CHEMICAL BIOLOGY
Phosphate Mimics, Cyclic Compounds as
Amanda C. Nottbohm and Paul J. Hergenrother, Department of Chemistry, Roger Adams Laboratory, University of Illinois, Urbana, Illinois
doi: 10.1002/9780470048672.wecb641
Compounds that contain phosphate and diphosphate moieties are not ideal biologic probes. Not only does their ionic character inhibit cell membrane permeability, but also, once inside a cell, the ester and the anhydride functionalities are likely targets for enzymatic cleavage. Thus, replacements for the phosphate motif are important as enzyme inhibitors, DNA or RNA analogs, phospholipid mimics, and phosphorylated metabolite analogs. To date, several classes of phosphate mimics have been developed that have been grouped into four categories: phosphorus-containing, sulfur-containing, dicarboxylates, and the novel cyclic mimics, which will be the focus of this review.
From nucleic acid phosphodiester linkages to reversible posttranslational modifications, the phosphate motif is one of life’s most versatile functional groups. Cellular energy is stored in phosphorylated compounds such as ATP, and coenzymes often contain the pyrophosphate moiety. Protein interactions, stability, and activity are often mediated by the controlled introduction or removal of phosphate groups; indeed, it has been estimated that over 30% of human cellular proteins contain covalently bound phosphate groups (1). This modification has been implicated in the regulation of cell cycle progression, transcription control, protein synthesis, glycogen metabolism, and intracellular transport (2, 3). A host of disease states including cancer, inflammation, diabetes, atherosclerosis, immunodeficiency, and the bubonic plague (4-7) have been associated with the disruption of the phosphorylation machinery within the cell.
Although the ionized state of the phosphate moiety at physiologic pH likely allows for more efficient retention of phosphorylated intermediates within the cell wall and organelles (8, 9) and slows hydrolysis greatly (10), this charged state also prevents compounds that contain phosphate and diphosphate moieties from being useful biologic probes. Not only does their ionic character hinder cell membrane permeability, but also, once inside a cell, both the phosphate-ester and -anhydride functionalities are likely targets for enzymatic degradation. Thus, much effort has been directed toward the development of novel replacements for the phosphate motif, as such compounds might become new enzyme inhibitors, DNA or RNA analogs, phospholipid mimics, and isosteres of phosphorylated metabolites.
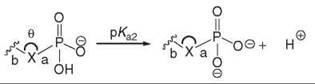
Therefore, the challenge is to design functional groups that mimic both the electronic properties and the spatial arrangement of the phosphate group while retaining cell permeability and stability. This task is extremely difficult considering the unique ability of the tetrahedral phosphate moiety to connect two modular subunits while possessing a negative charge. Additionally, this anion usually plays a major role in enzymatic binding and recognition events (11), as it often forms ionic contacts with both metal ions and lysine/arginine residues. Thus, phosphate replacements that lack ionization run the risk of not binding to their protein target. The ionization states and pKa values for free phosphate in solution are shown in Fig. 1 (12, 13). Interestingly, it has been demonstrated that the pK a of the phosphate group can change drastically on binding; in the binding of SH2 domains to phosphotyrosine-containing peptides, the phosphate in phosphotyrosine undergoes a large downward shift in pKa(from a free pKa of 6.1 to a bound pKa of 4.5) on binding, and additional studies indicated that 25% of the free energy of binding is caused by the second negative charge on the phospho monoester (14).
Current classes of phosphate isosteres have been developed to interact with target proteins in the same fashion as the phosphate moiety, but with increased bioavailability and stability in a physiologic environment. These phosphate motif replacements can be grouped broadly into four categories: three “traditional” replacement motifs (phosphorus-containing, sulfur-containing, and carboxylate linkages) that have been reviewed previously (3, 15), and the more unique cyclic mimics, which will be the major focus of this review (See Fig. 2 for general structures of mimics discussed in this review).

Figure 1. Designing mimics of phosphates and diphosphates is difficult because of their anionic character and tetrahedral geometry (as indicated by pKa values, bond lengths, and bond angles). (a) pKa values for inorganic phosphate (12). (b) pKa values for pyrophosphate (12). (c) P-O bond lengths of the hydrogen phosphate dianion (13). (d) P-O bond angles in the hydrogen phosphate dianion (13).

Figure 2. Traditional phosphorus-, sulfur-, and carboxylate-containing replacements for the phosphate group, along with the more recently discovered cyclic phosphate mimics.
Common Phosphate Isosteres: Phosphonates, Sulfones, and Carboxylates
Phosphonates, sulfones, and dicarboxylates are the most prevalent phosphate mimics in medicinal chemistry. Although the phosphonate moiety makes the smallest perturbation, these compounds often do not solve the problem of cell membrane permeability (16-21), and can be limited in their biologic activity. Because of the high ionization constant of phosphonates compared with phosphates (Table 1) (22), phosphonates are not di-ionic under physiologic conditions, and the oxygen-to-methylene substitution removes a potential hydrogen bonding moiety, which often leads to greatly reduced binding affinities (3). Nonetheless, development of phosphonate inhibitors of glycosyltransferases (23-25), fucosyltransferases (26), and squalene synthetase (27) among others have been pursued actively.
Of course, cell permeability is not always a prerequisite for biologic activity. In one example, Vaghefi et al. (24) proposed that selective phosphonate inhibitors of galactosyltransferase might exhibit activity at the surface of the tumor cell membranes; in such cases, a cell permeable inhibitor would be unnecessary. Although no in vivo testing was conducted, phosphonate analogs of UDP-galactose inhibited galactosyltransferase activity competitively with Ki’s of 62 to 96.9 μM (21).
As indicated by the relatively weak activities of phosphonates, often these compounds are not adequate mimics of the phosphate group because of their increased pKa2 relative to the parent phosphate (Table 1). Through the introduction of one or two fluorine atoms, one can decrease the phosphonate pKa2 slightly to match a standard phosphate more closely; increasingly potent inhibitors have been developed through the application of this concept. For example, when incorporated into a hexameric peptide sequence, phosphonodifluoromethyl phenylalanine inhibitors of protein tyrosine phosphatase 1B have been developed with IC50 values of 100 nM, whereas the same nonfluorinated analogs inhibit at 200 μM (28) (Fig. 3). In general, the biologic activity of (α-monofluoro) alkylphosphates remains less explored, which is surprising given the fact that their pKa2 is almost identical to that of the phosphate group (Table 1). Few studies have compared the activities of phosphonate-containing inhibitors with both their monofluorinated and difluorinated equivalents, yet it has been reported that the introduction of a new stereochemical center with monoflu- orophosphonates can potentially affect activity. For instance, the 7(S)-monofluorophosphonate analog of glucose 6-phosphate is 11 times less potent than the 7(R)-monofluorophosphonate derivative in studies of glucose 6-phosphate dehydrogenate substrates (22). Of course, because of the increased hydrophobicity of the fluorine-containing ligand relative to the parent phosphonate the effect of adding fluorine can also be deleterious (29, 30). For this reason, use of fluoromethylenephosphonates as phosphate mimics should be considered on a case-by-case basis.
Table 1. A comparison of bond angles, bond lengths, and pKa2 values for phosphates, phosphonates, and fluorinated analogs (9, 22) illustrates the problems with using phosphonates as phosphate mimics*
|
|
|||||
|
|
|
bond length A |
|
|
|
|
X |
bond angle θ |
a |
b |
pKa2 |
% dianion at pH 7.8 |
|
O |
118.7° |
1.59 |
1.43 |
6.4 |
95 |
|
CH2 |
121.1° |
1.81 |
1.51 |
7.5 |
62 |
|
CHF |
113.3° |
1.82 |
1.50 |
6.5 |
98 |
|
CF2 |
116.5° |
1.85 |
1.50 |
5.6 |
>99 |
*Only the monofluorophosphonates closely matches the pKa of the parent phosphate.
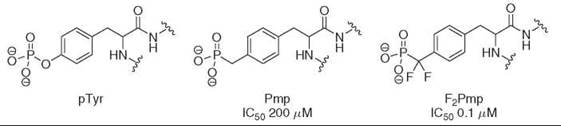
Figure 3. Phosphonomethyl phenylalanine (Pmp) and phosphonodifluoromethyl phenylalanine (F2Pmp) are nonhydrolyzable phosphotyrosyl (pTyr) mimetics. Hexameric peptide sequences that incorporate either Pmp or F2Pmp inhibit protein tyrosine phosphatase 1B. The difluoroanalogs are most potent, presumably because of their reduced pKa relative to the phosphonate.
To eliminate problems associated with the stability and the membrane impermeability of phosphates in vivo, incorporation of sulfur-associated functionalities has been explored. Oxidized forms of sulfur can provide isosteric replacements for a phosphate, but they will lack the full negative charge. Although a direct comparison with the phosphate moiety is difficult to make, studies have indicated that the oxygen of DMSO carries an effective charge of -0.63 (31), whereas the Mulliken atomic charge on a diaryl sulfone is -0.616 (32). On the other hand, computational examination of the ESP charges on the oxygens of phosphotyrosine molecules have found that they carry charges of -0.73 or -1.06 for the monoanionic and dianionic forms, respectively (33). It must be noted that these cannot be considered direct comparisons because Mulliken charges are calculated based on the linear combination of atomic orbitals method, and ESP charges represent the electrostatic potential around an atom.
Multiple efforts have been made to replace phosphorous- containing linkages with sulfur-containing isosteres in the context of enzyme inhibition. In a search for nonionic transition state analog inhibitors of restriction enzymes, Blattler et al. (34) found that nucleic acid duplexes that incorporate a dimethyl sulfone in place of a phosphodiester have distorted backbones similar to those in restriction enzyme bound DNA. Chimeric DNAs that incorporate sulfone linkages were synthesized, and depending on the location of the dimethylene sulfone linker, either between the first AT unit or the second AT unit in the Eco RV recognition site, Ki values were 20 nM and 120 pM, respectively (Fig. 4a). Other rationally designed phosphate surrogates that contain sulfur have also been successful. Sulfonylbenzoyl-nitrostyrenes (35), which are believed to mimic the ATP-tyrosine interaction in the transition state (Fig. 4b), are inhibitors of the EGF-receptor tyrosine protein kinase, and 5'-O-(N-salicylsulfamoyl) adenosines act as acyl-AMP analogs (36), which inhibit siderophore biosynthesis in M. tuberculosis and Y. pestis (Fig. 4c).

Figure 4. (a) Analogs of DNA that contain a dimethyl sulfone group have a distorted backbone compared with the natural biopolymer, which makes them useful as inhibitors of restriction enzymes. (b) A sulfonylbenzoyl nitrostyrene inhibitor of tyrosine protein kinase was designed based on a transition state model of the reaction of ATP with tyrosine residues and exhibits good activity. (c) Based on salicyl-AMP, a sulfamoyl-containing compound exhibits remarkable activity in the inhibition of siderophore biosynthesis.
Another commonly explored diphosphate replacement is the dicarboxylate moiety. For enzymes that contain divalent metal cations, it is believed that dicarboxylate-containing compounds might coordinate to the metal and form a complex similar to that of the metal-pyrophosphate (37). Additionally, as the pKa of dicarboxylates makes them dianionic under physiologic conditions (38), compounds that contain two carboxylate moieties, such as malonates and succinates, may mimic the negative charge of the phosphate adequately. For instance, modeling studies have shown that whereas malonate ethers are significantly larger than phosphates (39), their spacing of negatively charged oxygen atoms is within 0.1 A of the corresponding diphosphate (40).
Interestingly, the first nonphosphorus-containing mimics of farnesyl pyrophosphate were novel dicarboxylic acids isolated from the methyl ethyl ketone extract of the fungus Chaetomella acutiseta (Fig. 5a) (41, 42). Although it was known that these acids can exist either in the anhydride or in the open dicarboxylate form depending on the pH, extensive UV analysis indicated that the dicarboxylate form is the species responsible for inhibition of farnesyl-protein transferase activity. Modeling studies suggest that the hydrophilic head group of the dicarboxylate form is similar to the diphosphate moiety of farnesyl pyrophosphate both sterically and electronically (42). Tyrosine kinases have also been targeted with carboxylate-containing mimics of the phosphorylated tyrosine residue; although incorporation of an acetic acid moiety results in a 500-fold loss of binding affinity as compared with the corresponding phosphates, the oxamic acid moiety fares slightly better as an inhibitor of tyrosine kinase p56lck in the SH2 domain with KD values as low as 42 |lM (Fig. 5b) (21). Even though modeling studies have indicated that malonate ethers occupy approximately 20% more volume than a phosphate group (39), symmetric 3,5-disubstituted benzoates have also been identified as novel shikimate-3- phosphate mimics. 5-enolpyruvoyl-shikimate-3-phosphate synthase (EPSPS) catalyzes the transfer of the carboxyvinyl group of phosphoenolpyruvate to shikimate-3-phosphate (S3P), and it is an important herbicidal target. In this case, it is known that the 3-phosphate moiety contributes approximately 8 kcal/mol to the binding event with EPSPS, but the corresponding phosphonate mimics are extremely labile and impractical as inhibitors. Thus, Miller et al. (39) developed a 3,5-disubstituted benzoate analog of S3P that contains a 3-malonate ether with a Ki(app) value of 2.5 μM as compared with 7.0 μM and 1.0 μM (Kd values) for S3P (the natural substrate) and EPSP (the product), respectively (Fig. 5c). Although not always a successful (37, 40), evidence suggests that in certain cases the negative charge of the carboxylate moiety can mimic the anionic monophosphate or diphosphate.

Figure 5. (a) Chaetomellic acid A is a novel dicarboxylate-containing natural product that is thought to mimic farnesyl pyrophosphate and thus to inhibit the enzyme FTPase. This compound can exist in either the dicarboxylate or anhydride form. (b) As peptides that contain aromatic oxamic acids are good inhibitors of tyrosine kinase p56lck SH2 domains, it is believed that the oxamic acid moiety may mimic the phosphate group. (c) A symmetric 3,5-disubstituted benzoate analog of S3P (substrate) and EPSP (product) inhibits EPSPS.
Cyclic Phosphate Mimics
In an effort to improve cell permeability/bioavailability, recently developed phosphate isosteres often have decreased overall charge. In the past, it has been proposed that reduced potency is often a trade-off with increased cellular permeability of phosphate replacements (3), but these trends are beginning to change. A recent review of enzymatic phosphate recognition (11) pointed out that the phosphate-binding mode depends greatly on orientation within the protein. Although buried binding pockets often are filled with neutral amino acid residues, cationic residues play a larger role in phosphate recognition and binding closer to the surface. Additionally, over one third of phosphate binding sites do not contain a metal or other cationic phosphate binder (11); thus, the chances of using a neutral, more drug-like compound are increased in these cases. Next, cyclic phosphate mimics including tetronic acid-, squaric acid-, thiazolidinone-, rhodanine-, perfluorylaryl- and sulfhydantoin-based derivatives (see Fig. 2) will be discussed.
Cyclic phosphate mimics: tetronic acid derivatives
Using a 3-acyltetronic acid as a phosphate-mimicking core (Fig. 6a), potent inhibitors of cdc25B [a dual specificity phosphatase (DSP)] and vaccinia VH1-related phosphatase (VHR) were identified recently (43). Drawing on general natural product inhibitors of protein tyrosine phosphatases (PTPs), Sodeoka et al. (43) searched for a novel core structure that could interact specifically with the active site loop of DSPs. Although previous attempts to find PTP inhibitors focused on phosphonates and carboxylates as phosphate core isosteres (44), the acidic 3-acyltetronic acid group of the known VHR inhibitor RK-682 (Fig. 6b) looked promising as a general replacement for the phosphate moiety. A library of 36 tetronic acid derivatives was synthesized, and some selective inhibitors of both cdc25B and VHR were identified. In particular, a tetronic acid derivative with a diazomalonyl group was found to be extremely potent against cdc25B (Fig. 6b), with a 30-fold selectivity for cdc25B over VHR. In general, the library showed good selectivity for VHR and cdc25B over general tyrosine phosphatases. Molecular modeling studies indicated that the dissociated 3-acyltetronic acid anion can act as a hydrogen-bonding phosphate mimic, whereas the R and R1 groups provide two positions for additional derivatization, which allows for increased specificity.

Figure 6. (a) Because of their highly conjugated structure that distributes negative charge over several oxygens, 3-acyltetronic acid derivatives are believed to mimic the phosphate group, particularly that of phosphotyrosine. (b) RK-682 is a competitive and general phosphatase inhibitor that contains the 3-acyltetronic acid moiety. Additional derivatization of RK-682 with a diazomalonyl group resulted in the development of a selective phosphatase inhibitor for cdc25B versus VHR.
Cyclic phosphate mimics: squaric acid-based motifs
Over the years, squaric acid and its derivatives have found several uses in medicinal chemistry: Squaric acid inhibits glyoxylase I (45); semisquaric acid (3-hydroxy-3-cyclobutenedione) is an inhibitor of pyruvate dehydrogenase and transketolase (46); and other derivatives serve as antagonists of the N-methyl-D-aspartate receptor (47). Searching for an isostere with similar charge distribution to that of the phosphate group for the creation of oligodeoxynucleotide analogs, Sato et al. (48, 49) also examined squaric acid derivatives. Squaric acid contains two acidic hydroxyl groups (pKa values of 0.54 and 3.48, respectively) as well as two highly polarized carbonyl groups (50). Ab initio calculations of electrostatic charge distributions indicate that although a dissociated dimethyl phosphate has -0.84 charges on each of the oxygen atoms, N-isopropyl-N'-methylsquaryldiamide has similar polarizing patterns, with -0.47 and -0.51 charges on the oxygen atoms of the carbonyl groups (49). Furthermore, similar to phosphate functionality, the squaric acid motif contains carbonyls that can function as hydrogen-bonding partners and binding sites for divalent metal ions (Fig. 7a) (51, 52).
On the basis of these calculations, a modified 3'-5'thymidine dimer derivative (Fig. 7b, TsqT) was synthesized (49). Based on both UV and CD spectra of TsqT and the corresponding phosphate derivative (TpT), the dimer that contains the squaryldiamide linkage is similar structurally to the natural TpT. Thus, the TsqT dimer structural motif was incorporated into oligodeoxynucleotides using standard phosphoramidite chemistry, producing 5'-CGCATsqTAGCC-3' and 5'-G ACGCATsqTAGCCGAT-3'. Enzymatic digestion of squaryl-modified oligodeoxynucleotides, with both snake venom phosphodiesterase and calf intestine alkaline phosphatase, gave the corresponding deoxynucleotides and TsqT, which illustrates the enzymatic stability of these phosphate replacement motifs. Fluorescent derivatization and subsequent FRET experiments showed that duplexes formed from squaryl-modified oligodeoxynucleotides have a bent structure, and melting temperature (Tm) studies revealed destabilization of the overall structure as compared with the natural oligodeoxynucleotide. Nonetheless, hybridization of 5'-d(CGCATsqTAGCC)-3' and 3'-d(GGCTAATGCG) demonstrated A-T base pairing was preserved, which indicates that the squaryldiamide-modified oligodeoxynucleotides may have a variety of applications (49). In a similar manner, a 2'-5'-linked 3'-deoxyuridine thymidine dimer derivative (Fig. 7b, UsqT) was synthesized in an attempt to create a more robust oligodeoxynucleotide analog (48); it was hoped that a 2'-5'-linkage would eliminate the strain associated with the corresponding 3'-5' TsqT. Calculations and CD spectra analysis showed that the 2'-5'squaryl modified oligodeoxynucleotides have an overall structure that resembles that of TpT. Despite this similarity, UsqT derivative d(CGCAUsqTAGCC)-3' exhibits a decreased affinity for its complementary oligonucleotide, 3'-d(GCGTAATCGG), but demonstrates the unique ability to hybridize with 3'-d(GCGTA GTCGG), which is an oligodeoxynucleotide that contains a mismatched dG (48).
A final example illustrates the use of squaric acid derivatives in enzyme inhibition. In an effort to discover potent inhibitors (of Yersinia PTPs and PTP1B) with reduced charge and thus improved bioavailability, Xie et al. (53) also studied the squaric acid motif as a nonhydrolyzable phosphotyrosine mimic. 3-aryl-4-hydroxy-3-cyclobutene-1,2-diones based on the squaric acid structure were found to be inhibitors of Yersinia PTP (Fig. 7c). Additional analysis confirmed reversible competitive inhibition, which indicates that squaric acid binds to the active site of PTPs in a conformation that mimics that of the natural phosphate ester substrates (53).

Figure 7. (a) Dialkylphosphate similarity to squaric acid derivatives; both possess two negatively charged oxygen atoms in their tautomeric form. (b) Modified oligodeoxynucleotides that contain a squaryldiamide moiety are remarkably similar to natural oligodeoxynucleotides. 3'-5' linked dimers can form Watson-Crick base pairs with adenine, whereas the 2'-5' linked dimers can form both Watson-Crick base pairs with adenine and wobble base pairs with guanine. (c) Squaric acid-based inhibitors of PTPase from Yersinia are designed to mimic phosphotyrosine.
Cyclic phosphate mimics: thiazolidinones and rhodanines (thioxothiazolidinones)
MurB, an enzyme that reduces enolpyruvyl uridine diphosphate N-acetylglucosamine (EP-UNAG) to uridine diphosphate N-acetylmuramic acid (UNAM) (54), is an attractive antibacterial target (55). Using crystallographic data of MurB bound to EP-UNAG, Andres et al. (55) sought a suitable surrogate for the diphosphate moiety of the natural substrate, and focused eventually on 4-thiazolidinones. Modeling studies indicated that a carboxylic acid moiety at the R1 position potentially could mimic essential interactions of the diphosphate with a lysine residue (Fig. 8a). Additionally, it was believed that the thiazolidinone core would orient the side chains so that they might occupy the space normally reserved for the uridine and glucosamine motifs. Although it is unlikely that the thiazolidinone can mimic all interactions of the diphosphate, elimination of the corresponding rotatable bonds of the natural substrate was also predicted to have a positive entropic effect to binding of the thiazolidinone-based inhibitor. Only thiazolidinones that contained an n-butyl group at the R1 position exhibited any activity, with diastereomers derived from D-norleucine that exhibited a 4-fold improvement in activity over those synthesized from L-norleucine (Fig. 8b) (55).
In a recent screen of 8,000 compounds to identify potential drugs that target the synthesis of dTDP-rhamnose (from glucose-1-phosphate and dTTP), 11 inhibitors were identified; three of these contained a rhodanine (Fig. 2) structural motif (56). Inhibition was found for all three dTDP-rhamnose synthetic enzymes tested, including RmlB, RmlC, and RmlD. The fact that all three enzymes modify a phosphorylated rhamnose intermediate important for Mycobacterium tuberculosis (MTB) cell wall synthesis was a possible indication that substituted rho- danines might mimic the phosphate motif, but it was unknown whether such inhibitors actually bind in the same fashion as the natural substrates. A later computational follow-up study by Kantardjieff et al. (57) indicated that the three rhodanine inhibitors bind at or near the phosphate-binding region of MTB Rm1 C in a similar fashion to compounds that contain thiazolidinones. Core structure electron-density isosurfaces for these antimycobacterial compounds were generated, further hinting at the common features shared between substituted rhodanines or thiazolidinones and the diphosphate moiety.
High-throughput screening for compounds that might compete with the UDP-GlcNAc substrate of MurG also has led to the identification of a rhodanine core structure as a phosphate mimic (Fig. 8c) (58). Using a fluorescein derivative of UDP-GlcNAc, compounds that compete with the binding of the fluorescent substrate were identified on the basis of anisotropy changes on incubation with the putative inhibitors. Over 48,000 compounds were screened, and of the 277 compounds that were indicated to be possible inhibitors of MurG, 11 compounds exhibited greater inhibitory effects than the natural inhibitor UDP; 7 compounds contained a rhodanine core structure (58). A similar screen (59) also revealed several compounds with rhodanine-like structures. It should be noted that these 1,3-substituted compounds are conjugate acceptors and thus potentially reactive as electrophiles (60); however, additional kinetic analysis indicated that no covalent modification of the enzyme was occurring. Additionally, most compounds were selective for MurG over closely related enzymes that use similar substrates (59). The high percentage of inhibitors with a similar core may possibly hint at a common binding mode, which reveals the ability of rhodanines to potentially serve as diphosphate mimics.
Although it is unknown whether either thiazolidinones or rhodanines actually mimic the phosphate binding site or simply have shapes that occupy the nucleotide-sugar binding site favorably (as proposed by Carlson) (60), studies of systems that do not contain a nucleotide-sugar seem indicate to the former. Lysophosphatidic acid (LPA) is known to possess a range of biologic activities, which include the stimulation of cell proliferation, tumor cell invasion, induction of angiogenesis, and stimulation of phospholipase D activity (61-63). Gududuru et al. (64, 65) believed that LPA mimics might represent a novel class of anticancer compounds. A small library of serine amide phosphates (SAPs) was synthesized initially and exhibited low micromolar cytotoxicity against five human prostate cancer cell lines (65). Although SAPs were effective in killing prostate cancer cells, they exhibited little selectivity for noncancerous cells, and it was hypothesized that these compounds were hydrolyzed by lipid phosphate phosphatases under biologic conditions. Replacement of the phosphate group with a more robust pharmacophore was expected to enhance and prolong biologic activity. As Andres (55) had postulated previously, the thiazolidinone moiety possibly serves as a phosphate mimic, and a series of inhibitors was synthesized based on this design (Fig. 8d). Interestingly, oxidation of the thioether linkage to either a sulfoxide or sulfone functionality did not affect activity of the most potent antiproliferative compound. As these compounds are 2-5-fold more potent in the human prostate cancer cells as compared with the RH777 (control) cell line, the 4-thiazolidinone derivatives seem to provide a significant improvement over the phosphate-containing SAPs because of their increased stability (64).

Figure 8. (a) The substituted thiazolidinone core. (b) Sometimes, thiazolidinone-containing compounds make good inhibitors of enzymes that use nucleoside diphosphates. For example, MurB, which uses EP-UNAG as its natural substrate is inhibited by the thiazolidinone derivative pictured. (c) Rhodanine-containing compounds also serve as excellent inhibitors of enzymes with natural diphosphate-containing inhibitors. For comparison, UDP (an inhibitor of MurG) is pictured next to a rhodanine derivative that exhibits good antibacterial activity. (d) Thiazolidinones have proven their usefulness as general replacements for natural substrates that contain the phosphate group. For example, 2-aryl-4-oxo-thiazolidin-3-yl-amides have replaced serine amide phosphates as more stable mimics of lysophosphatidic acid, and they have been shown to be cytotoxic in prostate cancer cell lines.
Cyclic phosphate mimics: perfluoroaryl compounds
Modeling studies first indicated that particular perfluorylaryl compounds might mimic the overall spatial and electronic distribution of the diphosphate and triphosphate moiety (66). By appending a perfluorylaryl moiety onto a guanosine substrate, Barber et al. (67) hoped to mimic either guanosine 5'-triphosphate or guanosine 5'-diphosphate and thus modulate the activity of Ras. Barber et al. (67) succeeded in appending several perfluorylaryl phosphate replacement moieties onto guanosine. Although no IC50 values are given in the patent literature (66), the relative binding abilities of the guanosine nucleotide mimics were compared with that of GTP for the biding site of a mutant H-Ras protein using a cold chase experiment, in which either GTP or a perfluorylaryl compound was used as a competitor to tritiated GTP. When experiments were performed with the natural substrate, GTP (Fig. 9a), at a concentration of 25 μM, a 79% reduction in radioactivity was observed. Using the putative GDP mimic 5'-O-(2,3,5,6-Tetrafluoro-4-pyridyl) guanosine (Fig. 9a) at the same concentration, a 21% reduction in the mean c.p.m. was noted, which indicates that the perfluorylaryl compound likely inhibits binding of GTP to Ras to some extent. Interestingly, when using a compound designed to mimic GTP, 5'-O’-(2,3,6-Trifluoro-5-hydroxy-4-nitrophenyl) guanosine (Fig. 9a), no reduction occurred in the binding ability of tritiated GTP (66).
Taking this concept a step further, general inhibitors for protein farnesyltransferase (FTase) (67), geranylgeranyltransferase I (GGTase I), and squalene synthase (SqSase) (68) were also developed. In a posttranslational modification event, FTase transfers a farnesyl group to the Ras protein (69). Although not directly related to the function of Ras, SqSase uses the same substrate as FTase; thus, the compounds developed were also tested for SqSase inhibition (68). Finally, GGTase I controls the geranylgeranylation of Rho proteins and plays a role in cell motility and invasion, and also is a potential target for cancer therapy (70). Of 15 compounds tested, 5 compounds exhibited IC50 values less than 10 μM (68), with the best/most selective inhibitors for each enzyme illustrated in Fig. 9b-d.

Figure 9. (a) Some substituted perfluoroaryl groups are believed to be phosphate mimics as illustrated below. Although a perfluoroaryl GDP mimic could compete with tritiated GTP for binding to a mutant Ras protein, a similar GTP mimic showed no activity. (b) Perfluorylaryl groups have been shown to mimic the diphosphate groups of diphosphoterpenes, and thus inhibit enymes including FTase, (c) GGTase I, and (d) SqSase.
Cyclic phosphate mimics: sulfhydantoins
Relatively little information exists about sulfhydantoins in the literature. Although they have been described previously as peptidomimetic serine protease inhibitors (71), Saunders et al. (72) were the first to report the use of sulfhydantoins as phosphate mimics. In an attempt to develop novel inhibitors for SHP-2, which is a tyrosine phosphatase, it was noted that general phosphatase inhibitors either incorporate carboxylate groups or the fluorophosphonate moiety to mimic the negative charge of the phosphate at physiologic pH (71). Driven by the lack of drug-like SHP-2 inhibitors in the literature, they searched for heterocyclic modulators of SHP-2 activity based on sulfhydantoins. In vitro competition assays were completed to determine the IC50 values of several putative phosphate mimics, the most potent of which are illustrated in Fig. 10. Although exact IC50 values were not reported in the patent literature, the compounds depicted exhibit IC50 values between 1 and 100 μM (72).
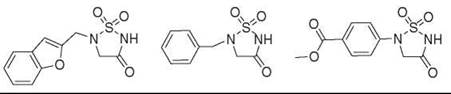
Figure 10. A sulfhydantoin motif serves as a phosphate group replacement in phosphotyrosine mimics, with IC50 values that range from 1 to 100 μM.
Cyclic phosphate mimics: hydroxytropolones
Hydroxytropolones derived from puberulonic acid, a natural product isolated from Penicillium, were investigated for their ability to inhibit inositol monophosphatase (IMPase). Although bisphosphonic acids were developed previously as potent inhibitors of IMPase, these compounds do not cross the blood-brain barrier and are therefore not useful antipsychotic agents. It was proposed that the hydroxyl groups combined with the planar structure of hydroxytropolones might allow chelation of the magnesium ions found in the active site of IMPase, which mimics the binding of the phosphate ester portion of D-myo-inositol-1-phosphate to the enzyme (73). Additional derivatization of the hydroxytropolone core revealed more potent inhibitors of IMPase with IC50 values as low as 4-5 μM (Fig. 11) (74). Inhibition of IMPase is observed even in the presence of 2 mmol/L Mg2+, which suggests that hydroxytropolones are not simply causing nonspecific inhibition through the reduction of the free metal concentration in the assay mixture. Approximately 18% of enzymes that interact with phosphate-containing substrates use essential metal ions (11); as inhibition with hydroxytropolones occurs though chelation of active site metal ions, other bimetallic, phosphate-binding enzymes might also be targets of hydroxytropolone inhibition. Indeed alkaline phosphatase, which contains two catalytic zinc ions, the tri-zinc-containing phospholipase C (75), and human immunodeficiency virus RT polymerase/RNase H/Integrase (76), which contains essential Mg2+ ions, are inhibited competitively by hydroxytropolones as well.
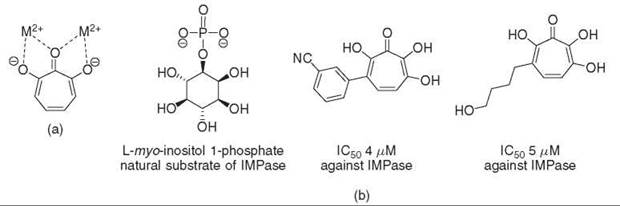
Figure 11. (a) Hydroxytropolones can mimic the interaction of a phosphate ester with enzymes that contain essential metal ions. (b) IMPase, a bimetallic enzyme that contains two Mg2+ ions, is inhibited by various polyhydroxytropolones. The mode of inhibition most likely occurs through the tropolones ability to chelate the active site metal ions.
Cyclic phosphate mimics: do they really mimic the phosphate group?
Many cyclic phosphate mimics have been developed recently, and only limited data exists about their ability to actually imitate the phosphate group. Thus, the term “phosphate mimic” simply refers to the ability of these compounds to act as potent inhibitors of enzymes that use phosphate-containing substrates. Nonetheless, a myriad of computational studies have attempted to showcase the structural and electrostatic similarities of certain cyclic phosphate replacements relative to their phosphate parents. Using the program SPARTAN 5.0 (Wavefunction, Inc., Irvine, CA), electrostatic charge distribution studies compared N-isopropyl-N’-methylsquaryldiamide with dissociated dimethylphosphate (Fig. 12a) (49). As one might expect, polarizing patterns of both compounds are similar, with a charge of -0.84 on each of the oxygens of dimethyl phosphate, and -0.47 and -0.51 on the carbonyl groups of the squaric derivative. Additionally, docking studies of aromatic squaric acid derivatives in the active site of the Yersinia PTP YopH (77) have indicated that interactions of PTP with its natural substrate are preserved when squaric acid derivatives are employed as inhibitors. Although the aromatic groups coming off the squaric acid can interact with a phenylalanine residue, a positively charged region of the protein is proposed to interact with the carbonyl moieties of the squaric derivative (Fig. 12b). Unfortunately, no direct comparison of the binding mode of phosphotyrosine versus the proposed binding of these squaric acid derivatives exists in the literature; no cocrystal structures or NMR structures of these compounds are bound to proteins.
Other docking studies have been completed with tetronic acid derivatives in the active site of VHR; results indicate that on dissociation, the 3-acyltetronic acid anion can bind tightly to the active site through several hydrogen bonds in a manner similar to that of phosphotyrosine (43). Finally, rhodanine and thiazolidinone cores have been shown to bind at or near the phosphate-binding site in modeling studies of various proteins (57), and they share many commonalities with compounds believed to mimic the pentavalent transition state of phosphate hydrolysis. Core structure electron-density isosurfaces for both rhodanine and thiazolidinone portray this well, as compared with the electron density structure of dissociated dimethyl phosphate. At this time, crystal structures of proteins complexed with these novel cyclic phosphate mimics have not been published.
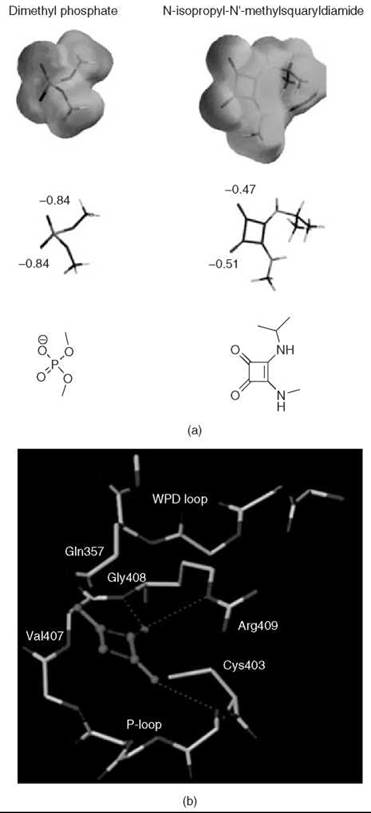
Figure 12. (a) Ab initio calculations show the similarity in charge distribution of dissociated dimethyl phosphate and N-isopropyl-N'-methylsquaryldiamide. Electrostatic charge distribution and charge localization are illustrated below (34, 49). Reprinted in part from Reference 47 with permission from the American Chemical Society. (b) Proposed interactions of squaric acid at the active site of YopH (77). Reprinted in part from Reference 75 with permission from Elsevier.
Choosing the Right Phosphate Isostere
With so many phosphate isosteres to choose from, where does one begin? Despite recent advances, it would seem that the ideal isostere must still be determined on a case-by-case basis. Ease of synthesis and compatibility of the designed synthetic route with production of a combinatorial library of compounds can be a major determining factor. Additionally, one must consider the history of each of these classes as a phosphate mimic. Much data has been generated on the “traditional” phosphate replacements, and many success stories exist with these classes of compounds. However, many published examples exist in which such replacements are biologically inactive (19, 40), and as highlighted by Fressigne et al. (78), the incorporation of phosphate replacements affects the conformational behavior of some systems strongly. In contrast, several recent publications have highlighted the potential of the cyclic compounds as phosphate mimics, but only time will tell whether as many failures exist as successes.
Choosing the right phosphate isostere: good
Although they have been around the longest, phosphorus- and sulfur-containing replacements for the phosphate moiety present the greatest synthetic challenges. The low yields associated with appending phosphonates and fluorophosphonates make the synthesis of combinatorial libraries more difficult, which has been an obstacle in the development of potent inhibitors from these classes of molecules. Additionally, although they are certainly more stable biologically than their parent phosphates, the highly polar nature of phosphonates hinders biologic activity because of limited cell permeability (17-21). For example, one of the most potent and selective inhibitors for protein tyrosine phosphatase 1B (PTP 1B) contains an aryl difluorophosphonate group (16). Because of its anionic character at physiologic pH, it is not cell permeable and exhibits no cellular activity; only when produced in a pro-drug form do such phosphonate inhibitors become useful (16). In general, the phosphonate inhibitors that exist in the literature have slightly reduced potencies compared with their phosphate parent (3); thus, other structural and electronic analogs of the phosphate moiety often will be superior.
Progress has been made in the development of potent sulfone-containing phosphate mimetic inhibitors, but it is difficult to predict in what cases a sulfone might resemble a phosphate. Whereas a select few sulfone-based inhibitors have demonstrated remarkable activity (79), others have shown no potency for the desired enzyme (19). Although they may be suitable analogs for DNA and/or RNA, at this point it is difficult to recommend the use of sulfone analogs as general phosphate isosteres. On the other hand, the acetylsulfamate group possesses considerable potential as a phosphate replacement; the highly acidic proton on the acetylsulfamate nitrogen might mimic some of the negative charge found on the phosphate, although more information is needed on this topic.
Most successful dicarboxylate-containing phosphate mimics have been based on carboxylate-rich natural products (42) that have been shown previously to have activity against the desired enzyme. Although modeling studies have shown that the negatively charged oxygens on maleate are spatially within 0.1 A of that in the equivalent diphosphate, some malonate-, succinate-, and tartaric acid-based phosphate mimics have not been potent inhibitors of enzymatic activity (35, 38). Although dicarboxylate-containing compounds have also been developed to mimic the six-membered ring pyrophosphate-divalent metal complex that is proposed to form in many enzymes (80, 81), in those cases monosaccharide linkages have been found to be much better mimics of this chair or boat conformation (Fig. 13) (37). Carboxylate moieties can be incorporated easily into compounds through amide linkers (37) or oxidation strategies (38), but their inability to consistently function as potent inhibitors limits their applicability.
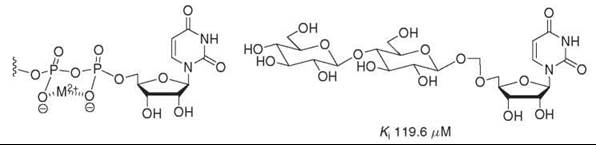
Figure 13. Monosaccharide units are putative mimics of the proposed phosphate-metal complex and inhibit some glycosyltransferases. An inhibitor of β-1,4-galactosyltransferase based on this concept is pictured below.
Choosing the right phosphate isostere: better
Although several phosphate isosteres discussed in the previous sections demonstrate great promise, little hard data exists about their ability to actually function as a phosphate replacement. For example, only one model system has been published for both the tetronic acid derivatives and the sulfhydantoins, and limited structural evidence exists that demonstrates phosphate-mimicking ability. Additionally, even though the sulfhydantoin or reverse sulfhydantoin core resembles the structure of biologically active the thiazolidinones and rhodanines, the only evidence for their activity is limited to the patent literature, in which no absolute IC50 values or crystal structures are presented (72). Finally, although the hydroxytropolones exhibit promising activity in a few model systems, labeling them as phosphate mimics rather than general inhibitors of bimetallic enzymes might be premature. Whereas it is possible that these three cores also function to some extent as a general phosphate replacement, more study must be completed before this can be confirmed.
Choosing the right phosphate isostere: best
Grouped in this final category are the highly desirable drug-like phosphate mimics, which include squaric acid derivatives, rho- danines/thiazolidinones, and perfluorylaryl compounds; a wide range of relatively simple methods exist for the synthesis of these derivatives, and they have been demonstrated to work as phosphate mimics for at least two different classes of enzyme substrates. Squaric acid derivatives have been used in two very different phosphate-mimicking situations: in squaryl-linked oligodeoxynucleotides (48, 49) and as inhibitors of PTPases (53). Additionally, computational studies that involve electrostatic charge distribution have added to the evidence that squaric acid derivatives might be similar spatially and electronically to the phosphate moiety. Thiazolidinone and rhodanine derivatives can simply be prepared through a one-pot cyclocondensation reaction, which has led to their relatively high occurrence as members of commercially available compound collections. Additionally, the thiazolidinone/rhodanine core has been shown to be a valid phosphate replacement for several classes of molecules; both nucleoside diphosphates (56, 58) and phosphorylated lipids (64) have been mimicked by these heterocyclic core structures, which hints at their possible potential to serve as a general phosphate mimetic. Perfuloroaryl compounds have been demonstrated to mimic both the diphosphate and triphosphate moiety of nucleoside phosphates (67) as well as phosphorylated isoprenoids (66, 68). Thus, it seems reasonable to believe that these strategies for phosphate mimicry could be applied successfully to other biologic targets. In the coming years, even more testing and crystallographic or computational evidence will hopefully illuminate the manner in which these compounds act as phosphate isosteres. For now, based on a few key examples these three derivatives all seem to have great potential.
Summary
Phosphonates, sulfones, and dicarboxylates are all valid replacements for the phosphate moiety, but it has become clear that several interesting alternatives to these functional groups exist. Although the squaric acid and thiazolidinone motifs have been best characterized in terms of their ability to actually mimic both the negative charge and the positional orientation provided by a phosphate group, other cyclic compounds have surfaced as potential phosphate replacements. As the synthesis of many of these compounds is relatively facile, these motifs should be exploited in greater frequency in the coming years. More studies are needed to determine whether these compounds actually mimic the phosphate group in an electrostatic and/or structural fashion or whether they simply bind to an allosteric site in the protein and thus inhibit activity. At this point, only crystal structures of enzymes bound to phosphate mimics can answer this question definitively. In the meantime, it is certainly worthwhile to continue exploration of tetronic acids, squaric acids, thiazolidinones, rhodanines, perfluoroaryl compounds, sulfhydantoins, and hydroxytropolones as general phosphate replacement groups for use as probes in chemical biology.
References
1. Cohen P. The regulation of protein function by multisite phospho rylation--a25 yearupdate. Trends Biochem. Sci. 2000; 25:596-601.
2. McCluskey A, Sakoff JA. Small molecule inhibitors of serine/threonine protein phosphatases. Mini Rev. Med. Chem. 2001; 1:43-55.
3. Rye CS, Baell JB. Phosphate isosteres in medicinal chemistry. Curr. Med. Chem. 2005; 23:3127-3141.
4. Al-Obeidi FA, Lam KS. Development of inhibitors for protein tyrosine kinases. Oncogene 2000; 19:5690-5701.
5. Chen YT, Seto CT. Parallel synthesis of a library of bidentate protein tyrosine phosphatase inhibitors based on the a-ketoacid motif. Bioorg. Med. Chem. 2004; 12:3289-3298.
6. Hunter T. Oncoprotein networks. Cell 1997; 88:333-346.
7. Szczepankiewicz BG, Liu G, Hajduk PJ, Abad-Zapatero C, Pei Z, Xin Z, Lubben TH, Trevillyan JM, Stashko MA, Ballaron SJ, Liang H, Huang F, Hutchins CW, Fesik SW, Jirousek MR. Discovery of a potent, selective protein tyrosine phosphatase 1B inhibitor using a linked-fragment strategy. J. Am. Chem. Soc. 2003; 125:4087-4096.
8. Davis BD. On the importance of being ionized. Arch. Biochem. Biophys. 1958; 78:497-509.
9. Gautier A. Phosphate mimic of nucleotides, conformational influences on the ribofuranose conformations. Heterocycles 2006; 67:823-837.
10. Westheimer FH. Why nature chose phosphates. Science 1987; 235:1173-1178.
11. Hirsch AKH, Fischer FR, Diederich F. Phosphate Recognition in Structural Biology. Angew. Chem. Int. Ed. 2007; 46:338-352.
12. Bjerrum J, Schwarzenbach G, Sillen LG. Stability constants. Part 1: Organic ligands. The Chemical Society, Spec. Pub. No. 6, London, 1957.
13. Schneider B, Kabelac M, Hobza P. Geometry of the phosphate group and its interactions with metal cations in crystals and ab initio calculations. J. Am. Chem. Soc. 1996; 118:12207-12217.
14. Grucza RA, Bradshaw JM, Futterer K, Waksman G. SH2 domains: From structure to energetics, a dual approach to the study of structure-function relationships. Med. Res. Rev. 1999; 19:273-293.
15. Burke TRJ, Yao Z-J, Liu D-G, Voigt J, Gao Y. Phosphoryltyrosyl mimetics in the design of peptide-based signal transduction inhibitors. Biopolymers 2001; 60:32-44.
16. Boutselis IG, Yu X, Zhang Z-Y, Borch RF. Synthesis and cell-based activity of a potent and selective protein tyrosine phosphatase 1B inhibitor prodrug. J. Med. Chem. 2007; 50:856-864.
17. Bertolino A, Altman LJ, Vasak J, Rilling HC. Polyisoprenoid amphiphilic compounds as inhibitors of squalene synthesis and other microsomal enzymes. Biochem. Biophys. Acta-Lipids and Lipid Metab. 1978; 530:17-23.
18. Castro A, Spencer TA. Formation and alkylation of anions of bis(methylsulfonyl.methane. J. Org. Chem. 1992; 57:3496-3499.
19. Castro A, Erickson SK, Shechter I, Spencer TA. Synthesis and biological evaluation of nonionic prenyl, geranyl, and farnesyl diphosphate surrogates. Bioorg. Chem. 1996; 24:242-250.
20. Zhu Y, Drueckhammer DG. An efficient synthesis of geminal di-sulfones. Tetrahedron Lett. 2002; 43:1377-1379.
21. Beaulieu PL, Cameron DR, FerlandJ-M, J., G, Ghiro E, Gillard J, Gorys V, Poirier M, Rancourt J, Wernic D, Llinas-Brunet M. Ligands for the tyrosine kinase p56lck SH2 domain: Discovery of potent dipeptide derivatives with monocharged, nonhydrolyzable phosphate replacements. J. Med. Chem. 1999; 42:1757-1766.
22. Berkowitz DB, Bose M, Pfannenstiel TJ, Doukov T. α-Fluorinated phosphonates as substrate mimics for glucose 6-phosphate dehydrogenase: the CHF stereochemistry matters. J. Org. Chem. 2000; 65:4498-4508.
23. Vaghefi MM, Bernacki RJ, Dalley NK, Wilson BE, Robins RK. Synthesis of glycopyranosylphosphonate analogues of certain natural nucleoside diphosphate sugars as potential inhibitors of glycosyltransferases. J. Med. Chem. 1987; 30:1383-1391.
24. Vaghefi MM, Bernacki RJ, Hennen WJ, Robins RK. Synthesis of certain nucleoside methylenediphosphonate sugars as potential inhibitors of glycosyltransferases. J. Med. Chem. 1987; 30:1391-1399.
25. Schmidt RR, Frische K. A new galactosyl transferase inhibitor. Bioorg. Med. Chem. Lett. 1993; 3:1747-1750.
26. Palcic MM, Heerze LD, Srivastava OP, Hindsgaul O. A bisubstrate analog inhibitor for α (1→2.-fucosyltransferase. J. Biol. Chem. 1989; 264:17174-17181.
27. Biller SA, Forster C, Gordon EM, Harrity T, Rich LC, Marretta J, Ciosek CPJ. Isoprenyl phosphinylformates: New inhibitors of squalene synthetase. J. Med. Chem. 1991; 34:1912-1914.
28. Burke TRJ, Kole HK, Roller PP. Potent inhibition of insulin receptor dephosphorylation by a hexamer peptide containing the phosphotyrosyl mimetic F2Pmp. Biochem. Biophys. Res. Commun. 1994; 204:129-134.
29. Boresch S, Leitgeb M, Beselman A, MacKerell ADJ. Unexpected relative aqueous solubilities of a phosphotyrosine analogue and two phosphonate derivatives. J. Am. Chem. Soc. 2005; 127:4640-4648.
30. Yao Z-J, King CR, Cao T, Kelley J, Milne GWA, Voigt J, Burke TRJ. Potent inhibition of Grb2 SH2 domain binding by non-phosphate-containing ligands. J. Med. Chem. 1999; 42:25-35.
31. Khurgin YI, Isaev AN. Geometry and electronic structure of the dimethyl sulfoxide dimer. Calculation by the MNDO method. Russian Chem. Bull. 1992; 41:1046-1049.
32. Morley JO. Theoretical studies on the structure and electronic properties of aryl sulfides and sulfones. Int. J. Quant. Chem. 1998; 66:141-147.
33. Wojciechowski M, Grycuk T, Antosiewicz JM, Lesyng B. Prediction of secondary ionization of the phosphate group in phosphotyrosine peptides. Biophys. J. 2003; 84:750-756.
34. Blattler MO, Wenz C, Pingoud A, Benner SA. Distorting duplex DNA by dimethylsulfone substitution: a new class of “transition state analog” inhibitors for restriction enzymes. J. Am. Chem. Soc. 1998; 120:2674-2675.
35. Traxler PM, Wacker O, Bach HL, Geissler JF, Kump W, Meyer T, Regenass U, Roesel JL, Lydon N. Sulfonylbenzoyl-nitrostyrenes: Potential bisubstrate type inhibitors of the EGF-receptor tyrosine protein kinase. J. Med. Chem. 1991; 34:2328-2337.
36. Ferreras JA, Ryu J-S, Di Lello F, Tan DS, Quadri LEN. Small- molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nature Chem. Biol. 2005; 1:29-32.
37. Wang R, Steensma DH, Takaoka Y, Yun JW, Kajimoto T, Wong C-H. A search for pyrophosphate mimics for the development of substrates and inhibitors of glycosyltransferases. Bioorg. Med. Chem. Lett. 1997; 5:661-672.
38. Dawson RMC, Elliott DC, Elliott WH, Jones KM. Data for Biochemical Research, 3rd edition. 1986. Clarendon Press, Oxford.
39. Miller MJ, Braccolino DS, Cleary DG, Ream JE, Walker MC, Sikorski JA. EPSP synthase inhibitor design iv: New aromatic substrate analogs and symmetrical inhibitors containing novel 3-phosphate mimics. Bioorg. Med. Chem. Lett. 1994; 4:2605-2608.
40. Boudreau MA, Vederas JC. Synthesis and biological evaluation of nucleoside dicarboxylates as potential mimics of nucleoside diphosphates. Org. Biomol. Chem. 2007; 5:627-635.
41. Gibbs JB, Pompliano DL, Mosser SD, Rands E, Lingham RB, Singh SB, Scolnick EM, Kohl NE, Oliff A. Selective inhibition of farnesyl-protein transferase blocks ras processing in vivo. J. Biol. Chem. 1993; 268:7617.
42. Singh SB, Zink DL, Liesch JM, Goetz MA, Jenkins RG, Nallin-Omstead M, Silverman KC, Bills GF, Mosley RT, Gibbs JB, Albers-Schonberg G, Lingham RB. Isolation and structure of chaetomellic acids A and B from Chaetomella acutiseta: Farnesyl pyrophosphate mimic inhibitors of ras farnesyl-protein transferase. Tetrahedron 1993; 49:5917-5926.
43. Sodeoka M, Sampe R, Kojima S, Baba Y, Usui T, Ueda K, Osada H. Synthesis of a tetronic acid library focused on inhibitors of tyrosine and dual-specificity protein phosphatases and its evaluation regarding VHR and Cdc25B inhibition. J. Med. Chem. 2001; 44:3216-3222.
44. Moran EJ, Sarshar S, Cargill JF, Shahbaz MM, Lio A, Mjalli AMM, Armstrong RW. Radio frequency tag encoded combinatorial library method for the discovery of tripeptide-substituted cinnamic acid inhibitors of the protein tyrosine phosphatase PTP1B. J. Am. Chem. Soc. 1995; 117:10787-10788.
45. Douglas KT, Nadvi IN. Inhibition of glyoxalase I: A possible transition-state analogue inhibitor approach to potential antineoplastic agents? FEBS Lett. 1979; 106:393-396.
46. Burka LT, Doran J, Wilson BJ. Enzyme inhibition and the toxic action of moniliformin and other vinylogous alpha-ketoacids. Biochem. Pharmacol. 1982; 31:79-84.
47. Kinney WA, Abou-Gharbia M, Garrison DT, Schmid J, Kowal DM, Bramlett DR, Miller TL, Tasse RP, Zaleska MM, Moyer JA. Design and synthesis of (2-(8,9-dioxo-2,6-diazabicyclo(5.2.0. non-1(7.-en-2-yl.-ethyl.phosphonic acid (EAA-090., a potent N-methyl-D-aspartate antagonist, via the use of 3-cyclobutene-1,2-dione as an achiral a-amino acid bioisostere. J. Med. Chem. 1998; 41:236-246.
48. Sato K, Tawarada R, Seio K, Sekine M. Synthesis and structural properties of new oligodeoxynucleotide analogues containing a 2/,5/-internucleotidic squaryldiamide linkage capable of formation of a Watson-Crick base pair with adenine and a wobble base pair with guanine at the 3/-downstream junction site. Eur. J. Org. Chem. 2004; 2142-2150.
49. Sato K, Seio K, Sekine M. Squaryl group as a new mimic of phosphate group in modified oligodeoxynucleotides: synthesis and properties of new oligodeoxynucleotide analogues containing an internucleotidic squaryldiamide linkage. J. Am. Chem. Soc. 2002; 124:12715-12724.
50. Schwartz LM, Howard LO. Conductance study of squaric acid aqueous dissociation. J. Phys. Chem. 1971; 75:1798-1803.
51. Schmidt AH. Reaktionen von quadratsaure und quadratsaure-derivaten. Synthesis 1980: 961-994.
52. Terao H, Sugawara T, Kita Y, Sato N, Kaho E, Takeda S. Proton relay in a one-dimensional hydrogen-bonded chain composed of water molecules and a squaric acid derivative. J. Am. Chem. Soc. 2001; 123:10468-10474.
53. Xie J, Comeau AB, Seto CT. Squaric acids: a new motif for designing inhibitors of protein tyrosine phosphatases. Org. Lett. 2003; 6:83-86.
54. Benson TE, Walsh CT, Massey V. Kinetic characterization of wild-type and S229A mutant MurB: evidence for the role of ser 229 as a general acid. Biochemistry 1997; 36:796-805.
55. Andres CJ, Bronson JJ, D’Andrea SV, Deshpande MS, Falk PJ, Grant-Young KA, Harte WE, Ho H-T, Misco PF, Robertson JG, Stock D, Sun Y, Walsh AW. 4-Thiazolidinones: novel inhibitors of the bacterial enzyme MurB. Bioorg. Med. Chem. Lett. 2000; 10:715-717.
56. Ma Y, Stern RJ, Scherman MS, Vissa VD, Yan W, Jones VC, Zhang F, Franzblau SG, Lewis WH, Mcneil MR. Drug targeting Mycobacterium tuberculosis cell wall synthesis: genetics of dTDP-rhamnose synthetic enzymes and development of a microtiter plate-based screen for inhibitors of conversion of dTDP-glucose to dTDP-rhamnose. Antimicrob. Agents Chemother. 2001: 1407-1416.
57. Kantardjieff KA, Kim C-Y, Naranjo C, Waldo GS, Lekin T, Segelke BW, Zemla A, Park MS, Terwilliger TC, Rupp B. Mycobacterium tuberculosis RmIC epimerase (Rv3465.: a promising drug-target structure in the rhamnose pathway. Acta Crystallogr., Sect. D Biol. Crystallogr. 2004; D60:895-902.
58. Helm JS, Hu Y, Chen L, Gross B, Walker S. Identification of active-site inhibitors of MurG using a generalizable high- throughput glycosyltransferase screen. J. Am. Chem. Soc. 2003; 125:11168-11169.
59. Hu Y, Helm JS, Chen L, Ginsburg C, Gross B, Kraybill B, Tiyanont K, Fang X, Wu T, Walker S. Identification of selective inhibitors for the glycosyltransferase MurG via high-throughput screening. Chem. Biol. 2004; 11:703-711.
60. Carlson EE, May JF, Kiessling LL. Chemical probes of UDP-galactopyranose mutase. Chem Biol. 2006; 13:825-837.
61. Zhou D, Luini W, Bernasconi S, Diomede L, Salmona M, Mantovani A, Sozzani S. Phosphatidic acid and lysophosphatidic acid induce haptotactic migration of human monocytes. J. Biol. Chem. 1995; 270:25549.
62. Qi C, Park JH, Gibbs TC, Shirley DW, Bradshaw CD, Ella KM, Meier KE. Lysophosphatidic acid stimulates phospholipase D activity and cell proliferation in PC-3 human prostate cancer cells. Cell Physiol. 1998; 174:261-272.
63. Imamura F, Horai T, Mukai M, Shinkai K, Sawada M, Akedo H. Induction of in vitro tumor cell invasion of cellular monolayers by lysophosphatidic acid or phospholipase D. Biochem. Biophys. Res. Commun. 1993; 193:497.
64. Gududuru V, Hurh E, Dalton J, Miller DD. Synthesis and antiproliferative activity of 2-aryl-4-oxo-thiazolidin-3-yl-amides for prostate cancer. Bioorg. Med. Chem. Lett. 2004; 14:5289-5293.
65. Gududuru V, Hurh E, Durgam GG, Hong SS, Sardar VM, Xu H, Dalton J, Miller DD. Synthesis and biological evaluation of novel cytotoxic phospholipids for prostate cancer. Bioorg. Med. Chem. Lett. 2004; 14:4919-4923.
66. Marriott JH, Jarman M, Neidle S. Patent number WO9740006. 1997.
67. Barber AM, Hardcastle IR, Rowlands MG, Nutley BP, Marriott JH, Jarman M. Solid-phase synthesis of novel inhibitors of farnesyl transferase. Bioorg. Med. Chem. Lett. 1999; 9:623-626.
68. Marriott JH, Moreno Barber AM, Hardcastle IR, Rowlands MG, Grimshaw RM, Neidle S, Jarman M. Synthesis of the farnesyl ether 2,3,5-trifluoro-6-hydroxy-4-((E,E.-3,7,11-trimethyldodeca-2,6,10-trien-1-yloxy.nitrobenzene, and related compounds containing a substituted hydroxytrifluorophenyl residue: novel inhibitors of protein farnesyltransferase, geranylgeranyltransferase I and squalene synthase. J. Chem. Soc. Perkin Trans. 2000; 1:4265-4278.
69. Hall A. A biochemical function for ras--at last. Science 1994; 264:1413-1414.
70. Lebowitz PF, Prendergast GC. Non-Ras targets of farnesyltransferase inhibitors: Focus on Rho. Oncogene 1998; 17:1439-1447.
71. Groutas WC, Kuang R, Venkataraman R, Epp JB, Ruan S, Prakash O. Structure-based design of a general class of mechanism-based inhibitors of the serine proteinases employing a novel amino acid-derived heterocyclic scaffold. Biochemistry 1997; 36:4739-4750.
72. Saunders JO, Miknis GF, Blake JF. Patent number WO2004/062664 A1, 2004.
73. Piettre SR, Ganzhorn A, Hoflack J, Islam K, Hornsperger J-M. α-Hydroxytropoloes: a new class of potent inhibitors of inositol monophosphatase and other bimetallic enxymes. J. Am. Chem. Soc. 1997; 119:3201-3204.
74. Piettre SR, Andre C, Chanal M-C, Ducep J-B, Lesur B, Piriou F, Raboisson P, Rondeau J-M, Schelcher C, Zimmerman P, Ganzhorn A. Monoaryl- and bisaryldihydroytropolones as potent inhibitors of inositol monophosphate. J. Med. Chem. 1997; 40:4208-4221.
75. Martin SF, Follows BC, Hergenrother PJ, Franklin CL. A novel class of zinc-binding inhibitors for the phosphatidylcholine-preferring phospholipase C from Bacillus cereus. J. Org. Chem. 2000; 65:4509-4514.
76. Didierjean J, Isel C, Querre F, Mouscadet J-F, Aubertin A-M, Valnot J-Y, Piettre SR, Marquet, R. Inhibition of human immunodeficiency virus type 1 reverse transcriptase, RNase H, and integrase activities by hydroytropolones. Antimicrob. Agents Chemother. 2005; 49:4884-4894.
77. Hu X, Stebbins CE. Molecular docking and 3D-QSAR studies of Yersinia protein tyrosine phosphatase YopH inhibitors. Bioorg. Med. Chem. 2005; 13:1101-1109.
78. Fressigne C, Piettre SR, Condamine E, Altona C, Gautier A. A C3 modified nucleotide. The difluorophosphonate function, a phosphate mimic, governs the conformational behaviour of the ribofuranose. Tetrahedron 2005; 61:4769-4777.
79. Blattler MO, Wenz C, Pingoud A, Benner SA. Distorting duplex dna by dimethylsulfone substitution: a new class of “transition state analog” inhibitors for restriction enzymes. J. Am. Chem. Soc. 1998; 120:2674-2675.
80. Murray BW, Takayama S, Schultz J, Wong C-H. Mechanism and specificity of human α-1,3-fucosyltransferase V. Biochemistry 1996; 35:11183.
81. Murray BW, Wittmann V, Burkart MD, Hung S-C, Wong C-H. Mechanism of human α-1,3-fucosyltransferase V: glycosidic cleavage occurs prior to nucleophilic attack. Biochemistry 1997; 36:823-831.
See Also
Approaches to Enzyme Inhibition
Chemical Libraries: Screening for Biologically Active Small Molecules
Small Molecule Inhibitors, Design and Selection of
Small Molecules in Nature, Tools to Identify Biologically Active
Small Molecules to Elucidate Biological Function