CHEMICAL BIOLOGY
Protein Misfolding and Disease, Chemical Biology of
Johanna C. Scheinost, Grant E. Boldt, and Paul Wentworth, Jr. The Scripps-Oxford Laboratory, Department of Biochemistry, University of Oxford, UK Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute La Jolla, CA
doi: 10.1002/9780470048672.wecb647
The amyloidoses are a class of conformational diseases that arise from the conversion of normally unfolded or globular proteins into fibrillar aggregates that are either pathogenic or non-functional. At present there are more than 20 proteins that are associated with human amyloid diseases. This review focuses on three natively unfolded proteins that form fibrillar aggregates; amyloid-fi, islet amyloid polypeptide ('amylin'), and α-synuclein, the diseases they contribute to and chemical and biophysical approaches that are used to investigate these proteins' aggregation.
Introduction
Conversion of native conformational folded proteins or peptides into highly-ordered insoluble fibrillar aggregates termed ‘amyloid’ has been demonstrated to be clinically-relevant in several diseases. Misfolded proteins are deposited in a variety of tissues, leading to serious and even fatal human diseases. To date there are over 20 proteins and peptides that are known to induce pathological conditions associated with protein misfolding (Table 1). These proteins include the amyloid-fi peptides, prion proteins, α-synuclein, transthyretin and polyglutamine-containing peptides (1). The hypothesis that these diseases are in fact caused by misfolded protein and the misfolded protein does not arise as a side effect of the disease is supported by familial disease, where proteins containing mutations that facilitate aggregation result in more severe and early onset disease variants.
Diseases arising as a result of protein misfolding are classified based on the location of the protein deposition. Thus, the amyloidoses are generally classified as either neuronal or systemic. The majority of conditions arise sporadically, with only 10% of the cases being linked to hereditary causes. The familial forms of the diseases are caused by mutations that make a protein more prone to aggregation, or by mutations in processing proteins like proteases. Under rare circumstances (5% of protein misfolding diseases), protein aggregation can be initiated by infection and transmission. All diseases caused by misfolded protein share the problem of definitive diagnosis as they are difficult to identify in vivo, especially during early stages of symptoms.
Precursors of amyloid are quite diverse, ranging from small unstructured peptides like amyloid-β peptides to large oligomeric multi-domain proteins like p53. Despite the differences in structure of the monomers, the structures of the misfolded β-sheet rich forms share similar features. In recent years it has become apparent that the proteins that form amyloid share common features, and it has been suggested that many if not all proteins share the intrinsic predisposition to form amyloid under the right conditions (2, 3). This hypothesis is affirmed by the absence of a correlation between the propensity of a peptide or protein to misfold and its sequence. Precursor proteins share neither homology in sequence nor structure. However, certain factors such as hydrophobic regions and/or unstable globular protein conformations contribute to a predisposition to form amyloid.
In this review, we will discuss the aggregation of three intrinsically unstructured peptides and proteins and the diseases to which they contribute. Specifically we will concentrate on amyloid-fi (Afi), islet amyloid polypeptide (IAPP), and α-synuclein.
Table 1. List of Protein Misfolding Diseases and the Implicated Proteins and Peptides
|
Clinical Syndrome |
Fibril Component |
|
Alzheimer’s Disease |
A|3 peptide, 1-40, 1-42 |
|
Spongiform encephalopathies |
Full-length prion and fragments |
|
Primary systemic amyloidosis |
Intact light chain and fragments |
|
Secondary systemic amyloidosis |
76-residue fragment of amyloid A protein |
|
Familial amyloidotic polyneuropathy I |
Transthyretin variants and fragments |
|
Senile systemic amyloidosis |
Wild-type transthyretin and fragments |
|
Hereditary cerebral amyloid angiopathy |
Fragments of cystatin-C |
|
Haemodialysis-related amyloidosis |
β2-macroglobulin |
|
Familial amyloidotic polyneuropahy II |
Fragments of apolipoprotein A-1 |
|
Finnish hereditary amyloidosis |
71-residue fragment of gelsolin |
|
Type II diabetes |
Islet-associated polypeptide |
|
Medullary carcinoma of the thyroid |
Fragments of calcitonin |
|
Atrial amyloidosis |
Atrial natriuretic factor |
|
Lysozyme amyloidosis |
Full-length lysozyme variants |
|
Insulin-related amyloid |
Full-length insulin |
|
Fibrinogen α-chain amyloidosis |
Fibrinogen α-chain variants |
|
Synucleinopathies |
α-synuclein |
Biological Background of Protein Misfolding Diseases
Amyloid-β (Aβ) and Alzheimer's disease
Alzheimer’s disease (AD) is the most common neurodegenerative disease in humans. AD is a mostly sporadic, late-onset disease (90%). The risk of developing AD increases exponentially with age. In the early stage, AD is often overlooked as it manifests in mildly impaired short-term memory. The long-term memory is unaffected. With progression of the disease, the impairment of memory both short and long-term becomes apparent. Patients undergo a change in personality from having paranoid delusions to hallucinations and anxiety. At the late stages of AD, the affected person loses his/her ability to participate in daily life and fails to perform the most basic actions such as eating or walking.
A key hallmark of AD is the misfolding of two proteins, Aβ and tau, whose extra- and intracellular aggregates in the brain are coincident with and most likely causative of the disease. Therefore aggregates of these proteins are used as histological markers for diagnosis of AD post mortem. Tau is a protein needed for microtubule stabilization. However, upon hyperphosphorylation it forms neurofibrillar tangles (NFTs) that perturb the integrity of microtubules leading to impaired neuronal function. The presence of NFTs correlates to the severity of AD. However, NFTs are absent in 10% of AD patients and in up to 50% of mild of AD cases (4).
Aβ is the principal constituent of senile plaques found in AD patients’ brains and as such has made it a key suspect in the search for causative agents of AD. Aβ is a 39-43 residue peptide (Fig. 1) that is derived from the proteolytic cleavage of the amyloid precursor protein APP, a type I integral membrane protein. APP is processed by three proteases α-, β-, and γ-secretases. Cleavage of APP by α- and γ-secretases releases the non-amyloidogenic fragment p3. Upon cleavage of APP by β- and γ-secretases, Aβ is released. The peptide is characterized by a hydrophilic N-terminus and a hydrophobic C -terminus, corresponding to parts of the extracellular and intramembrane domains of APP, respectively. Aβ exists mainly as a 40 or 42 residue long peptide (Aβ(1-40) and Aβ(1-42)) (Fig. 1). The C-terminally extended, more hydrophobic sequences like AP(1-42) aggregate faster in vitro than their shorter partners, which might explain why they are more abundant in senile plaques.

Figure 1. Sequences of Aβ, IAPP and α-synuclein.
Native Aβ is unstructured and its function is still uncertain. Several possible roles have been suggested, including cholesterol transport, antioxidative protection and TGF-β activity (5-7). Similarly, a definite function for APP is still unidentified. APP is a putative Notch-like receptor for an unknown ligand (8) and has been suggested to play a role in intercellular adhesion (9).
The presence of extracellular plaques consisting of misfolded Aβ in the brain of AD patients led to the widely accepted hypothesis that insoluble aggregates of Aβ are the toxic conformation of the native peptide. However, there is a poor correlation between the number of mature fibrils found in brain and neuronal death (10). Studies have now shown that soluble AP oligomers are able to damage and kill neurons in central nervous system cultures (11), providing a direct link between Aβ and AD. More recently, dodecamers of Aβ have been identified as the causative agent in impairment of spatial memory in mice (12), although the relevance of this result in the context of other memory changes is unknown (13). These oligomeric species are most likely precursors or folding intermediates and do not emerge by a distinct pathway (14). It has already been shown that oligomers in the absence of fibrils and monomers can inhibit hippocampal long-term potentiation in rats in vivo (15).
Developing animal models for AD is an ongoing challenge due to the multifactorial nature of the syndrome. Existing murine models, such as Tg2576, express mutated human APP (the Swedish mutation) and develop amyloid plaques, but lack other important characteristics of AD, such as NFTs. These mice develop memory loss but are devoid of neurodegeneration. Recently, a triple transgenic mouse model has been developed that expresses the Swedish mutant APP, a human tau mutant, as well as a human presenilin-1 (PS1) mutant (16). These triple transgenic mice build up misfolded AP and tau and more accurately mimic human AD pathology.
To date, there is no generally accepted theory that accounts for the development of AD pathology. The multifactorial basis of the disease makes such a theory unlikely to be possible in the foreseeable future. In addition to the NFTs and amyloid-hypothesis of the disease, oxidative stress, systemic levels of redox active metal ions, cardiovascular disease, the apoEε4 allele and type 2 diabetes are all clear risk factors for development of AD. However, recent research supports the notion that the Aβ buildup may be a key event in AD and that other manifestations of the disease, like NFT formation, result from an imbalance between Aβ production and AP clearance (17).
Islet amyloid polypeptide (IAPP) and type 2 diabetes
Deposition of amyloid in pancreatic islets is a feature of type 2 diabetes in man, but the factors that contribute to this phenomenon are unknown. Progressive amyloidosis results in loss of insulin-producing cells and increased disease state. The presence of amyloid depositions in pancreatic tissue was first described over a century ago (18). However, it took much effort and time to identify the peptide component of these accumulations as islet amyloid polypeptide (IAPP, also called amylin) (19, 20). IAPP is a 37 residue long peptide (Fig. 1) which is cosecreted with insulin. It originates from a longer precursor, pro-IAPP (67 amino acids). The peptide is co-stored with insulin in β-cell secretory granules and secreted together with insulin in response to β-cell stimulation, primarily glucose intake. Its native structure both intra- and extracellular prior to aggregation in vivo is still unclear; under physiological concentrations in vitro it adopts a stable random coil structure. During maturation of β-cell secretory granules, the N- and C-termini of pro-IAPP are enzymatically cleaved by pro-hormone convertases 1/3 and 2. The IAPP peptide is naturally present in humans and other species and is usually excreted via the kidney. It is unclear if IAPP has any function in vivo although multiple possible roles have been suggested.
IAPP forms insoluble aggregates, confined within the islets of Langerhans, which are usually incidental with the non-insulin- dependent type 2 diabetes. In contrast to the autoimmune disease type 1 diabetes, which is characterized by the destruction of insulin secreting cells, type 2 diabetes is a late-onset disease with decreased insulin secretion and reduced sensitivity towards insulin of peripheral tissue. Misfolded IAPP is present in over 90% of type 2 diabetes patients (as diagnosed post mortem), but also in 15% of non-diabetics, indicating that other factors that are not diabetes related may play a role in the aggregation of the 37mer. Unlike Aβ, IAPP is present in concentrations above the critical concentration even in healthy individuals, but aggregation is prevented. It has been shown that insulin but not pro-insulin forms a complex with IAPP that stabilizes the peptide in vitro and possibly in vivo.
The role of amyloid in type 2 diabetes appears to be complex: severe islet dysfunction correlates well with extensive amyloidosis; however, the degree of amyloidosis can vary extremely in long term patients. The factors that trigger the onset of the disease are both genetic and environmental. On the other hand, a common qualification of misfolding diseases is that they occur mostly sporadic, without the necessity of mutations.
Mature fibrils formed from IAPP demonstrate β-cell toxicity in vitro (21). Analogous to the toxic-conformation-discussion with Aβ, it has also been proposed that smaller, soluble oligomers of IAPP have cytotoxic effects (22) and show membrane-disrupting activity.
A link between AD and diabetes?
It has long been suggested that diabetes is a risk factor for AD, and a cohort study on people older that 55 years revealed that patients suffering from diabetes have a 65% increase in the risk of developing AD compared to the group that did not suffer from diabetes (23). Moreover, the two diseases share similar physiological processes, most notably degeneration and age dependence. Interestingly, insulin receptors are not only found in the peripheral system, but also in neurons in the CNS (24). Therefore, the role of these receptors is not limited to the regulation of blood sugar levels but they are also involved in neuronal differentiation and cell proliferation (25, 26), implicating an intricate connection between insulin and AD. This unexpected link of AD and diabetes has led to the postulation of a new, brain specific ’’type 3 diabetes” (27), which was further elucidated recently when it was shown that soluble oligomers of Ab disrupt insulin signaling by binding to the insulin receptors on the neuronal surface (28). Given that synaptic failure and memory dysfunction are characteristic for AD, this link is another plausible explanation for the observed symptoms of the disease.
α-Synuclein and synucleopathies
α-Synuclein (Fig. 1) is the protein component associated with synucleinopathies, protein misfolding diseases that are characterized by the presence of α-synuclein deposits. Synucle-inopathies include Parkinson disease, Lewy body (LB) dementia, diffuse LB dementia, and also the LB variant of AD.
α-Synuclein has been found to be the major constituent of Lewy bodies (LBs). Intriguingly, these deposits are not extracellular as with Aβ and IAPP, but intracellular inclusion bodies found in dopaminergic and non-dopaminergic neurons and glia. The protein morphology within the inclusions can be distinct; at least five different conformations have been described: LBs, Lewy neurites, glial cytoplasmic inclusions, neuronal cytoplasmic inclusions, and axonal spheroids.
α-Synuclein exits in three isoforms of varying length in humans. The best characterized is the full length, 140 amino acid long protein; the other isoforms are shorter and derived by splicing. The N-terminus of the 140mer is characterized by the presence of a seven eleven amino acid long repeat containing highly conserved hexameric motifs (KTKEGV). These repeats are thought to be implicated in the interaction with lipids and adopt α-helical structure upon contact with lipid vesicles. The central region of the protein is highly prone to aggregation.
α-Synuclein is natively unstructured and a definitive function remains elusive. Possible roles include the regulation of vesicular release and other synaptic tasks as α-synuclein has been found to exist in equilibrium of free protein with vesicle-bound protein. α-Synuclein interacts in vitro with synthetic or purified phospholipid membranes, altering their integrity and suggesting participation in membrane lipid component organization. Moreover, it may also act as inhibitor of lipid organization. An intriguing function of α-synuclein is that it may operate as a molecular chaperone.
α-Synuclein is known to bind many proteins; over 500 proteins interacted in a proteomic analysis of a neuroblastoma cell line. Contact with some proteins or protein complexes promotes aggregation, e.g. histones and the tau protein, while others, particularly the shorter synucleins (β and γ), inhibit misfolding. Due to a high plasticity of the protein, it can adopt many different conformations. This conformational diversity of α-synuclein has had the protein labeled as a “protein chameleon”. Under physiological conditions in vitro the protein is unstructured. In the presence of lipid vesicles, its N-terminus can acquire α-helical structure in vitro. It can form non-covalent dimers and higher order oligomers leading to insoluble aggregates. Interestingly, α-synuclein can also form covalent dimers that are crosslinked via a dityrosine bond, under oxidative conditions.
Evidence points towards the oligomeric state of α-synuclein as the toxic conformation causing neuronal death. It has been suggested that fibril formation is merely an escape route or protection mechanism in the brain to prevent further neuronal damage.
Biophysical Characterization of Fibrils and Molecular Mechanism of their Formation
Although precursor proteins of amyloid are extremely diverse in sequence, the end-product fibrils in general share several structural features such as cross-P sheet assemblies, non-branched fibrils of similar length and structural organization.
Key intermediates in fibril formation
In general, there is a simple picture of the events that occur during oligomerization of natively unstructured protein and peptides: unfolded monomers undergo organized self-assembly via several intermediate assemblies on the pathway to fibrils. Many of these intermediates have been characterized and named based on in vitro studies, but there is no defined nomenclature as of yet. The following intermediates have been identified so far in the fibril polymerization process:
Monomers of the native peptide or protein can either be unfolded or folded. However, even natively unstructured peptides, e.g. Aβ, can adopt some secondary structure depending on the physiological environment, and some proteins exist in a native oligomerized state, like transthyretin. Monomers can arrange into oligomers, which are small globular, possibly spherical, micelle-like assemblies that contain some secondary structure (29). It is still unclear whether these oligomers are direct intermediates of the fibril formation pathway, or whether they occur as part of an “off-pathway”. For the misfolding of Aβ, evidence points towards oligomers being precursors of fibrils (29). Moreover, these oligomers are thought to be the neurotoxic conformation in a variety of misfolding diseases, most importantly AD (11). Structural instabilities of folding intermediates make it difficult to determine the structure of these intermediates, but not impossible (24).
Two unfortunate names that can easily lead to confusion have been given to the “in-pathway” conformations protofibrils and protofilaments. Protofibrils are, similar to oligomers, mostly unstructured but linear (31). However, in most cases the term “protofibrils” is used interchangeably with the term oligomer. The name protofilaments refers to linear aggregates with β-sheet structure that have a diameter of 3 nm and are usually 50-100 nm in length. Mature, elongated protofilaments, also called filaments, intertwine to form protein fibrils. Fibrils are generally unbranched, approx. 10 nm in diameter and can be several μm long.
Nucleated polymerization and other models of fibril formation
The actual mechanism of self-association of monomeric protein into amyloid is complex and three mechanisms of structure conversion have been proposed (32): In templated assembly, a monomeric native state peptide binds to an existing nucleus. Upon binding, there is a change in the secondary structure of the monomer as it is added to the growing chain. Monomer directed conversion involves the presence of a misfolded monomer that templates the structure conversion of a native monomer, followed by disassociation and chain formation. The third model is nucleated polymerization, which is the most widely accepted model for the fibril growth.
Nucleated polymerization is a crucial process in cells, involved in providing a rigid cytoskeleton by organization of tubulin into microtubules and actin into thin filaments. These processes are under tight control; however, nucleated polymerization of proteins can also be “accidental” and occur to proteins that, in healthy individuals, remain in their native structure. The kinetics of misfolding are characterized by the presence of a lag phase, a rapid growth or elongation phase, and a plateau phase. The overall rate of amyloid aggregation is restricted by the formation of a nucleus, a process that is thought to have either slow kinetics or is driven by an unfavorable equilibrium. A nucleus is defined as the least stable conformer in the aggregation process that is in equilibrium with monomeric protein (33). The formation of a nucleus, a “stochastic” event, is concentration dependent. Nuclei only occur above a critical protein monomer concentration (for AP this concentration is approx. 15 μM). However, in the present of preformed seeds the lag phase is completely abolished. Once a nucleus or seed is present, it acts as a template for the conversion of monomers into P-sheet rich conformers that assemble rapidly into protofibrils and fibrils. Upon consumption of monomers there is a plateau phase where no more fibrils are formed. This general model of fibril formation is able to explain the spontaneous onset of most amyloidoses as well as their rapid progression. However, the kinetics of this mechanism can be influenced to a great extent by quite a few factors both in vitro and in vivo: pH, temperature of the sample, presence of preformed seeds, presence of metal ions, and oxidative damage.
Mathematical descriptions of the kinetics of protein misfolding have largely been attempted using non-complex simulations and algorithms. It has been suggested that a complete mathematical analysis of these processes is currently beyond our capability (34).
Structure of fibrils
No high resolution structure of fibrils has been published yet, as fibrils are insoluble, non-crystalline material, and therefore the classic structure determination methods like NMR and X-ray crystallography fail.
First experiments to elucidate the structure of the proteins in the fibrillar organization were done by X-ray diffraction. Amyloid fibrils show a characteristic diffraction pattern, the so called β-cross pattern (35), which is indicative of β-sheets parallel to the fibril axis with the protein strands perpendicular to the fibril’s long axis (36, 37). The pattern of amyloid is characterized by reflections at 4.75 A (along the fibril axis) and 10 A (perpendicular to the fibril axis) which occur from regular repeats and stacking of monomers.
Further characterization of Aβ fibrils involved solid-state NMR to determine the structure of the peptide subunits. Depending on the experimental constraints applied, several models for full length (e.g. (38)) and fragments of Aβ (e.g. (39, 40)) have been developed, most of them proposing an antiparallel, in-register β-sheet organization. However, it has since been proposed that the monomers composing a fibril associate in parallel, not antiparallel β-sheets (41) with full length Aβ, and a model based on these constraints has been determined (42) (Fig. 2).
Within the IAPP polypeptide, three key regions have been identified that are responsible for its misfolding: residues 8-20 (43), 20-29 (44), and 30-37 (45). Residues 20-29 were initially found to be the amyloidogenic chore of IAPP, as peptide fragments corresponding to that sequence was found to form fibrils in vitro. Taken together, these three regions might participate in the formation of an intramolecular β-sheet with both parallel and antiparallel organization (43). Electron paramagnetic resonance spectroscopy of spin-labeled IAPP fibrils suggested a structure similar to that of AP: in-register, parallel β-sheet structure with units showing a disordered N-terminal segment (46).
Solid-state NMR studies of α-synuclein fibrils have shown that the β-sheet rich domain is located between at least residues 38-95, with the N-terminal residues being unstructured and a rigid backbone starting at residue 22 (47). These results are consistent with data obtained from electron paramagnetic resonance (EPR) of spin-labeled protein, showing an ordered core around residues 31-109, with parallel, in-register β-sheets (42, 43).

Figure 2. Structural model for Aβ 1-40 fibrils, consistent with solid state NMR constraints on the molecular conformation and intermolecular distances and incorporating the cross-β motif common to all amyloid fibrils. Residues 1-8 are considered fully disordered and are omitted. (a) Schematic representation of a single molecular layer, or cross-β unit. The yellow arrow indicates the direction of the long axis of the fibril, which coincides with the direction of intermolecular backbone hydrogen bonds. The cross-β unit is a double-layered structure, with in-register parallel β-sheets formed by residues 12-24 (light grey ribbons) and 30-40 (dark grey ribbons). (b) Central API-40 molecule from the energy-minimized, five-chain system, viewed down the long axis of the fibril. Taken from Petkova, A.T., et al.. Proceedings of the National Academy of Sciences, 2002. 9926: p. 16742-16747.
Lessons for drug development
To date, there is no successful treatment for misfolding diseases. The development of structural models for fibrils and the elucidation of intermediates of fibril formation give insights into possible drug targets. Several approaches are possible using small molecules: the use of chemical chaperones to stabilize the native structures of the protein, possibly combined with another method, the use of P-sheet breakers to destabilize β-sheet intermediates. Other approaches include the competitive inhibition of protein-protein interactions, either for the monomer to prevent oligomerization, or for oligomers to prevent further monomer addition. An interesting, but problematic option is the use of molecules that increase the turnover or degradation of fibrils. Immunotherapy for AD was developed by injecting Aβ(1-42) into mice to immunize young animals against the disease and to reduce neuropathology in older animals (50). Initial clinical trials were performed but needed to be aborted as 6% of individuals developed meningoencephalitis (51), although beneficial effects can also be seen in these individuals (52).
Small molecule inhibitors of Ab assembly
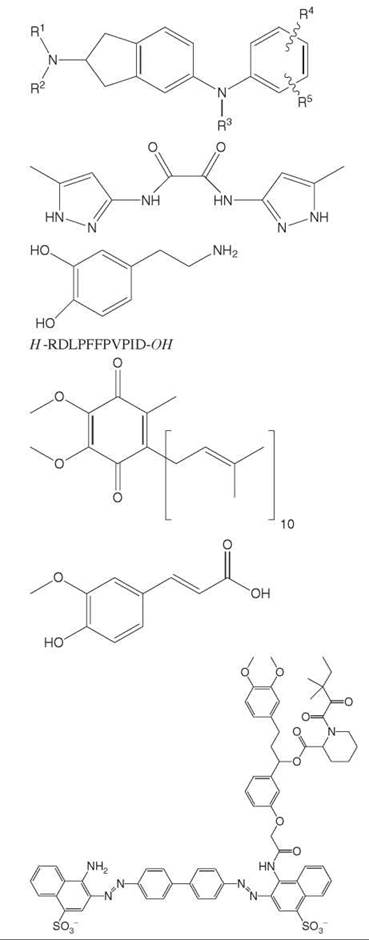
Several small molecule inhibitors that prevent assembly of Aβ have been reported, however, the literature is far from systematic. Most of the studies listed in Table 2 (references for studies found in (53)) test only a few compounds and do not attempt to derive a structure-activity profile. Moreover, the chemical structures do not lend themselves to good starting points for medicinal chemistry in order to improve upon their properties. To date, the known compounds that inhibit Aβ assembly are weakly potent and are poorly bioavailable, particularly to the brain. Furthermore, progress towards development of aggregation inhibitors in the clinical setting remains elusive due to a clear disconnect between the in vitro and in vivo fibril-formation process. These points are further demonstrated by the fact that the only amyloid-aggregation inhibitors that have advanced to late-stage clinical trials, Alzhemed™, Cerebril™ and Kiacta™ do not fit the classical drug-like profile and their in vivo disposition and mechanism of action is not well understood.
Table 2. Inhibitors of Aβassemblya
|
|
Compound |
Mode of action |
Example |
|
|
Aminoindanes |
Fibrils |
|
|
|
Aminopyrazoles |
Fibrils |
|
|
|
Anti-parkinsonian agents |
Fibrils |
|
|
|
Beta-sheet breakers peptides |
Fibrils |
|
|
|
Coenzyme Q10 |
Fibrils |
|
|
|
Ferulic Acid |
Fibrils |
|
|
|
FK506 complexes |
Fibrils |
|
|
Hydroxyindoles |
Fibrils |
|
|
|
Hydroxyphenyl pyridazine diamine |
Fibrils |
||
|
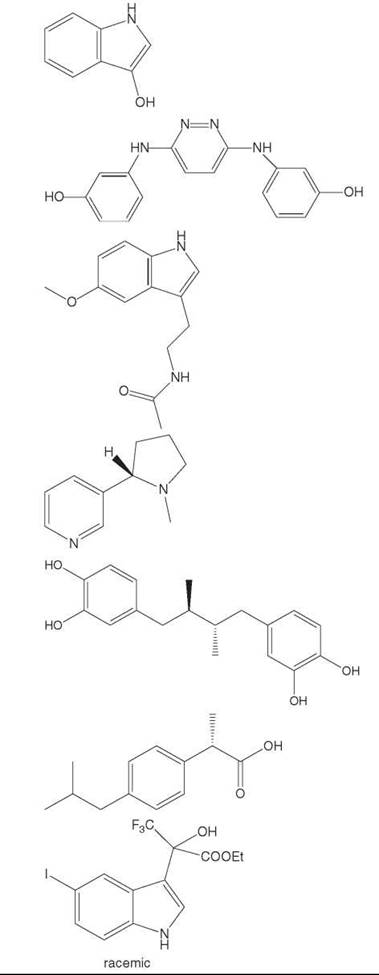
Melatonin |
Fibrils |
||
|
Nicotine |
Fibrils |
||
|
Nordihydroguaiaretic acid |
Fibrils |
||
|
NSAIDs |
Fibrils |
||
|
Organofluorine indol-3-yl derivatives |
Fibrils |
||
|
|
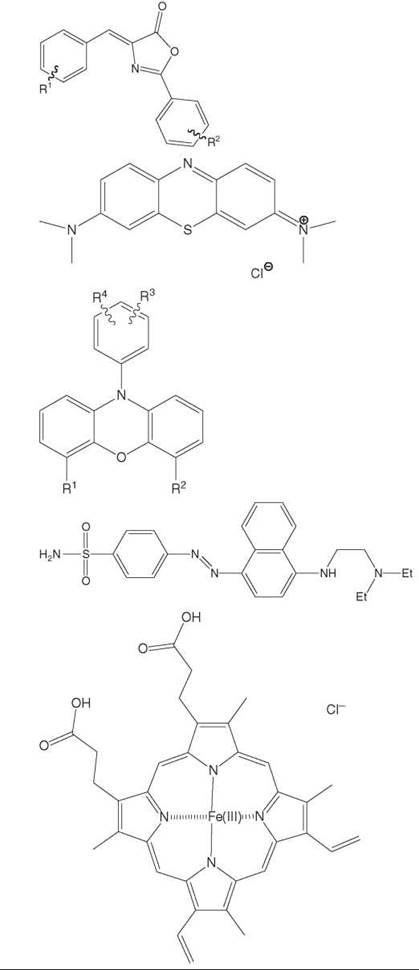
Oxazalones |
Fibrils |
|
|
|
Phenothiazines |
Fibrils |
|
|
|
Phenoxazines |
Fibrils |
|
|
|
Phenylazobenzene sulfonamides |
Fibrils |
|
|
|
Porphyrins |
Fibrils |
|
|
|
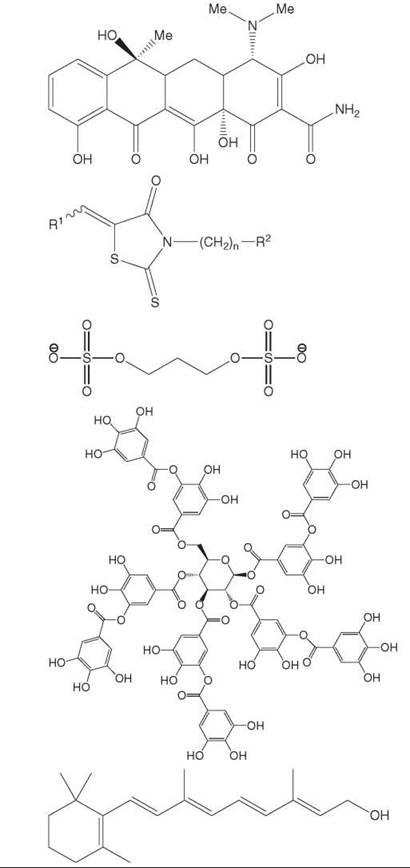
Rifampicin and tetracycline |
Fibrils |
|
|
|
Rhodanines |
Fibrils |
|
|
|
Small molecule anionic sulfonates of sulfates |
Fibrils |
|
|
|
Tannic acid |
Fibrils |
|
|
|
Vitamin A |
Fibrils |
|
|
|
Wine-related polyphenols |
Fibrils |
|
|
|
Curcumin and rosmarinic acid |
Fibrils, Oligomers |
|
|
|
Cyclohexanehexol |
Fibrils, Oligomers |
|
|
|
Hydroxyphenyl triazine diamine |
Oligomers |
|
|
|
Phenols |
Oligomers |
|
|
|
Tricyclic pyrones
|
Oligomers
|
|
aReferences for studies found in 47
Methods Used for Fibril Structure Determination and Aggregation Assays
Research on amyloid fibrils was facilitated by the fact that fibrils similar to the ones found in vivo can be formed in vitro from peptides and denatured proteins. However, structure determination of amyloid fibrils has proven to be difficult due to the lack of methods and the complexity of the aggregates. Standard methods for structure determination, primarily solution-state NMR and X-ray diffraction, fail for amyloid fibrils due to the lack of soluble material or the inability to form classic protein crystals. Only recently we have acquired an in-depth understanding of the detailed conformation of fibrils.
The techniques to follow aggregation of amyloidogenic proteins and to determine their molecular conformation range from spectrometric assays (thioflavin T, Congo red) over spectroscopic assays (FTIR and CD) and visualization techniques (AFM and TEM) to methods that provide detailed insight into the atomic coordinates (solid-state NMR and X-ray diffraction).
The principal difficulty in studying amyloid formation in vitro is doubtlessly sample preparation and protein quality, as impurities, aggregation conditions (buffer, pH, temperature, etc.) and the chosen deseeding method can have a major impact on the fibril formation kinetics as well as the structure of the fibrils.
Spectrophotometric assays
Congo red is a diazo dye with the name-giving characteristic red color. Amyloid stained with Congo red depicts green birefringence under polarized light and this method has been used to diagnose amyloid diseases in histological samples. To date, it is still used to classify proteins aggregates as amyloid. However, due to subjective interpretation and many incidents of “false negatives”, this method is to be used carefully and should be accompanied by a parallel verification method. Congo red can also be utilized in an absorbance assay to quantify the amount of misfolded material (54).
A straightforward method used frequently to follow the misfolding kinetics of previously deseeded protein is the thioflavin T assay (55). Thioflavin T is a molecule that binds rapidly to protein aggregates but not to monomeric or oligomeric intermediates. Upon binding, a red shift of the excitation and emission wavelength is monitored, allowing observing the kinetics of protein misfolding with a fluorescence microscope or by fluorescence spectroscopy. As this assay detects only β-sheet rich conformers, it is not suitable to identify unstructured intermediates of the misfolding process.
Spectroscopic assays
To determine the presence of β-sheet in amyloid samples, spectroscopic techniques such as circular dichroism (CD) and Fourier transform infrared (FTIR) spectroscopy are routinely used. In far U.V. CD spectroscopy, the sample is examined before and after aggregation to record any change in secondary structure. Samples of soluble protein are subjected to left and right handed circular polarized light and the difference in its absorbance at far-UV wavelengths is recorded. Obtained spectra are then compared to a reference library to allow an assessment of secondary structural elements. Aβ is unstructured under physiological conditions and exhibits a classic random coil far U.V. CD spectra. However, as aggregation of AP progresses, and the monomers fold into protofibrils and fibrils, the CD spectra change to reflect β-sheet formation.
The presence of β-sheet in a protein solution can also be determined using FTIR by inspecting the amide I band, which occurs in the region between 1600 cm-1 and 1700 cm-1, and taking into account possible contributions from side chains. Amyloid fibrils typically have β-sheet peaks below 1620 cm-1. FTIR is not suitable to determine atomic coordinates, but it has given detailed insights into protein structure, and its easy sample preparation and the applicability to most molecules have led to the development of many experimental techniques (reviewed in (56)).
Visualization techniques (AFM and TEM)
Interesting information about the organization of fibrils has been obtained from atomic force microscopy (AFM) and transmission electron microscopy (TEM). Both methods give high resolution images of fibrils and show that fibrils are unbranched, twisted, and several μm long. These techniques are usually used to support data obtained from Thioflavin T assays, as this method gives no information about the presence of fibrils (it only confirms presence of β-sheets).
Molecular structure determination (solid-state NMR and x-ray diffraction)
Initial X-ray experiments on amyloid fibrils defined the classic b-sheet diffraction pattern with reflections at 4.75 A and 10 A, indicative of b-sheets parallel to the fibril axis, and the protein strand perpendicular to the fibril’s long axis (29-31).
A more detailed view into the 3D organization of a fibril can be obtained by solid-state NMR. Solid-state NMR has the advantage that it can define the spatial arrangement of both the intrachain and intermolecular configurations. Using fibrillar, lyophilized protein the molecular packing of amyloid can be defined (57, 58). Solid-state NMR spectroscopy revealed a more detailed structure of several Aβ analogs and truncated sequences as well as the conformation of other peptides like α-synuclein, truncated peptides of transthyretin and the prion protein (Helmus J et al Molecular conformation and dynamics of the Y145Stop variant of human prion protein in amyloid fibrils. Proc Natl Acad Sci USA. 2008 Apr 29;105(17):6284-9. in (59)). The data obtained from AP fibrils allowed the development of a structural model of the fibrillar form.
References
1. Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Ann. Rev. Biochem. 2006; 75:333-366.
2. Chiti F, et al. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Nat. Acad. Sci. 1999; 96:3590-3594.
3. Bucciantini M, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002; 416:507.
4. Tiraboschi P, et al. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 2004; 62:1984-1989.
5. Yao ZX, Papadopoulos V. Function of {beta}-amyloid in cholesterol transport: a lead to neurotoxicity. FASEB J. 2002; 16:1677-1679.
6. Zou K, et al. A novel function of monomeric amyloid beta -protein serving as an antioxidant molecule against metal-induced oxidative damage. J. Neurosci. 2002; 22:4833-4841.
7. Huang SS, et al. Amyloid beta-peptide possesses a transforming growth factor-beta activity. J. Biol. Chem. 1998; 273:27640-27644.
8. Annaert W, De Strooper B. Presenilins: molecular switches between proteolysis and signal transduction. Trends Neurosci. 1999; 22:439-443.
9. Soba P, et al. Homo- and heterodimerization of APP family members promotes intercellular adhesion. Embo. J. 2005; 24:3624-3634.
10. Terry RD, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991; 30:572-80.
11. Lambert MP, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 1998; 95:6448-6453.
12. Lesne S, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006; 440:352-357.
13. Walsh DM, Selkoe DJ. Abeta Oligomers - a decade of discovery. J. Neurochem. 2007; 101:1172-1184.
14. Walsh DM, et al. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999; 274:25945-25952.
15. Walsh DM, et al. Naturally secreted oligomers of amyloid (beta) protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416:535.
16. Oddo S, et al. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003; 24:1063.
17. Hardy J, DJ Selkoe. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002; 297:353-356.
18. Opie E. The relation of diabetes mellitus to lesions of the pancreas. Hyaline degeneration of the islands of Langerhans. J. Exp. Med. 1901; 5:527-540.
19. Westermark P, et al. Islet amyloid in type 2 human diabetes mellitus and adult diabetic cats contains a novel putative polypeptide hormone. Am. J. Pathol. 1987; 127:414-417.
20. Cooper GJS, et al. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 Diabetic patients. Proc. Nat. Acad. Sci. 1987; 84:8628-8632.
21. Lorenzo A, et al. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994; 368:756.
22. Demuro A, et al. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005; 280:17294-17300.
23. Arvanitakis, Z., et al., Diabetes Mellitus and Risk of Alzheimer Disease and Decline in Cognitive Function. Arch Neurol, 2004.61(5):p.661-666.
24. Havrankova, J., J. Roth, and M. Brownstein, Insulin receptors are widely distributed in the central nervous system of the rat. Nature, 1978.272(5656):p.827.
25. Northam, E.A., D. Rankins, and F.J. Cameron, Therapy insight: the impact of type 1 diabetes on brain development and function. Nat Clin Pract Neurol, 2006.2(2):p.78-86.
26. Gispen, W.H. and G.-J. Biessels, Cognition and synaptic plasticity in diabetes mellitus. Trends in Neurosciences, 2000.23(11):p.542.
27. Steen, E., et al., Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J Alzheimers Dis, 2005.7(1):p.63-80.
28. Zhao, W.-Q., et al., Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J., 2008.22(1):p.246-260.
29. Yong W, et al. Structure determination of micelle-like intermediates in amyloid beta-protein fibril assembly by using small angle neutron scattering. Proc. Nat. Acad. Sci. 2002; 99:150-154.
30. Huang THJ, et al. Structural studies of soluble oligomers of the alzheimer (beta)-amyloid peptide. J. Molec. Biol. 2000; 297:73.
31. Murphy RM. Kinetics of amyloid formation and membrane interaction with amyloidogenic proteins. Biochim. Biophys. Acta 2007; 1768:1923.
32. Kelly JW. Mechanisms of amyloidogenesis. Nat. Struct. Mol. Biol. 2000; 7:824.
33. Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol. 1999; 309:256-274.
34. Wetzel R. Kinetics and thermodynamics of amyloid fibril assembly. Acc. Chem. Res. 2006; 39:671-679.
35. Pauling L, Corey RB. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. USA. 1951; 37:251-256.
36. Eanes ED, Glenner GG. X-ray diffraction studies on amyloid filaments. J. Histochem. Cytochem. 1968. 16:673-677.
37. Sunde M, et al. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Molec. Biol. 1997; 273:729.
38. Tjernberg LO, et al. A molecular model of Alzheimer amyloid beta-peptide fibril formation. J. Biol. Chem. 1999; 274:12619-12625.
39. Benzinger TLS, et al. Two-dimensional structure of beta-Amyloid (10-35) fibrils. Biochem. 2000; 39:3491-3499.
40. Benzinger TLS, et al. Propagating structure of Alzheimer’s beta-amyloid (10-35) is parallel beta-sheet with residues in exact register. PNAS 1998; 95:13407-13412.
41. Antzutkin ON, et al. Multiple quantum solid-state NMR indicates a parallel, not antiparallel, organization of beta -sheets in Alzheimer’s beta-amyloid fibrils. Proc. Nat. Acad. Sci. 2000; 97:13045-13050.
42. Petkova AT, et al. A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Nat. Acad. Sci. 2002; 99:16742-16747.
43. Jaikaran ETAS, et al. Identification of a novel human islet amyloid polypeptide (beta)-sheet domain and factors influencing fibrillogenesis. J. Molec. Biol. 2001. 308:515.
44. Westermark P, et al. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc. Nat. Acad. Sci. 1990; 87:5036-5040.
45. Nilsson MR, Raleigh DP. Analysis of amylin cleavage products provides new insights into the amyloidogenic region of human amylin. J. Molec. Biol. 1999; 294:1375.
46. Jayasinghe SA, Langen R. Identifying structural features of fibrillar islet amyloid polypeptide using site-directed spin labeling. J. Biol. Chem. 2004; 279:48420-48425.
47. Heise H, et al. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc. Natl, Acad. Sci. USA 2005; 102:15871- 15876.
48. Der-Sarkissian A, et al. Structural organization of {alpha}-synuclein fibrils studied by site-directed spin labeling. J. Biol. Chem. 2003; 278:37530-37535.
49. Chen M, et al. Investigation of alpha-synuclein fibril structure by site-directed spin labeling. J. Biol. Chem. 2007; 282:24970-24979.
50. Schenk D, et al. Immunization with amyloid-(beta) attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999; 400:173.
51. Schenk D. Amyloid-(beta) immunotherapy for Alzheimer’s disease: the end of the beginning. Nat. Rev. Neurosci. 2002; 3:824.
52. Hock C, et al. Antibodies against (beta)-amyloid slow cognitive decline in Alzheimer’s Disease. Neuron 2003; 38:547.
53. Levine H. Small molecule inhibitors of A&b.beta; assembly. Amyloid 2007; 14:185-197.
54. Klunk WE, Jacob RF, Mason RP. Quantifying amyloid beta-Peptide (Abeta) aggregation using the congo red-abeta (CR-Abeta) spectrophotometric assay. Anal. Biochem. 1999; 266:66.
55. Levine-III H. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993; 2:404-410.
56. Hiramatsu H, Kitagawa T. FT-IR approaches on amyloid fibril structure. Biochim. Biophys. Acta 2005; 1753:100.
57. Ritter C, et al. Correlation of structural elements and infectivity of the HET-s prion. Nature 2005; 435:844.
58. Nelson R, et al. Structure of the cross-(beta) spine of amyloid-like fibrils. Nature 2005; 435:773.
59. Naito A, Kawamura I. Solid-state NMR as a method to reveal structure and membrane-interaction of amyloidogenic proteins and peptides. Biochim. Biophys. Acta 2007; 1768:1900.