med. Chem.Lett. 2006; 16:5968- 5972.
16. Nanda KK, Nolt MB, Cato MJ, Kane SA, Kiss L, Spencer RH, Jixin Wang J, Lynch JL, Regan CP, Stump GL, Li B, White R, Yeh S, Bogusky MJ, Bilodeau MT, Dinsmore CJ, Lindsley CW, Hartman GD, Wolkenberg SE, Trotter BW. Potent antagonists of the atrial lKur/Kvl.5 potassium channel: synthesis and evaluation of analogous N,N-diisopropyl-2-(pyridine-3-yl)acetamides. Bioorg. Med. Chem. Lett. 2006; 16:5897-5901.
17. Wolkenberg SE, Zhao Z, Kapitskaya M, Webber AL, Pertukhin K, Tang YS, Dean DC, Hartman GD, Lindsley CW. Identification of potent agonists of Photoreceptor-Specific Nuclear Receptor (NR2E3) and preparation of a radioligand. Bioorg. Med. Chem. Lett. 2006; 16:5001-5004.
18. Lindsley CW, Zhao Z, Leister WH, O’Brien JA, Lemiare W, Williams DL Jr, Chen T-B, Chang RSL, Burno M, Jacobson MA, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Tsou NN, Duggan ME, Conn PJ, Hartman GD. Design, synthesis and in vivo efficacy of novel glycine transporter-1 (GlyT1) inhibitors derived from a series of [4-Phenyl-1-(propylsulfonyl)piperidin-4-yl] methyl benzamides. ChemMedChem. 2006; 1:807-811.
19. Zhao Z, Wisnoski DD, O’Brien JA, Lemiare W, Williams DLJr, Jacobson MA, Wittman M, Ha S, Schaffhauser H, Sur C, Pet- tibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. Challenges in the development of mGluR5 positive allosteric modulators: the discovery of CPPHA. Bioorg. Med. Chem. Lett. 2007; 17:1386-1391.
20. Lindsley CW, Wolkenberg SE, Shipe W. Accelerating lead development by microwave-enhanced medicinal chemistry. Drug Discov. Today: Technol. 2005; 2:155-161.
21. Lindsley CW, Weaver D, Conn PJ, Marnett L. Preclinical drug discovery research and training at Vanderbilt. ACS Chem. Biol. 2007; 2:17-20.
22. Ueki T. High throughput screening in the process of drug discovery. Nippon Yakurigaku Zasshi 2007; 129:276-280.
23. Macarron R. Critical review of the role of HTS in drug discovery. Drug Discov. Today 2006; 11:277-279.
24. DeSimone RW. The use of combi chem, high-speed analog chemistry and HTS in drug discovery. Drug Discov. Today 2003; 8:156.
25. For information on Hamamatsu FDSS. www.hamamtsu.com.
26. For information on BlueShift Isocyte. www.blueshiftbiotech.com.
27. Rodriguez A, Williams R, Jones C, Niswender C, Meng X, Lindsley CW, Conn PJ. Novel positive allosteric modulators of mGluR5 discovered in HTS have in vivo activity in rat behavioral models of antipsychotic activity. Nat. Chem. Bio. In press.
28. Dorwald FZ. Organic Synthesis on Solid Phase. 2000. Wiley-VCH, Weinheim.
29. Ellingboe JW Solid-phase synthesis in lead optimization and drug discovery. Curr. Opin. Drug Disc. Devel. 1999; 2:350-357.
30. Kuroda N, Hird N, Cork DG. Further development of a robust workup process for Solution-Phase High-Throughput Library Synthesis to address environmental and sample tracking issues. J. Comb. Chem. 2006; 8:505-512.
31. Altorfer M, Ermert P, Faessler J, Farooq S, Hillesheim E, Jeanguenat A, Klumpp K, Maienfisch P, Martin JA, Merrett JH, Parkes KEB, Obrecht J-P, Pitterna T, Obrecht D. Applications of parallel synthesis to lead optimization. Chimia. 2003; 57:262-269.
32. Booth RJ, Hodges JC. Solid-supported reagent strategies for rapid purification of combinatorial synthesis products. Acc. Chem. Res. 1999; 32:18-26.
33. Kaldor SW, Siegel MG. Combinatorial chemistry using polymer-supported reagents. Curr. Opin. Chem. Bio. 1997; 1:101-106.
34. Siegel MG, Hahn PJ, Dressman BA, Fritz JE, Grunwell JR, Kaldor StW. Rapid purification of small molecule libraries by ion exchange chromatography. Tetrahedron Lett. 1997; 38:3357-3360.
35. Leister WH, Strauss KA, Wisnoski DD, Zhao Z, Lindsley CW. Development of a custom high-throughput preparative liquid chromatography/mass spectrometer platform for the preparative purification and analytical analysis of compound libraries. J. Comb. Chem. 2003; 5:322-329.
36. Kyranos JN, Lee H, Goetzinger WK, Li LYT. One-minute full-gradient HPLC/UV/ELSD/MS analysis to support high-throughput parallel synthesis. J. Comb. Chem. 2004; 6:796-804.
37. Gladysz JA, Curran DP, Horvath IT, Eds. Handbook of Fluorous Chemistry. 2004. Wiley-VCH, New York.
38. Shipe WD, Yang F, Zhao Z, Wolkenberg SE, Nolt MB, Lindsley CW. Convenient and general microwave-assisted protocols for the expedient synthesis of heterocycles. Heterocycles. 2006; 70:665-689.
39. Kappe CO, Dallinger D. The impact of microwave synthesis on drug discovery. Nat. Rev. Drug Disc. 2006; 5:51-63.
40. Blom KF. Two-pump at-column-dilution configuration for preparative liquid chromatography-mass spectrometry. J. Comb. Chem. 2002; 4:295-301.
41. Curran SA, Williams DE. Design and optimization of an NMR flow cell for a commercial NMR spectrometer Appl. Spectros. 1987; 41:1450-1454.
42. Kariv I, Rourick RA, Kassel DB, Chung TDY. Improvement of “hit-to-lead” optimization by integration of in vitro HTS experimental models for early determination of pharmacokinetic properties. Comb. Chem. High Throughput Screen. 2002; 5:459-472.
43. Kenakin T. A guide to drug discovery: predicting therapeutic value in the lead optimization phase of drug discovery. Nat. Rev. Drug Discov. 2003; 2:429-438.
Further Reading
Ardrey A. Liquid Chromatography-Mass Spectrometry: An Introduction. 2005. John Wiley & Sons, New York.
Burgess KD, Ed. Solid Phase Organic Synthesis. 2000. John Wiley & Sons, New York.
Sucholeiki I, Ed. High-Throughput Synthesis. 2001. Marcel Dekker, Inc., New York.
Tierney JP, Lidstrom P Microwave Assisted Organic Synthesis. 2005. CRC Press, Boca Raton, Florida.
See Also
ADMET Properties of Drugs
High Throughput Screening (HTS) Techniques: Overview of Applications in Chemical Biology
Solution-Phase Synthesis of Biomolecules
Synthetic Chemistry: Building Molecules to Modulate Biological Systems
Target Family-Biased Compound Library: Optimization, Target Selection and Validation