CHEMICAL BIOLOGY
Roles of Water in Biological Recognition Processes
Martin F. Chaplin, London South Bank University, London, United Kingdom
doi: 10.1002/9780470048672.wecb636
Water is central to biology, providing much more than the means to allow the movement of biologic molecules. It is an excellent solvent, a substrate and facilitator for reactions, and it provides a means for ionizations and extends the intricate three-dimensional surface geometry and complex charge distribution of biomolecules out towards the bulk solution. Water's hydrogen bonds and dipoles are strong enough to present structural information concerning surfaces to distant diffusing molecules and to fine tune their orientation to match potential recognition sites, but they are weak enough to break to make way for direct binding processes. Water occupies potential interaction sites until they are recognized by suitable molecules, with the release of the water providing free energy to help compensate for the entropy loss on binding. Also, water molecules may determine the conformation of the biological molecule and directly assist and take part in the recognition process. Among the techniques available, X-ray crystallography has provided the most information on these roles for water.
Aqueous Hydrogen Bonding in Biology
Water is suited highly to its role as the medium of life and clearly has guided molecular evolution. It is the third smallest molecule known, and so it can penetrate the smallest of structural crevices. It is polar and capable of hydrating and shielding ions and dissolving a wide range of materials, both polar and small nonpolar. It can bind and release hydrogen ions to create a rich ionic environment with long-range interactions. Most importantly, water can form highly directional hydrogen bonds that allow interactions with electronegative atoms and link together to form chains and clusters. All biomolecules function within this aqueous environment; with much of biology being guided by water that facilitates highly specific recognition processes (1).
The water molecule
The water molecule (H2O) consists of an electronegative oxygen atom holding tightly to the electrons from two hydrogen atoms, which leaves their positively charged nuclei partially exposed (Fig. 1a). Thus, water has a dipole that will interact with charged molecules and ions to solvate and to shield them from other charged species. Water ionizes to give hydrated protons and hydroxide ions. Importantly it can also carry excess protons and act as a proton wire to transfer protons rapidly from one site to another in biomolecular systems (2).
The disparity in the mass of the oxygen and hydrogen atoms allows the hydrogen nuclei to vibrate with considerable movement, which takes them closer to interact with other atoms and endows them with more freedom of movement. Because of water’s small size, a high density of water molecules exists in solutions that produce a high density of molecular interactions, which make large and important enthalpic and entropic contributions. In many circumstances, the free energies of interactions are relatively small because of the compensation between these terms; stronger binding to water is associated with both lower enthalpy and lower entropy (3). In other cases, such interactions can provide substantial entropic or enthalpic drive to direct a recognition process.

Figure 1. (a) The structure of an H2O molecule in liquid water, which shows the average bond length and angle, the approximate outline shape, and the surface electrostatic potential. (b) A typical tetrahedral water cluster found in liquid water that shows typical dimensions of its hydrogen bonds. Donating (d) and accepting (a) hydrogen bonds are indicated. Individual bond lengths and angles vary considerably around those given.
Hydrogen bonding
Water molecules interact with each other and with electronegative atoms by means of roughly, tetrahedrally positioned hydrogen bonds (Fig. 1b). These are modest, mainly electrostatic, directed interactions. Individually, they are strong compared with thermal fluctuations, although far weaker than covalent bonds, giving water its powerful hydration properties. The small molecular size and the extensive vibration of the hydrogen nuclei in water causes individual hydrogen bonds to have only short lifetimes (~ps), although they often will reform after breaking. Thus, water possesses significant thermodynamic hydration power sufficient to affect the structure of the hydrated biomolecules, but the rapid kinetics for hydrogen bond formation and breakage causes the hydrating water molecules to exchange rapidly, which otherwise might be taken for weak binding. This counterintuitive behavior is key to much of the use and the properties of water within biomolecular interactions. In particular, it allows water to orient molecules strongly according to neighboring surface characteristics but to move rapidly out of the way when required to allow biomolecular binding.
The directional nature of the hydrogen bonds is responsible for local correlations between the orientations of the molecules. In turn, it endows liquid water with its high dielectric and its consequent ability to shield electrostatic interactions. Also, it allows water molecules to transfer information concerning the structure of surfaces outward towards the bulk of the solution. The strength of water’s hydrogen bonds depends on the strength and the direction of the other hydrogen bonding that involves the same water molecules. The preference of water molecules for balanced numbers of donor and acceptor hydrogen bonds results in both cooperative and anticooperative effects: an accepted hydrogen bond strengthening a water molecule’s hydrogen bond donating ability but weakening its ability to accept a second hydrogen bond (Fig. 1b) (4). Some strong hydrogen bonds in biological systems are formed when water donates a hydrogen bond to charge-dense oxygen atoms in carboxylate and phosphate groups, or when water accepts a hydrogen bond from positively charged amino groups or oxonium ions.
The high density of hydrogen bonding sites in liquid water, together with their flexibility, allows water to act as a lubricant for translational movement between biomolecular surfaces while retaining hydrogen bonding and consequent control over the orientation of the moving molecules.
The heterogeneous nature of water
Under circumstances that favor hydrogen bonding, the density of water expands toward that of fully tetrahedral hydrogen-bonded ice (5). Such a structure has more negative enthalpy and lower entropy. When ice melts, this expanded structure collapses somewhat to the liquid state, but it still retains much of its hydrogen bonding. Liquid water is heterogeneous with fluctuating volumes of lower density, in which more hydrogen bonds are changing back and forth, into volumes of higher density where more broken hydrogen bonds and greater van der Waals interactions exist (3). Thus, aqueous systems are in constant flux with the local equilibria between the low-density and high-density states shifted by different surfaces, groups, and ions (6). Where the less dense structuring is found, water is more structured and viscous, whereas denser patches with greater dangling hydroxyl groups are more reactive, less viscous, and contain more rapidly diffusing molecules. The characteristics of the biomolecular structure are responsible principally for controlling the structuring in the surface water layers. Thus, the ease of diffusion for visiting molecules is controlled both toward and away from areas of biomolecular surfaces.
The hydrophobic effect
When water lies next to hydrophobic surfaces, it cannot form as many hydrogen bonds as when it is in the bulk of the liquid. It can maximize its van der Waals contacts and hydrogen bonding to itself and to the bulk by forming loose, transient networks of pentagonal rings over some areas. Otherwise, these surfaces are energetically unfavored. Water molecules next to such surfaces can translate sideways and away from the surface more easily than they can rotate, and many hydroxyl groups will be left without hydrogen-bonding partners. Hence, biomolecular hydrophobic-binding sites usually are hydrated poorly, with the water molecules they contain unable to form their optimum number of hydrogen bonds, and often they possess moderately high entropy when compared with bulk water molecules. When displaced by a complementary-shaped hydrophobic ligand, the entropy loss on binding between the biomolecules will be compensated by the binding free energy due to the complementary surface dispersion enthalpy and both enthalpic and entropic contributions from the released water.
Water binding
The strength of binding of water to and between biomolecules is difficult to predict because of the varied and complex nature of hydration interactions and the static and dynamic effects of the local environment. Binding consists of enthalpic and entropic terms and includes the effects of the rearrangement, removal, or addition of water within its surroundings. Enthalpy changes include hydrogen bonds, electrostatic terms, and dispersion terms, whereas entropy changes include conformational restrictions in the biomolecules and changes in both the rotational and the translational freedom of the water molecules reflecting differences in the organization between the bulk and interfaces. The process of binding (exemplified by protein-ligand binding) can be described by the following equilibria:
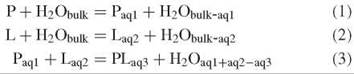
where the subscripted terms “bulk” and “aq” represent different amounts of water. Although the organization of the water at the interfaces differs on binding, most remaining interfacial water molecules retain their original anchoring hydrogen bond(s). The last equation (Equation 3) determines the binding free energy, which in turn differentiates recognition in terms of the specificity of different ligands. As clearly an entropic cost (T∆S is negative) exists in binding P to L, then compensation must exist in terms of any combination of entropy gain (T∆S is positive) by the water released, entropy gain by remaining water at the interfaces (but usually entropy is lost here), enthalpy gain (∆H is negative) in the water released, enthalpy gain by remaining water at the interfaces, and enthalpy gain in the interaction between P and L. It should be noted that apart from carboxylate and phosphate oxygen atoms, water energetically prefers to hydrogen bond to itself rather than to biomolecules, if it can.
It is known that water mediates protein folding by orienting the polypeptide strands and manages their interactions as they move from being solvent separated to forming the direct peptide hydrogen bonds found in their secondary structures (7). Although it has not been shown with the same degree of proof, it is likely that similar processes operate in macromolecular binding. Water has both dynamic and structural roles. The ease with which the water molecules move relative to each other and the biomolecular surfaces determines the dynamics, whereas the strength of the hydrogen bonding linking areas on this surface and between surfaces determine its structural effects. Dynamically, it can control and guide the process by its gradual expulsion from between surfaces, whereas structurally it contributes to the binding thermodynamics.
Biological Recognition
Our appreciation of biological recognition is different today from that of a few years ago. Then, the focus was concerned primarily with the complementary molecular surfaces (e.g., the lock-and-key mechanism), together with appropriately placed hydrogen bonding and electrostatic interactions between hydrophilic groups. Water was assumed simply to make way for the binding. Today, we know that water controls the kinetics and thermodynamics totally and must be included in any accurate description of these processes.
Biomolecules and water
Water molecules coat biomolecular, ionic, and solute surfaces in aqueous solution, minimizing the system free energy using van der Waals contacts, electrostatic interactions, and hydrogen bonds to compensate partially for the entropic and enthalpic cost of their removal from the bulk phase. They even enter hydrophobic spaces, which maximizes their van der Waals surface contacts and hydrogen bonds as far as possible.
Water molecules hydrogen bond to several biomolecular groups. Of particular note are the donated hydrogen bonds to the oxygen atoms in carboxylate, phosphate, and peptide carbonyl oxygen atoms and the accepted hydrogen bonds from peptide amines, other neutral and positively charged amines, and alcohol groups. When bridging within or between biomolecules, water usually forms three or four hydrogen bonds and rarely (if ever) forms just two hydrogen bonds of the same form, accepting or donating, because of their anticooperativity (4). Usually, only bridges that consist of single water molecules are considered, because bridging by chains of water molecules are more difficult to determine in structural studies because of their kinetic instability. However, such extensive bridges are important in the overall thermodynamics and stereospecificity of binding. Together, these aqueous interactions probably are decisive in guiding conformational rearrangements, differentiating potential binding sites, and selecting those eventually chosen (8). Normally, a mixture of direct and water-mediated links exist in about equal amounts with the direct links between the biomolecules being relatively more directed and inflexible and kinetically more difficult to reverse without the help from neighboring hydration.
The role of water in mediating binding differs between the biomolecules because of variations in the strength of the natural hydration of the interfaces, which approximates roughly to DNA > proteins > ligands.
Protein-protein recognition
The protein surface is varied in shape, structure, and charge, so it is no surprise that its hydration is varied equally with both strong interactions and weak interactions in about equal measure. Water molecules form an extensive network around proteins, which extend its surface characteristics out into the bulk. Often, this extended network seems rather thin to many analytical methods and current modeling, but it is thought likely that its influence extends somewhat further to several water layers. The amount of structured water around a protein controls the protein’s dynamics, which in turn may control its binding to other biomolecules. Water is important for the structure of individual proteins, their linking to form quaternary structures, and binding to form molecular clusters. Although such structuring may help recognition by hydrophilic sites, it will mask complementary structures where dehydration is required. In general, the hydrogen bonding to hydrating water molecules in binding sites is poor so that it may be removed easily both kinetically and thermodynamically.
Proteins may make specific links through their side chains or less specific links through their peptide links. Using water molecules to connect these groups, rather than having direct interactions, gives a greater degree of freedom in terms of the relative orientation and separation of the protein surfaces. Also, it allows far greater freedom of movement while remaining linked.
The role of water in protein-protein recognition was established first between trypsin and pancreatic trypsin inhibitor in the earliest X-ray analysis of such complexes (9). Trypsin has an aspartic acid at the bottom of its specificity binding pocket that is found to bind an arginine in soybean trypsin inhibitor or a lysine in pancreatic trypsin inhibitor. However, a water molecule is required to mediate the interaction with the lysine, as this side chain is somewhat the shorter of the two.
Analysis of 115 homodimeric protein and 46 protein-protein interfaces from crystals gave an average of about one water molecule per square nanometer of surface interface (10). Most water molecules were bound singly with 30% bridging between the proteins. Many hydrogen bonds made use of charged acid side chains or main chain peptide carbonyl groups. A separate study observed the atomic structure of the recognition sites in 75 protein-protein complexes, including 24 protease-inhibitor, 19 antibody-antigen, and 11 that are involved in signal transduction. This analysis indicated that water molecules contributed to the close packing of atoms that ensured complementarity between the protein surfaces and that provided suitably situated polar interactions (11). Both studies showed that some interfaces were essentially dry with water molecules distributed around the periphery of the contact, whereas others were hydrated within the central region of the interfaces. As an example, the E9 deoxyribonuclease (DNase) and immunity protein Im9 protein-protein binding interaction involves a 16 nm2 largely anhydrous interface with 12 direct hydrogen bonds connecting nine residues on each of the proteins stabilized by five water bridges totaling 14 hydrogen bonds that involve six residues on the DNase and four residues on the Im9 (12).
Protein-ligand recognition
The binding sites of proteins are, or are surrounded by, relatively rich areas for binding water. Many biological ligands, on the other hand, contain relatively few groups that can hydrogen bond to water. An extreme example of these ligands is phospholipase A2 (13), which has a hydrophobic catalytic site held some distance from the substrate phospholipid surface and requires rigidly ordered water molecules to surround the active site and to maintain this interfacial binding.
Analysis of 392 crystal ligand-protein structures (14) showed an average of 4.6 ligand-bound water molecules, with 76% of them bridging between the ligand and the protein. On average, three interactions existed that involved each of these linking water molecules with the protein-ligand complex: two to the protein and one to the ligand. A separate analysis of 251 protein-ligand active-site complexes gave an average of 34 water molecules and 118 aqueous hydrogen bonds per active site, with average coordination of just over two strong hydrogen bonds per water molecule (15). In addition, these water molecules were found to lie close to carbon-bound hydrogen atoms, which increased their van der Waals contacts. Most aqueous hydrogen bonds were to the protein rather than to the ligand, with an intermediate number of water-water hydrogen bonds.
Using water to link the ligand and the protein can broaden the specificity due to the extra stereochemical flexibility. Thus, the peptide-binding protein OppA uses several flexibly adaptive water molecules to hydrogen bond and to shield charges when binding lysine framed tripeptides KXK, where K represents lysine and X is any one of the 20 common amino acids (16). Before such binding, these bridging water molecules are held by two or more hydrogen bonds to the unliganded protein, and they are conserved generally in the liganded protein crystals.
The hydrophobic interaction is an important factor in molecular recognition (17). Thus, the streptavidin-binding site for biotin contains a five-membered cyclic pentameric ring of water molecules stabilized by additional hydrogen bonds to coplanar amino acid side chains from the streptavidin and enclosed top and bottom by hydrophobic groups. When these highly ordered water molecules are expelled to make way for biotin, there is an entropy gain that more than compensates for the entropy loss on the strong streptavidin-biotin binding.
Peptide inhibitors are used in highly active antiretroviral therapy (HAART) to bind to the HIV-1 protease. Using six different peptide inhibitors, it was found that five water molecules were conserved at the binding site and similarly mediate a bridge between the dimeric protease and the inhibitor (18). All have a full complement of four hydrogen bonds and stabilize the extended form of the peptide inhibitors by binding to their peptide carbonyls; thus being somewhat independent of their amino acid sequence. Many other water molecules are mostly conserved between the majority of these complexes, with most mediating their internal hydrogen bonding.
More than one-third of the ribose interactions in ATP, ADP, and FAD-protein complexes are mediated by water linking from the 2' and/or 3' hydroxyls to the protein (19). Although water-mediated links can be used to broaden ligand specificity, they may also reduce it, as in the water-assisted asparagine recognition by asparaginyl-tRNA synthetase (Fig. 2) (20).

Figure 2. Cartoon that shows water-assisted asparagine recognition and aspartate discrimination by asparaginyl-tRNA synthetase. (a) Among the bound water molecules in the unliganded enzyme, one (Wat1) is bound between the Leu229 peptide carbonyl and the Glu230 carboxylate and another (Wat4) between the side chains of Tyr333 and Arg364. (b) These water molecules link to the amide group on the asparagine-AMP substrate on binding and two water molecules are released (Wat2 and Wat3). Aspartate-AMP cannot bind at the same site as the Wat1 water molecule is not able to donate three hydrogen bonds. Some hydrogen atoms have been added to clarify the hydrogen bonding. Figure partially redrawn from Reference 17 with permission.
DNA-DNA recognition
Although the structure of the DNA double helix is well known to be caused by base pairing and base stacking, the form of the helix is also highly dependent on its state of hydration. Hydration is crucially important for the conformation and use of nucleic acids and for its recognition by itself and other molecules (21). The strengths of many aqueous interactions are greater than those for proteins because of their highly ionic character. The DNA double helix can take up several conformations with differing hydration. The predominant natural DNA, B-DNA, has a wide and deep major groove as well as a narrow and deep minor groove and requires the greatest hydration. Partial dehydration converts it to A-DNA (with a narrow and deep major groove and a very wide but shallow minor groove) by decreasing the free energy required for A-DNA deformation and twisting, which is employed usefully by encouraging supercoiling but eventually leads to denaturation.
Hydration is greater and is held more strongly around the phosphate groups that run along the inner edges of the major grooves, but it is more ordered and more persistent around the bases with their more directional hydrogen-bonding ability and restricted space. Because of the regular structure of DNA, hydrating water forms chains along the double helix in both the major and the minor grooves. The cooperative nature of this hydration aids both the zipping (annealing) and unzipping (unwinding) of the double helix. Water motion within the grooves is slowed down compared with the bulk water, with the greatest reduction within the more restricting minor groove (22). On separation of the double helix (i.e., melting), about four water molecules per base pair are released despite extra hydration sites being released by the previously hydrogen-bonded base pairing (23), which confirms the importance of the cooperative nature of the water binding within the grooves.
DNA-protein recognition
The major problem in DNA protein recognition is how the protein can rapidly find a particular DNA base sequence from the enormous choice available when the specificity of the bases is somewhat hidden by the hydrogen bond linked and stacked base pairs. In an analysis of 109 protein-DNA complexes, an average of about nine water molecules per complex were bridged between the protein and the DNA, with another 125 binding to one or the other (24). Most water molecules were useful in screening unfavorable electrostatics from phosphates to allow binding (Fig. 3). The most common link found was where the water molecule donated hydrogen bonds to both a side chain acid group on the protein and a phosphate oxygen atom on the nucleic acid with the water molecule accepting one or two hydrogen bonds from elsewhere. Also in evidence were water links between donating protein-charged amino or guanidinium groups and accepting phosphate oxygen atoms. Another analysis of 39 crystallographically characterized protein-DNA complexes indicated that 46% of bridging waters (32 per complex) linked the protein to the nucleotide bases and the remainder was useful in screening out the unfavorable electrostatics, being linked very strongly to the DNA phosphate groups (25).
The trp repressor-operator complex exhibits no direct hydrogen bonds from the protein to the DNA bases but has three water molecules that mediate the necessary contacts for specific recognition between protein and DNA, so exemplifying well the importance water has in the process of recognition (26). Water-mediated links are also important in nuclear hormone receptors such as the estrogen receptor-DNA complex.
A major question concerns how a protein detects speedily a specific DNA sequence-binding site from the many closely related sites available. Restriction endonucleases have to find rapidly their specific hydrolytic sites in DNA molecules among the many similar sites, which differ by as little as a single base pair. They do this first by scanning the DNA rapidly at a distance, held at low affinity by favorable electrostatic interactions but separated by a significant but lubricating hydration layer. This process is then followed by partial dehydration and binding at their specific sites (27). Thus, the complexes of the restriction endonucleases BamHI and EcoRI with nonspecific sections of DNA have about 100-150 more bound water at the interface than the specific complexes (28, 29), which distances the protein from the DNA. The large amount of these water molecules released subsequently to the bulk liquid on specific binding contributes favorably to the binding thermodynamics. The specificity is almost total with a single substitution of thymine at the beginning of the specific nucleic acid sequence GAATTC (to give TAATTC) removing nearly all binding (5000 x less) by EcoRI (29).
The restriction endonuclease MspI makes specific contacts with all eight bases in the four base pair recognition sequence (CCGG), by six direct and five water-mediated hydrogen bonds and 13 water-mediated links to the phosphates (30). Numerous van der Waals contacts exist as water molecules contribute to the close packing, which mediates shape complementation. A key feature of direct protein DNA links in bound complexes is that they can change to and from water-mediated links to allow movement within binding cleft (31) while retaining contact information.

Figure 3. Cartoon that shows how water molecules shield the phosphate charges from the protein by donating hydrogen bonds (shown as dashed lines) to acceptor atoms on both the DNA and the protein, to attenuate their electrostatic repulsion. A need exists for these water molecules to accept hydrogen bonds from other water molecules (as shown) or for protein groups to stabilize such links.
Investigative Methods
No clear-cut method exists that gives complete or unambiguous information concerning the biomolecular binding processes in aqueous solution. The main problem is that although water’s hydrogen bonds are significant thermodynamically, water molecules are very mobile individually, as described earlier. Water molecules, therefore, have significant thermodynamic effects on recognition that are difficult to pin down by diffraction or NMR, because of their constant movement. A hydrogen bond only has a lifetime measured in picoseconds, and even an individual H2O molecule exists for less than a millisecond. In addition, explicit hydration is not included easily within molecular simulations because of the complexity of the inter-water and water-surface interactions. Many computer models for water have been developed, but they have very limited use in forecasting accurately the properties of bulk water. However, their predictions are probably more accurate close to surfaces than at intermediate distances where greater complexity exists because of the bulk aqueous milieu. Therefore, if care is taken, simulations can be used to help determine the structure and the properties at biologic interfaces (32).
The most useful data in many of the above studies is from X-ray and neutron diffraction, which provides the average positions of some water molecules in the free and the bound complexes. Although the structural information concerns crystals only, it is believed that the stereochemistries of many biomolecules within crystals are often close to those found in biological situations. Both methods detect water molecules in favored sites but fail to detect exactly where the most water molecules are present at any given time. Also, X-ray data cannot provide direct information that concerns the hydrogen bonding as the hydrogen atoms cannot be observed. The data does, however, provide a solid foundation and can be used as a starting point in molecular simulations.
NMR can be used to determine the presence and the movement of water molecules next to biomolecular surfaces (33). Sometimes it is particularly useful to detect the links to buried water and to water molecules that mediate between biomolecules.
Occasionally, vibrational spectroscopy can provide information that concerns specific water molecules or clusters when they are distinct sufficiently from the wide range of such structures found in the bulk liquid (34).
Calorimetry determines the heat generated (AH) during binding and solvation processes. Together with the binding constant for the binding equilibrium (providing AG), the entropy changes during binding (AS) can also be determined. These values are interesting and useful but fail to provide precise or accurate molecular details concerning the binding even when a range of similar molecules are compared.
Determination of the osmotic pressure of solutions in the presence of other solutes and biologic-binding processes can indicate clearly changes in the amount of water bound or, more exactly, the amount of water that can no longer interact freely with the solutes. Such methods have been useful particularly to determine the DNA-protein interactions (29).
Conclusion: Knowns and Unknowns
The more information is discovered concerning the role of water in biological recognition processes, the more its importance is confirmed. Where diffraction data is available we have, or can get, a good appreciation of the biological recognition process and the importance of the water molecules at the interface. However, currently we lack the means to determine the importance of water molecules removed slightly from the interface or details of how they guide the binding process, if indeed they do. While retaining the structural base from diffraction studies, the development of more accurate molecular simulations together with more powerful computers and concentrating on the interfaces and neighboring water without wasting too much resource on inconsequential distant bulk water (for example, Reference 35) is clearly a way forward.
References
1. Ben-Naim A. Molecular recognition—viewed through the eyes of the solvent. Biophys. Chem. 2002; 101-102:309-319.
2. Wraight CA. Chance and design—Proton transfer in water, channels and bioenergetic proteins. Biochim. Biophys. Acta 2006; 1757: 886-912.
3. Ben-Naim A. Water and Aqueous Solutions. 1974. Springer, New York.
4. Luck WAP. The importance of cooperativity for the properties of liquid water. J. Mol. Struct. 1998; 448:131-142.
5. Cho CH, Singh S, Robinson GW. Understanding all of water’s anomalies with a nonlocal potential. J. Chem. Phys. 1997; 107:7979-7988.
6. Wiggins PM. High and low-density water in gels. Prog. Polymer. Sci. 1995; 20:1121-1163.
7. Fernandez A, Kardos J, Goto Y. Protein folding: could hydrophobic collapse be coupled with hydrogen-bond formation? FEBS Lett. 2003; 536:187-192.
8. Levy Y, Onuchic JN. Water mediation in protein folding and molecular recognition. Annu. Rev. Biophys. Biomol. Struct. 2006; 35:389-415.
9. Janin J. Wet and dry interfaces: the role of solvent in protein- protein and protein-DNA recognition. Structure 1999; 7:R277-R279.
10. Rodier F, Bahadur RP, Chakrabarti P, Janin J. Hydration of protein-protein interfaces. Proteins 2005; 60:36-45.
11. Lo Conte L, Chothia C, Janin J. The atomic structure of protein- protein recognition sites. J. Mol. Biol. 1999; 285:2177-2198.
12. Kuhlmann UC, Pommer AJ, Moore GR, James R, Kleanthous C. Specificity in protein-protein interactions: the structural basis for dual recognition in endonuclease colicin-immunity protein complexes. J. Mol. Biol. 2000; 301:1163-1178.
13. Zhao L, Pal SK, Xia T, Zewail AH. Dynamics of ordered water in interfacial enzyme recognition: bovine pancreatic phospholipase A2. Angew. Chem. Int. Ed. 2004; 43:60-63.
14. Lu Y, Wang R, Yang C-Y, Wang S. Analysis of ligand-bound water molecules in high-resolution crystal structures of protein-ligand complexes. J. Chem. Inf. Model. 2007; 47:668-675.
15. Panigrahi S, Desiraju GR. Strong and weak hydrogen bonds in the protein-ligand interface. Proteins 2007; 67:128-141.
16. Sleigh SH, Seavers PR, Wilkinson AJ, Ladbury LE, Tame JRH. Crystallographic and calorimetric analysis of peptide binding to OppA protein. J. Mol. Biol. 1999; 291:393-415.
17. Young T, Abel R, Kim B, Berne BJ, Friesner RA. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:808-813.
18. Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure 2002; 10:369-381.
19. Babor M, Sobolev V, Edelman M. Conserved positions for ribose recognition: importance of water bridging interactions among ATP, ADP and FAD-protein complexes. J. Mol. Biol. 2002; 323:523-532.
20. Iwasaki W, Sekine S-I, Kuroishi C, Kuramitsu S, Shirouzu M, Yokoyama S. Structural basis of the water-assisted asparagine recognition by asparaginyl-tRNA synthetase. J. Mol. Biol. 2006; 360:329-342.
21. Chaplin, MF. Opinion: do we underestimate the importance of water in cell biology? Nature Rev. Mol. Cell Biol. 2006; 7:861-866.
22. Pal S, Maiti PK, Bagchi B. Anisotropic and sub-diffusive water motion at the surface of DNA and of an anionic micelle CsPFO. J. Phys. Condens. Matter 2005; 17:S4317-S4331.
23. Spink CH, Chaires JB. Effects of hydration, ion release, and excluded volume on the melting of triplex and duplex DNA. Biochemistry 1999; 38:496-508.
24. Reddy CK, Das A, Jayaram B. Do water molecules mediate protein-DNA recognition? J. Mol. Biol. 2001;314:619-632.
25. Wibowo FR, Rauch C, Trieb M, Wellenzohn B, Liedl KR. Water-mediated contacts in the trp-repressor operator complex recognition process. Biopolymers 2004; 73:668-681.
26. Spyrakis F, Cozzini P, Bertoli C, Marabotti A, Kellogg GE, Mozzarelli A. Energetics of the protein-DNA-water interaction. BMC Struc. Biol. 2007; 7:4.
27. Fuxreiter M, Mezei M, Simon I, Osman R. Interfacial water as a “Hydration fingerprint” in the non-cognate complex of BamHI. Biophys. J. 2005; 89:903-911.
28. Siderova NY, Muradymov S, Rau DC. Differences in hydration coupled to specific and non-specific competitive binding and to specific DNA binding of the restriction endonuclease BamHI. J. Biol. Chem. 2006; 281:35656-35666.
29. Siderova NY, Rau DC. Differences between EcoRI non-specific and “star” sequence complexes revealed by osmotic stress. Bio- phys. J. 2004; 87:2564-2576.
30. Xu QS, Kucera RB, Roberts RJ, Guo H-C. An asymmetric complex of restriction endonuclease MspI on Its palindromic DNA recognition site. Structure 2004; 12:1741-1747.
31. Nguyen B, Hamelberg D, Bailly C, Colson P, Stanek J, Brun R, Neidle S, Wilson WD. Characterization of a novel DNA minor-groove complex. Biophys. J. 2004; 86:1028-1041.
32. Zhao X, Huang X, Sun C. Molecular dynamics analysis of the engrailed homeodomain-DNA recognition. J. Struct. Biol. 2006; 155:426-437.
33. Huang H, Melacini G. High-resolution protein hydration NMR experiments: probing how protein surfaces interact with water and other non-covalent ligands. Analyt. Chim. Acta 2006; 564:1-9.
34. Jalkanen KJ, Jiirgensen VW, Claussen A, Rahim A, Jensen GM, Wade RC, Nardi F, Jung C, Degtyarenko IM, Nieminen RM, et al. Use of vibrational spectroscopy to study protein and DNA structure, hydration, and binding of biomolecules: a combined theoretical and experimental approach. Int. J. Quantum Chem. 2006; 106:1160-1198.
35. Praprotnik M, Matysiak S, Site LD, Kremer K, Clementi C. Adaptive resolution simulation of liquid water. J. Phys. Condens. Matter 2007; 19:292201.
Further Reading
Chaplin MF. Water Structure and Science. http://www.lsbu.ac.uk/water/.
Coulocheri SA, Pigis DG, Papavassiliou KA, Papavassiliou AG. Hydrogen bonds in protein-DNA complexes: where geometry meets plasticity. Biochimie. 2007; 89:1291-1303.
See Also
Biointeractions at Solid-Water Interfaces
NMR to Study Molecular Recognition
Water in Protein Folding, Role of
Water, Properties of; Water Channels