CHEMICAL BIOLOGY
Pharmaceutical Industry, Biocatalysts and Chemocatalysts
David J. Ager, DSM Pharmaceutical Products, Raleigh, North Carolina
Oliver May, DSM Pharmaceutical Products, Advanced Synthesis, Catalysis and Development, Geleen, The Netherlands
doi: 10.1002/9780470048672.wecb651
Catalytic reactions provide the opportunity to perform more environmentally friendly reactions. As the pharmaceutical industry produces a large amount of waste for a relatively small amount of drug product manufactured, the use of catalytic reactions is becoming more important. Catalysts can be biological or chemical in nature and can be used to effect a wide variety of transformations.
The pharmaceutical industry employs a wide variety of chemical transformations to prepare the active components of drugs. Cost and environmental pressures encourage the use of catalytic reactions for both bond-forming reactions and the creation of stereogenic centers. As the pharmaceutical industry generates a large amount of waste in the preparation of a relatively small amount of drug product, catalytic reactions will only increase in importance in this industrial sector (1). The E-factor, which is the amount of waste produced (Kg) to make a Kg of product, is high for the fine chemical and pharmaceutical industries. The use of catalytic methods, rather than stoichiometric ones, can help reduce waste (2). The development of a “green” process, however, has to be weighed against the speed of developing the process to the target molecule.
The purpose of this article is to provide an overview of the different types of chemical and biological catalysis currently available to the pharmaceutical industry in the process area. In other words, these transformations can be performed at scale. The types of catalysts that have been used are given together with systems that show potential for future application. The chemocatalytic area has addressed the synthesis of aromatic and heterocyclic compounds, which are common classes in pharmaceutically active compounds, whereas biocatalyst applications tend to be aimed toward the production of chiral molecules.
The uses of catalysts for asymmetric pharmaceutical synthesis have been reviewed by others (see the Further Reading section).
Types of Catalysts
Catalysts can be classified in many ways. A summary of the methods discussed in this article is given in Table 1. For enzyme catalysts, the recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (IUBMB) have been followed (3). Enzymes are classified into six general groups, and the first digit of the enzyme commission number corresponds to the following general categories: 1) oxidoreductases, 2) transferases, 3) hydrolases, 4) lyases, 5) isomerases, and 6) ligases. The number of large-scale applications differs significantly among these enzyme types. Most commercial applications use hydrolases or oxidoreductases, which can be attributed to the broad range of enzymes available in these two classes (4).
Table 1. Catalysts useful for pharmaceutical applications
|
Type |
Class |
Catalyst |
Example of transformation |
|
Biological |
Living whole cell |
Many within the cell |
Preparation of secondary metabolite |
|
|
Enzyme |
Oxidoreductases |
Oxidations and reductions |
|
|
|
Transferases |
Methylation, glycosylation and amino group transfers |
|
|
|
Hydrolases |
Ester hydrolysis |
|
|
|
Lyases |
C-C and C-N bond formations |
|
|
|
Isomerases |
Racemization |
|
|
|
Ligases |
Coupling reactions |
|
Chemical |
Transition metal |
Hydrogenations |
Alkene reductions |
|
|
|
Asymmetric hydrogenations |
Generation of new stereogenic center |
|
|
|
Aryl coupling reactions |
Preparation of biaryl compounds |
|
|
|
Coupling reactions |
Aniline preparation |
|
|
|
Isomerizations |
Chiral imines from allyl amines |
|
|
|
Metathesis |
Ring formation |
|
|
Organocatalysis |
Carbon-carbon bond formation |
Aldol reaction |
|
|
|
Oxidations |
Epoxidations |
Biological Catalysts
In a few cases, biocatalysts have the advantage that no chemo-catalytic alternative exists. It usually occurs when the exquisite stereoselection of a biocatalyst is used to distinguish between two equally reactive groups within a molecule based on stereochemistry; the stereoselective oxidations of steroids and aromatic compounds are examples (5). Another instance in which biocatalysts are very powerful and no chemocatalyst equivalent is available is for glycosylation reactions and the stereocon-trolled synthesis of polysaccharides. In many areas, however, biological and chemical catalysts compete; examples of this competition include the reduction of ketones (vide infra) and the desymmetrization of cyclic anhydrides (6, 7). In these cases, the choice of which catalyst system to use will depend on accessibility and on process performance in such areas as selectivity, activity, and consumption, as well as cost. These parameters are highly product specific and often are difficult or impossible to predict. For the development of syntheses of new products, the fast screening of highly diverse libraries, be they biocatalytic or chemocatalytic, is, therefore, an important tool to determine the best choice of a catalytic system (8, 9). The use of molecular biological methodologies do allow for highly selective and efficient biocatalysts to be developed in a relatively short period of time (7). Without precedence, the development of a chemocatalyst is a long-term option.
Enzymes
Enzymes can be used in different formulations, immobilized or soluble, and with different degrees of purity, such as cell preparations and crude or enriched isolates. Isolation to a purified form takes time and effort and is usually avoided unless absolutely necessary. In many cases, molecular biology allows for an enzyme to be highly enriched (overproduced) in an organism, which reduces the need for purification (10). Such recombinant cells are, therefore, often used as cell preparations except if the cell needs to be treated to make the substrate accessible to the biocatalyst. More than 50 different enzyme subclasses are commercially available and can be used to prepare chiral molecules. A summary (11-14) of the most often used classes of enzymes that have been used in chemical synthesis is given in Table 2 (16-45). Reactions do not have to be performed in totally aqueous media as some enzymes can tolerate organic solvents (15).
Table 2. Enzymes used in the preparation of pharmaceuticals
|
Enzyme subclass |
Substrate |
Product |
Reference(s) |
|
Racemases |
α-Hydroxy acids |
α-Hydroxy acids |
16 |
|
|
α-Amino acids |
α-Amino acids |
16, 17 |
|
Oxidases |
Alcohols |
Carbonyl compounds |
18, 19 |
|
Dehydrogenases |
Carbonyl compounds |
Alcohols, hydroxy acids, amino acids |
20, 21 |
|
Lipases |
Esters, amides |
Alcohols, carboxylic acids, alcohols, amines |
22-26 |
|
Aldolases |
Carbonyl compounds |
Hydroxy carbonyl compounds |
27-30 |
|
Hydroxynitrile lyases |
Carbonyl compounds |
Cyanohydrins |
27, 31, 32 |
|
Esterases |
Esters |
Alcohols, carboxylic acids |
33, 34 |
|
Nitrilases |
Nitriles |
Carboxylic acids |
35 |
|
N-Acetylamino acid hydrolase |
N -Acetyl amino acids |
Amino acids |
36, 37 |
|
Amidases |
Amino acids |
Amino acids |
38-40 |
|
Hydantoinases |
5’-Monosubstituted hydantoins |
Amino acids |
41, 42 |
|
Halohydrin dehalogenases |
Halohydrins, epoxides |
Diols, epoxides, P-hydroxynitriles |
43 |
|
Ammonia lyases |
Cinnamic acid derivatives |
Phenylalanine derivatives |
44 |
|
Proteases |
Amino acids |
Peptides |
45 |
Enzymatic processes are now being applied to a wide range of pharmaceutical product syntheses (46). Examples are given for the preparation of cyanohydrins, which can then be used to prepare α-hydroxy acids and α-amino acids.
Cyanohydrins are a very useful class of compounds as they can be transformed into a wide variety of compounds while retaining the stereogenic center (32, 35). Hydroxy nitrilases are available from natural sources (13), which can give access to either enantiomer of the product cyanohydrin (Fig. 1) (47).
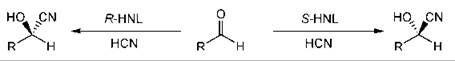
Figure 1. Cyanohydrin formation with hydroxy nitrilases.
An example of an acylase to perform a resolution is provided by the Degussa process to L-methionine (1). The racemic acetylmethionine (2) is prepared by a chemical synthesis. The acylase hydrolyses only the L-isomer (Fig. 2). The D-isomer is racemized by base and put back into the process stream (48).
The most powerful approaches, which can be used with several different enzyme systems, lead to a single enantiomer as the product in high yield and do not rely on a classic resolution approach in which the unwanted enantiomer is discarded. These approaches include dynamic kinetic resolutions, deracemizations, and asymmetric and desymmetrization reactions (49, 50). In some cases, a chemical catalyst may be available to “recycle” the unwanted isomer in the same reactor (vide infra). It is sometimes possible to racemize the unwanted isomer of the substrate and then to perform the reaction again (51).

Figure 2. Synthesis of L-methionine.
Whole cells
When chemical transformations were performed by whole cells, such as the reduction of carbonyl compounds by baker’s yeast, low asymmetric induction could result as two enzymes are present in the organism that provide the antipodes of the product (52). This result has now been circumvented by the use of genetically modified microorganisms so that the desired enzyme is overproduced (53, 54).
The use of a whole cell allows for a required enzyme cofactor to be regenerated. In other cases, it allows for several enzymes to work in parallel and to perform many complex transformations. An example is provided by the synthesis of D-amino acids from hydantoins (Fig. 3). The carbomylase drives the reaction to completion as carbon dioxide and ammonia are evolved. The same approach has been used with the L-versions of the enzymes to synthesize L-amino acids (14, 42, 55).
Several complex antibiotics are prepared by whole-cell fermentations. Examples are the pencillin antibiotics in which the side chain can be removed and replaced with a synthetic one to enhance activity or stability. Other examples include the macrolide antiobiotics, such as avermectin (56) and erythromycin (57), in which the organism uses an enzyme “cassette” to build up the seco-chain before cyclization.
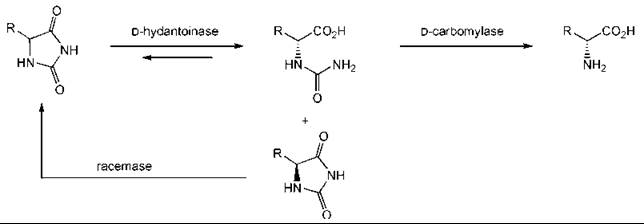
Figure 3. Synthesis of D-amino acids from hydantoins.
In some instances, metabolic engineering of an organism can provide the desired compound. As an example, shikimic acid is used as the starting material in the synthesis of Tamiflu (Roche Laboratories, Inc., Nutley, NJ), which is an antiviral drug. Bacteria produce shikimic acid as an intermediate on the biosynthetic route to chorismic acid, itself an intermediate for several essential products such as phenylalanine, tyrosine, and ubiquinone that the cell needs to function (58). By knocking out or controlling the genes that develop the enzymes that use shikimic acid as the substrate, the organism can be persuaded to overproduce this valuable starting material (Fig. 4).
Chemists have also taken lessons from nature and often use biomimetic syntheses or approaches to complex molecules; here, reactions used in an organism are mimicked in the laboratory (59, 60). In addition, catalytic transformations can be coupled, and it could be two chemocatalysts (vide infra) (61, 62).
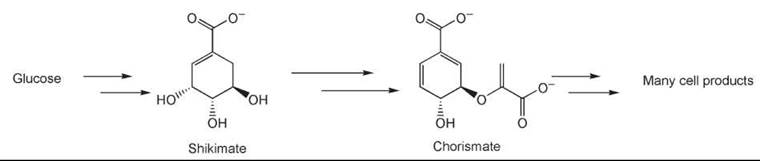
Figure 4. Biosynthetic access to chorismic and shikimic acids.
Transition metal-based catalysts perform a wide variety of reactions. Many useful reactions can be used to build the carbon-carbon framework of the target molecule or to introduce functional groups into complex molecules. Many achiral methods exist; they often are named after the person who discovered or popularized them (see Table 3) (64-170). In some instances, the achiral reaction has been adapted to provide an asymmetric method; the latter examples are included in Table 4 (93, 118, 120, 124, 142, 149-202). The use of metal catalysts that act as Lewis acids or bases have been omitted as numerous examples can be described (63).
Table 3. Transition metal catalyzed reactions
|
Reaction type |
Substrate |
Product |
Reference(s) |
|
Aryl coupling reactionsa |
|
|
64-71 |
|
Heck reaction |
Alkene |
Arylalkene |
71-83 |
|
Suzuki reaction |
Boronic acid or ester |
Biaryl |
71, 73, 84-86 |
|
Buchwald-Hartwig reaction |
Amine |
Aniline |
87-89 |
|
Cyclizations |
Alkene |
Various |
90-92 |
|
Metathesis |
Alkene or alkyne |
Alkene or alkyne |
93-116 |
|
Hydrogenations |
Alkenes |
Alkanes |
117-120 |
|
Hydroformylations |
Alkenes |
Aldehydes |
121-123 |
|
Hydrovinylations |
Alkene |
Alkene |
124 |
|
Hydroaminations |
Alkene |
Amine |
125-127 |
|
Protecting group removal |
Various |
Various |
128 |
|
Pauson-Khand reaction |
Alkenes |
Ketones |
129-133 |
|
Oxidations |
Alcohols |
Carbonyl compounds |
134-136 |
|
|
Alkene |
Epoxide |
137 |
|
|
Alkene |
Diol |
138, 139 |
|
|
Alkene |
Carbonyl compounds |
140 |
aThese include couplings such as the Kumada, Sonogashira, Negishi, and Stille reactions.
Table 4. Transition metal-based catalytic reactions that generate a new stereogenic center
|
Reaction type |
Substrate |
Product |
Reference(s) |
|
Hydrogenation |
Enamides |
α-Amino acid derivatives |
118, 120, 149-153 118, 142,152-154 118, 152, 153 |
|
|
α,β-Unsaturated carboxylic acid derivatives Enamines |
α-Substituted carboxylic acid derivatives Amines |
|
|
|
118, 152, 153 |
||
|
|
Imines |
Amines |
118, 152, 153 |
|
|
Ketones |
Alcohols |
118, 152, 153, 155, 156 |
|
|
Alkenes |
Alkanes |
118,157 |
|
Reductions |
Ketones |
Alcohols |
158 |
|
Hydroamination |
Alkene |
Amine |
159 |
|
Hydrovinylation |
Alkene |
Alkene |
124 |
|
Hydrosilylations |
Carbonyl compounds |
Alcohols |
160 |
|
Alkylations |
Carbonyl compounds |
α-Substituted carbonyl compounds Alcohols |
161 |
|
|
Carbonyl compounds |
162-164 |
|
|
Strecker reaction |
Carbonyl compounds |
α-Amino nitriles |
165 |
|
Cyanohydrin formation |
Carbonyl compounds |
α-Hydroxy nitriles |
166-168 |
|
Allylic alkylations |
Allyl esters or similar |
Alkene |
169-172 |
|
Aldol and related reactions |
Carbonyl compounds |
β-Hydroxy carbonyl compounds |
173 |
|
Conjugate additions |
α,β-Unsaturated compounds |
β-Substituted compounds |
174-176 |
|
Halogenations |
Carbonyl compounds |
α-Halocarbonyl compounds |
177 |
|
Isomerizations |
Alkene |
Alkene |
178, 153 |
|
Hydrolysis |
Epoxides |
Diols |
179,180 |
|
Oxidations |
Alkenes |
Epoxides |
181 182-188 |
|
|
Alkenes |
Aziridines |
189 |
|
|
Allyl alcohols |
Epoxy alcohols |
190-192 |
|
|
Sulfides |
Sulfoxides |
193 |
|
|
Alkenes |
Diols |
192, 194, 195 |
|
|
Alkenes |
Amino alcohols |
196-198 |
|
C-H activation |
Various |
Various |
199, 200 |
|
Heck reaction |
Alkene |
Arylalkene |
71, 201, 202 |
|
Metathesis |
Alkenes |
Alkenes |
93 |
When implementing a transition metal-catalyzed step at scale, many factors have to be considered, some of which also relate to biological and organocatalytic reactions. The one factor that does not overlap with these other types of systems is the price of the metal. Although cheaper metals such as iron, nickel, and copper can be used for some transformations, often the metal required is precious, such as palladium, platinum, rhodium, iridium, or ruthenium. The use of gold catalysis has recently become an area of intense research (141). These precious metals are expensive; usage needs to be minimal, and they must be recycled either for reuse in the reaction or through recovery. Refining has to be a topic of considerable economic concern. For some reactions, especially asymmetric transformations, the ligands needed to perform the reaction may be more expensive than the metal! Here, the catalyst has to be extremely efficient to achieve the required cost benefits. The economics of the transformation not only depend on the cost of the catalyst and how much is used (usually defined by turnover number, which is the number of times the catalyst goes round the catalytic cycle), but also the duration of the reaction. The turnover frequency is the number of times the catalyst completes a catalytic cycle per hour. Reactor time can be expensive, and time needs to be minimized but not at the cost of making the reaction so fast that it becomes unsafe or reagents, such as hydrogen, cannot be delivered at an appropriate rate.
An example of a metal-catalyzed reaction to form a biaryl product is the Suzuki reaction. The coupling can be performed without any phosphorus ligands for the metal and with only a small amount of the metal (0.05 mol %) (Fig. 5) (9). A reaction that has become popular is the preparation of aromatic amines by a palladium-catalyzed coupling reaction (Fig. 6). This methodology is general (89).
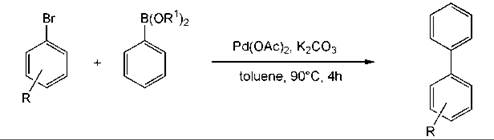
Figure 5. Biaryl compounds by Suzuki coupling.
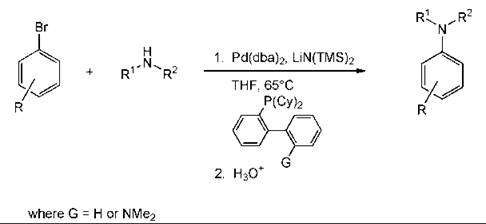
Figure 6. Anilines by palladium catalysed coupling.
In addition to carbon-carbon bond formation, transition metal catalysts can also generate a stereogenic center. The first reaction of this type in which useful amounts of asymmetric induction were observed was an asymmetric hydrogenation to make phenylalanine and the method has been used for many years to synthesize the anti-Parkinsons drug, L-Dopa (3) (Fig. 7) (142, 143).

Figure 7. Asymmetric hydrogenation route to L-Dopa.
This transformation was important as it showed that a chemical catalyst could perform with similar asymmetric integrity to that of a biological system. Today, literally thousands of ligands and catalysts can be used to perform asymmetric hydrogenations as well as other reactions; see Table 4.
Many aspects must be considered in finding a catalyst to perform a step in the synthesis of a drug. The main aspect is the time required to find suitable catalyst systems. If a closely analogous reaction has been reported in the literature, then it may not be a large problem or concern. In most instances, however, this is not the case. In addition to enantioselectivity or diastereoselectivity, the factors necessary to find an efficient achiral catalyst must also be fulfilled.
Stereogenic centers can also be prepared by carbon-carbon bond-forming reactions or reductions of functional groups other than alkenes. Some reactions are also summarized in Table 4 (144); for a comprehensive work on asymmetric catalysts, see Reference 145. In some cases, two stereogenic centers can be created. This result can be achieved either in a single step as with the asymmetric reduction of a tetrasubstituted alkene, or by coupling two reactions together as with a conjugate addition followed by trapping the resultant enolate with an electrophile (146, 147). An illustration of this strategy is the synthesis of the bicyclic ketone 4 (Fig. 8) (147, 148). The allyl group is a good electrophile and is then converted to the analogous ketone by a Wacker oxidation.

Figure 8. Bicyclic enone synthesis by conjugate addition and aldol reaction.
An example of an asymmetric hydrogenation used in the preparation of a pharmaceutical intermediate is provided by a synthesis to carbapenems (5) (178). Reduction of the β-keto ester occurs under equilibrating conditions so that the erythro-product is formed in high yield and selectivity (203). Another catalytic step with ruthenium is used to introduce the acetoxy group (Fig. 9) (153).
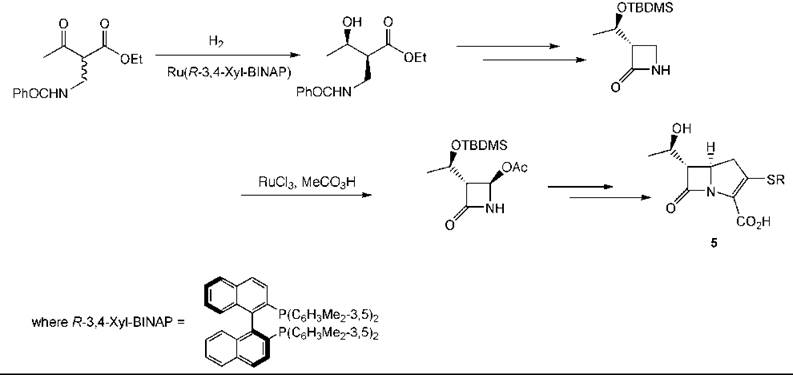
Figure 9. Carbapenem synthesis by an asymmetric hydrogenation.
An asymmetric oxidation is used in the synthesis of esomeprazole (6), a proton pump inhibitor, which has therapeutic advantages over the racemic mixture omeprazole (Fig. 10) (204).

Figure 10. Esomeprazole synthesis by an asymmetric oxidation.
Chemocatalysts sometimes have an advantage over biological systems. Often the antipode of a ligand is accessible, although if a natural product is used as the source of the stereogenicity, then it may be less abundant and more expensive. As a last resort, and as ligands are relatively small molecules, an achiral synthesis and resolution might be used. This latter option is not available with a biological catalyst.
One of the main concerns of using a transition or heavy metal catalyst, especially toward the end of the synthetic sequence, is the removal of the metal. A wide variety of methods is known to accomplish this task. Metal-specific sequestering agents are now available. An alternative is to immobilize the catalyst, but it may not be a cost-effective solution for small volumes (205, 206). Of course, a heterogeneous catalyst can be used in the first place (207, 208).
Organocatalysts
This class of catalysts covers chemocatalysts that do not contain a transition metal. The class has been known for many years, but it is relatively recently that the term “organocatalyst” has been used (209). A wide variety of transformations can be performed, which is currently an area of intense research (209-218). Table 5 (220-252) summarizes some key transformations in which organocatalysis can be useful. Reactions range from the asymmetric epoxidation of alkenes, which need not be conjugated to another functional group, to aldol reactions and other carbon-carbon forming transformations. Some progress has also been made to couple two reactions together (219).
L-Proline catalyzes the aldol reaction. This approach has been applied to the synthesis of carbohydrate derivatives as illustrated by the glucose derivative 7 (Fig. 11) (237). The three-component Mannich reaction can be used to prepare β-amino and β-amino α-hydroxy carbonyl compounds in a single step (Fig. 12) (233). As with other types of catalysts, organocatalysts can be immobilized to aid recovery (253).
Table 5. Examples of transformations catalyzed by organocatalysts
|
Reaction type |
Substrate |
Catalyst type |
Reference(s) |
|
Epoxidation |
Alkene |
Carbohydrate derivatives |
220, 221 |
|
|
Carbonyl compounds |
Ylides |
222, 223 |
|
Alkylations |
Carbonyl compounds |
Alkaloids, amines |
224, 225 |
|
|
Amino acid derivatives |
Alkaloids |
225-230 |
|
Aldol reaction |
Carbonyl compounds |
Amino acid derivatives |
231-240 |
|
Mannich reaction |
Carbonyl compounds |
Amino acid derivatives |
233, 240, 241 |
|
Conjugate additions |
Unsaturated carbonyl compounds |
Various |
242 |
|
Baylis-Hillman reaction |
α,β-Unsaturated carbonyl compounds |
Nucleophilic |
243-245 |
|
Acylations |
Alcohol |
Ester |
246-249 |
|
Hydroxylations |
Carbonyl compounds |
Amino acid derivatives |
250 |
|
Reductions |
Carbonyl compounds |
Dihydropyridines |
251 |
|
Stetter and benzoin reactions |
Aldehydes |
Carbenes |
249, 252 |
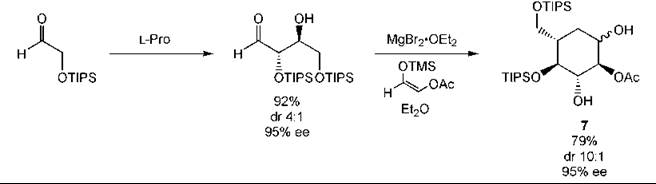
Figure 11. Carbohydrate synthesis by an organocatalytic aldol reaction.
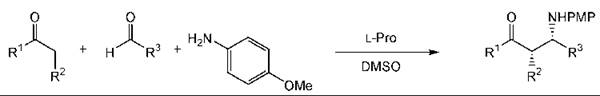
Figure 12. A three-component Mannich reaction.
Systems with Biocatalysis and Chemocatalysis
As enzymes usually only accept one enantiomer or isomer as substrate, many enzymatic reactions are resolutions; the unaffected isomer is waste. One way to circumvent this problem, which can have significant economical consequences, is to include a racemization or isomerization step with a second catalyst so that the substrate for the desired transformation can be accepted as the correct isomer (254, 255). This method allows dynamic kinetic resolutions to be performed with the desired product isomer being produced in high yield rather than with the 50% maximum available from a classic resolution approach (256).
A chemical catalyst can be used to racemize an alcohol, whereas an enzyme is used to prepare an ester of one of the enantiomers of that alcohol. In this example, reduced pressure was used to remove the isopropanol by-product and drive the reaction to completion whereas the Shvo catalyst was used to racemize the alcohol (Fig. 13) (257).

Figure 13. Dynamic kinetic resolution method to the ester of a chiral alcohol.
Both biological and chemical-based catalysts are useful for a wide variety of reactions that range from carbon-carbon bond formation to the generation of a new stereogenic center. With the increasing awareness of green chemistry and the need to reduce waste in the pharmaceutical industry where this problem has been particularly bad, the use of catalytic reactions will surely continue to increase.
Biocatalysts are being applied widely in the industry, including the preparation of carbon-carbon bonds. Stereoselective oxidation with biocatalysts is an area where chemistry will find it hard to compete. A need still exists for new catalysts to replace stoichiometric reagents, as in the reduction of an amide to an amine, amide formation, and substitution of an alcohol (Mitsunobu reaction) (258). In both arenas of catalysis, the overall goal for green chemistry and stereoselectivity must be carbon-hydrogen bond activation.
References
1. Sheldon RA. The E factor: fifteen years on. Green Chem. 2007; 9:1273-1283.
2. Poliakoff M, Fitzpatrick JM, Farren TR, Anastas PT. Green chemistry: science and politics of change. Science 2002; 297:807-810.
3. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology. Enzyme Nomenclature. 1992. Academic Press: New York.
4. Straathof AJJ, Panke S, Schmid A. The production of fine chemicals by biotransformations. Curr. Opin. Biotechnol. 2002; 13:548-555.
5. Hudlicky T, Gonzalez D, Gibson DT. Enzymatic dihydroxylation of aromatics in enantioselctive synthesis: expanding asymmetric methodology. Aldrichim. Acta 1999; 32:35-62.
6. Chen Y, McDaid P, Deng L. Asymmetric alcoholysis of cyclic anhydrides. Chem. Rev. 2003; 103:2965-2983.
7. Pollard DJ, Woodley JM. Biocatalysis for pharmaceutical intermediates: the future is now. Trends Biotechnol. 2007; 25:66-73.
8. Reetz M. New methods for the high-throughput screening of enantioselective catalysts and biocatalysts. Angew. Chem. Int. Ed. 2002; 41:1335-1338.
9. de Vries JG, de Vries AHM. The power of high-throughput experimentation in homogeneous catalysis research and fine chemicals. Eur. J. Org. Chem. 2003; 799.
10. Reetz M. Combinatorial and evolution-based methods in the creation of enantioselective catalysts. Angew. Chem. Int. Ed. 2001; 40:284-310.
11. Whitesides GM, Wong C-H. Enzymes as catalysts in organic synthesis. Aldrichim. Acta 1983; 16:27-33.
12. Wong CH, Whitesides GM. Enzymes in Organic Synthesis. 1994. Pergamon Press, Oxford, UK.
13. Drauz K, Waldman H, eds. Enzyme Catalysis in Organic Synthesis: A Comprehensive Handbook. 2002. VCH, New York.
14. Pantaleone DP. Biotransformations: “green” processes for the synthesis of chiral fine chemicals. In: Handbook of Chiral Chemicals, 2nd edition. Ager DJ, ed. 2006. CRC Press, Boca Raton, FL. pp. 359-403.
15. Klibanov AM. Asymmetric transformations catalyzed by enzymes in organic synthesis. Acc. Chem. Res. 1990; 23:114-120.
16. Schnell B, Faber K, Kroutil W. Enzymatic racemisation and its application to synthetic biotransformations. Adv. Synth. Catal. 2003; 345:653-666.
17. Alfonso I, Gotor V. Biocatalytic and biomimetic aminolysis reactions: useful tools for selective transformations on polyfunctional substrates. Chem. Soc. Rev. 2004; 33:201-209.
18. Dembitsky VM. Oxidation, epoxidation and sulfoxidation reactions catalysed by haloperoxidases. Tetrahedron 2003; 59:4701-4720.
19. Kroutil W, Mang H, Edegger K, Faber K. Biocatalytic oxidation of primary and secondary alcohols. Adv. Synth. Catal. 2004; 346:125-142.
20. Nakamura K, Yamanaka R, Matsuda T, Harada T. Recent developments in asymmetric reduction of ketones with biocatalysts. Tetrahedron: Asymmetry 2003; 14:2659-2681.
21. Krix G, Bommarius AS, Drauz K, Kottenhahn M, Schwarm M, Kula M-R. Enzymatic reduction of α-keto acids leading to L-amino acids, D- or L-hydroxy acids. J. Biotechnol. 1997; 53:29-39.
22. Ema T. Rational strategies for highly enantioselective lipase-catalyzed kinetic resolutions of very bulky chiral compounds: substrate design and high-temperature biocatalysis. Tetrahedron: Asymmetry 2004; 15:2765-2770.
23. Ghanem A, Aboul-Enein HY. Lipase-mediated chiral resolution of racemates in organic solvents. Tetrahedron: Asymmetry 2004; 15:3331-3351.
24. Ghanem A, Aboul-Enein HY. Application of lipases in kinetic resolution of racemates. Chirality 2005; 17:1-15.
25. Ghanem A. Trends in lipase-catalyzed asymmetric access to enantiomerically pure/enriched compounds. Tetrahedron 2007; 63:1721-1754.
26. Thiel F. Enhancement of selectivity and reactivity of lipases by additives. Tetrahedron 2000; 56:2905-2919.
27. Sukumaran J, Hanefeld U. Enantioselective C-C bond synthesis catalysed by enzymes. Chem. Soc. Rev. 2005; 34:530-542.
28. Whalem LJ, Wong C-H. Enzymes in organic synthesis: aldolase-mediated synthesis of iminocyclitols and novel heterocycles. Aldrichimica Acta 2006; 39:63-71.
29. Dean SM, Greenberg WA, Wong C-H. Recent advances in aldolase-catalyzed asymmetric synthesis. Adv. Synth. Catal. 2007; 349:1308-1320.
30. Machajewski TD, Wong CH. The catalytic asymmetric aldol reaction. Angew. Chem. Int. Ed. 2000; 39:1352-1374.
31. North M. Synthesis and applications of non-racemic cyanohydrins. Tetrahedron: Asymmetry 2003; 14:147-176.
32. Gregory RJH. Cyanohydrins in nature and the laboratory: biology, preparations, and synthetic applications. Chem. Rev. 1999; 99:3649-3682.
33. Hermann H, Kietzmann MU, Ivancic M, Zenzmaier C, Luiten RGM, Skranc W, Wubbolts M, Winkler M, Birner-Gruenberger R, Pichler H, Schwab H. Alternative pig liver esterase (APLE) - cloning, identification and functional expression in Pichia pastoris of a versatile new biocatalyst. J. Biotechnol. 2008; 133:301-310.
34. Zhu L-M, Tedford MC. Applications of pig liver esterases (PLE) in asymmetric synthesis. Tetrahedron 1990; 46:6587-6611.
35. Groger H. Enzymatic routes to enantiomerically pure aromatic α-hydroxy carboxylic acids: a further example for the diversity of biocatalysts. Adv. Synth. Catal. 2001; 343:547-558.
36. McCague R, Taylor SJC. Integration of an acylase biotransformation with process chemistry: a one-pot synthesis of N-t-Boc-L-3-(4-thiazolyl)alanine and related amino acids. In: Chirality in Industry II. Collins AN, Sheldrake GN, Crosby J, eds. 1997. John Wiley & Sons, New York. pp. 184-206.
37. Bommarius AS, Drauz K, Klenk H, Wandrey C. Operational stability of enzymes: acylase-catalyzed resolution of N-acetyl amino acids to enantiomerically pure L-amino acids. Ann. N.Y. Acad. Sci. 1992; 672:126-136.
38. Schoemaker HE, Boesten WHJ, Kaptein B, Hermes HFM, Sonke T, Broxterman QB, van den Tweel WJJ, Kamphuis J. Chemo-enzymic synthesis of amino acids and derivatives. Pure Appl. Chem. 1992; 64:1171-1175.
39. Crosby J. Synthesis of optically active compounds: a large scale perspective. Tetrahedron 1991; 47:4789-4846.
40. Bruggink A, Roos EC, de Vroom E. Penicillin acylase in the industrial production of β-lactam antibiotics. Org. Proc. Res. Develop. 1998; 2:128-133.
41. May O, Verseck A, Bommarius AS, Drauz K. Development and dynamic kinetic resolution processes for biocatalytic production of natural and nonnatural L-amino acids. Org. Proc. Res. Develop. 2002; 6:452-457.
42. Syldatk, C.; Pietzsch, M. Hydrolysis and formation of hydantoins. In: Enzyme Catalysis in Organic Synthesis; A Comprehensive Handbook. Drauz K, Waldman H, eds. 1995. VCH, New York.
43. Suzuki T, Kasai N, Yamamoto R, Minamiura N. A novel generation of optically active 1,2-diols from the racemates by using halohydrin dehalogenase. Tetrahedron: Asymmetry 1994; 5:239-246.
44. Poppe L, Retey J. Friedel-crafts-type mechanism for the enzymatic elimination of ammonia from histidine and phenylalanine. Angew. Chem. Int. Ed. 2005; 44:3668-3688.
45. Bordusa F. Proteases in organic synthesis. Chem. Rev. 2002; 102:4817-4867.
46. Patel RN. Enzymatic synthesis of chiral intermediates for drug development. Adv. Synth. Catal. 2001; 343:527-546.
47. Poechlauer P, Skranc W, Wubbolts M. The large-scale biocatalytic synthesis of enantiopure cyanohydrins. In: Asymmetric Catalysis on Industrial Scale. Blaser H-U, Schmidt E, eds. 2004. Wiley-VCH: Weinheim, Germany. pp. 151-164.
48. Groger H, Drauz K. Methods for the enantioselective biocatalytic production of L-amino acids on an industrial scale. In: Asymmetric Catalysis on Industrial Scale. Blaser H-U, Schmidt E, eds. 2004. Wiley-VCH: Weinheim, Germany. pp. 131-147.
49. Schoffers E, Golebiowski A, Johnson CR. Enantioselective synthesis through enzymatic asymmetrization. Tetrahedron 1996; 52:376-3826.
50. Garcia-Urdiales E, Alfonso I, Gotor V. Enantioselective enzymatic desymmetrizations in organic synthesis. Chem. Rev. 2005; 105:313-354.
51. Ebbers EJ, Ariaans GJA, Houbiers JPM, Bruggink A, Zwanen- burg B. Controlled racemization of optically active organic compounds: prospects for asymemtric transformations. Tetrahedron 1997; 53:9417-9476.
52. Csuk R, Glaenzer BI. Baker’s yeast mediated transformations in organic chemistry. Chem. Rev. 1991; 91:49-97.
53. Stewart JD. A chemist’s perspective on the use of genetically engineered microbes as reagents for organic synthesis. Biotechnol. Genetic Engineer. Rev. 1997; 14:67-143.
54. Stewart JD. A genomic approach to investigating baker’s yeast reductions. In: Biocatalysts in the Pharmaceutical and Biotechnology Industries. Patel RN, ed. 2007. CRC Press, Boca Raton, FL. pp. 333-350.
55. Ager DJ. Amino acids. In: Handbook of Chiral Chemicals: 2nd ed. 2006. Ager DJ, ed. CRC Press, Boca Raton, FL. pp. 11-30.
56. Ikeda H, Omura S. Avermectin biosynthesis. Chem. Rev. 1997; 97:2591-2609.
57. Staunton J, Wilkinson B. Biosynthesis of erythromycin and rapamycin. Chem. Rev. 1997; 97:2611-2629.
58. Walsh CT, Liu J, Rusnak F, Sakaitani M. Molecular studies on enzymes in chorismate metabolism and the enterobactin biosynthesis pathway. Chem. Rev. 1990; 90:1105-1129.
59. de la Torre MC, Sierra MA. Comments on recent achievements in biomimetic organic synthesis. Angew. Chem., Int. Ed. 2004; 43:160-181.
60. Yoder RA, Johnson JN. A case study in biomimetic total synthesis: polyolefin carbocyclizations to terpenes and steroids. Chem. Rev. 2005; 105:4730-4756.
61. Lee JM, Na Y, Han H, Chang S. Cooperative multi-catalyst systems for one-pot organic transformations. Chem. Soc. Rev. 2004; 33:302-312.
62. Pellissier H. Asymmetric domino reactions. Part B: Reactions based on the use of chiral catalysts and biocatalysts. Tetrahedron 2006; 62:2143-2173.
63. Corma, A.; Garcia, H. Lewis acids: from conventional homogeneous to green homogeneous and heterogenous catalysis. Chem. Rev. 2003; 103:4307-4365.
64. Christmann U, Vilar R. Monoligated palladium species as catalysts in cross-coupling reactions. Angew. Chem., Int. Ed. 2005; 44:366-374.
65. Frisch AC, Beller M. Catalysts for cross-couplng reactions with non-activated alkyl halides. Angew. Chem., Int. Ed. 2005; 44:674-688.
66. Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Palladium-catalyzed cross-coupling reactions in total synthesis. Angew. Chem., Int. Ed. 2005; 44:4442-4489.
67. de Meijere, A.; F., D.; Eds. Metal-Catalyzed Cross-Coupling Reactions, 2nd ed. 2004. Wiley-VCH, Weinheim, Germany.
68. Corbet J-P, Mignani G. Selected patented cross-coupling technologies. Chem. Rev. 2006; 106:2651-2710.
69. Stanforth S. P. Catalytic Cross-coupling reactions in biaryl synthesis. Tetrahedron 1998; 54:263-303.
70. Littke AF, Fu GC. Palladium-catalyzed coupling reactions of aryl chlorides. Angew. Chem., Int. Ed. 2002; 41:4176-4211.
71. Tietze LF, Ila H, Bell HP. Enantioselective palladium-catalyzed transformations. Chem. Rev. 2004; 104:3453-3516.
72. Heck RF. Transition metal-catalyzed reactions of organic halides with carbon monoxide, olefins, and acetylenes. Adv. Catal. 1977; 26:323-349.
73. Zapf A. Novel substrates for palladium-catalyzed coupling reactions of arenes. Angew. Chem., Int. Ed. 2003; 42:5394-5399.
74. Farina V. High-turnover palladium catalysts in cross-coupling and Heck chemistry: a critical overview. Adv. Synth. Catal. 2004; 346:1553-1582.
75. Heck RF. Cobalt and palladium reagents in organic synthesis: the beginning. Synlett 2006; 2855-2860.
76. Heck RF. Palladium-catalyzed reactions of organic halides with olefins. Acc. Chem. Res. 1979; 12:146-151.
77. Heck RF. Palladium-catalyzed vinylation of organic halides. Org. Reactions 1982; 27:345-390.
78. Whitcombe NJ, Hii KK, Gibson SE. Advances in the Heck chemistry of aryl bromides and chlorides. Tetrahedron 2001; 57:7449-7476.
79. Chinchilla R, Najera C. The Shonogashira reaction: a booming methodology in synthetic organic chemistry. Chem. Rev. 2007;107:874-922.
80. Netherton MR, Fu GC. Nickel-catalyzed cross-couplings of unactivated alkyl halides and pseudohalides with organometallic compounds. Adv. Synth. Catal. 2004; 346:1525-1532.
81. Negishi E-I, Hu Q, Huang Z, Qian M, Wang G. Palladium-catalyzed alkenylation by the Negishi coupling. Aldrichimica Acta 2005; 38:71-88.
82. Negishi E-I. Palladium- or Nickel-Catalyzed Cross Coupling. A New Selective Method for Carbon-Carbon Bond Formation. Acc. Chem. Res. 1982; 15:340-348.
83. Denmark SE, Ober MH. Organosilicon reagents: synthesis and application to palladium-catalyzed cross-coupling reactions. Aldrichimica Acta 2003; 36:75-85.
84. Miura M. Rational ligand design in constructing efficient catalyst systems for Suzuki-Miyaura coupling. Angew. Chem., Int. Ed. 2004; 43:2201-2203.
85. Bellina F, Carpita A, Rossi R. Palladium catalysts for the Suzuki cross-coupling reaction: an overview of recent advances. Synthesis 2004; 2419-2440.
86. Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995; 95:2457-2483.
87. Nakamura I, Yamamoto Y. Transition-metal-catalyzed reactions in heterocyclic synthesis. Chem. Rev. 2004; 104:2127-2198.
88. Schlummer B, Scholz U. Palladium-catalyzed C-N and C-O coupling—A practical guide from an industrial vantage point. Adv. Synth. Catal. 2004; 346:1599-1626.
89. Mauger CC, Mignani G. Synthetic applications of Buchwald’s phosphines in palladium-catalyzed aromatic-bond-forming reactions. Aldrichimica Acta 2006; 39:17-24.
90. Zeni G, Larock RC. Synthesis of heterocycles via palladium n-olefin and n-alkyne chemistry. Chem. Rev. 2004; 104:2285-2309.
91. Montgomery J. Nickel-catalyzed reductive cyclizations and couplings. Angew. Chem. Int. Ed. 2004; 43:3890-3908.
92. Hyland C. Cyclisations of allylic substrates via palladium catalysis. Tetrahedron 2005; 61:3457-3471.
93. Connon SJ, Blechert S. Recent developments in olefin crossmetathesis. Angew. Chem., Int. Ed. 2003; 42:1900-1923.
94. Schrock RR, Hoveyda AH. Molybdenum and tungsten imido alkylidene complexes as efficient olefin-metathesis catalysts. Angew. Chem. Int. Ed. 2003; 42:4592-4633.
95. Schmidt B. Ruthenium-catalyzed cyclizations: more than just olefin metathesis! Angew. Chem. Int. Ed. 2003; 42:4996-4999.
96. Vernall AJ, Abell AD. Cross metathesis of nitrogen-containing systems. Aldrichim. Acta 2003; 36:93-105.
97. Diver ST, Giessert AJ. Enyne metathesis (enyne bond reorganization). Chem. Rev. 2004; 104:1317-1382.
98. Dieters A, Martin SF. Synthesis of oxygen- and nitrogen- containing heterocycles by ring-closing metathesis. Chem. Rev. 2004; 104:2199-2238.
99. Buchmeiser MR. Recent advances in the synthesis of supported metathesis catalysts. New J. Chem. 2004; 28:549-557.
100. Grubbs RH. Olefin metathesis. Tetrahedron 2004; 60:7117-7140.
101. Astruc D. The metathesis reactions: from a historical perspective to recent developments. New J. Chem. 2005; 29:42-56.
102. Wallace DJ. Relay ring-closing metathesis—a strategy for achieving reactivity and selectivity in metathesis chemistry. Angew. Chem. Int. Ed. 2005; 44:1912-1915.
103. Katz TJ. Olefin metathesis and related reactions initiated by carbene derivatves of metals in low oxidation states. Angew. Chem. Int. Ed. 2005; 44:3010-3019.
104. Nicolaou KC, Bulger PG, Sarlah D. Metathesis reactions in total synthesis. Angew. Chem. Int. Ed. 2005; 44:4490-4527.
105. Donohoe TJ, Orr AJ, Bingham M. Ring-closing metathesis as a basis for the construction of aromatic rings. Angew. Chem. Int. Ed. 2006; 45:2664-2670.
106. Zhang W, Moore JS. Alkyne metathesis: catalysts and synthetic applications. Adv. Synth. Catal. 2007; 349:93-120.
107. Chauvin Y. Olefin metathesis: the early days (Nobel Lecture 2005). Adv. Synth. Catal. 2007; 349:27-33.
108. Grubbs RH. Olefin-metathesis catalysts for the preparation of molecules and materials (Nobel Lecture 2005). Adv. Synth. Catal. 2007; 349:34-40.
109. Schrock, R. R. Multiple Metal-Carbon Bonds for Catalytic Metathesis Reactions (Nobel Lecture 2005). Adv. Synth. Catal. 2007; 349:41-53.
110. Schrock RR, Czekelius C. Recent advances in the syntheses and applications of molybdenum and tungsten alkylidene and alkylidyne catalysts for the metathesis of alkenes and alkynes. Adv. Synth. Catal. 2007; 349:55-77.
111. Grubbs RH, Miller SJ, Fu GC. Ring-closing metathesis and related processes in organic synthesis. Acc. Chem. Res. 1995; 28:446-452.
112. Grubbs RH, Chang S. Recent advances in olefin metathesis and its application in organic synthesis. Tetrahedron 1998; 54:4413-4450.
113. Phillips AJ, Abell AD. Ring-closing metathesis of nitrogen-containing compounds: applications of heterocycles, alkaloids, and peptidomometics. Aldrichim. Acta 1999; 32:75-89.
114. Furstner A. Olefin metathesis and beyond. Angew. Chem., Int. Ed. 2000; 39:3012-3043.
115. Trnka TM, Grubbs RH. The development of L2X2Ru = CHR olefin metathesis catalysts: an organometallic success story. Acc. Chem. Res. 2001; 34:18-29.
116. Schrodi Y, Pederson RL. Evolution and applications of second- generation ruthenium olefin metathesis catalysts. Aldrichim. Acta 2007; 40:45-52.
117. Birch AJ, Williamson DH. Homogeneous hydrogenation catalysts in organic synthesis. Org. Reactions 1976; 24:1-186.
118. de Vries JG, Elsevier CJ, eds. The Handbook of Homogeneous Hydrogenation. 2007. Wiley VCH Verlag, Weinheim, Germany.
119. Muetterties EL, Bleeke JR. Catalytic hydrogenation of aromatic hydrocarbons. Acc. Chem. Res. 1979; 12:324-331.
120. Blaser H-U, Malan C, Pugin B, Spindler F, Steiner H, Studer M. Selective hydrogenation for fine chemicals: recent trends and new developments. Adv. Synth. Catal. 2003; 345:103-151.
121. Agbossou F, Carpentier J-F, Mortreux A. Asymmetric hydroformylation. Chem. Rev. 1995; 95:2485-2506.
122. Breit B, Seiche W. Recent advances on chemo-, regio-, and stereoselective hydroformylation. Synthesis 2001; 1-36.
123. Breit B. Synthetic aspects of stereoselective hydroformylation. Acc. Chem. Res. 2003; 26:264-275.
124. RajanBabu TV. Asymmetric hydrovinylation reaction. Chem. Rev. 2003; 103:2845-2860.
125. Widenhoefer RA, Han X. Gold-catalyzed hydroamination of C-C multiple bonds. Eur. J. Org. Chem. 2006; 4555-4563.
126. Muller TE, Beller M. Metal-initiated amination of alkenes and alkynes. Chem. Rev. 1998; 98:675-703.
127. Seayad J, Tillack A, Hartung CG, Beller M. Base-catalyzed hydroamination of olefins: an environmentally friendly route to amines. Adv. Synth. Catal. 2002; 344:795-813.
128. Kocienski P. Protecting Groups. 2003. Thieme Verlag, Stuttgart, Germany.
129. Bonaga LVR, Krafft ME. When the Pauson-Khand and Pauson-Khand type reactions go awry: a plethora of unexpected results. Tetrahedron 2004; 60:9795-9833.
130. Gibson SE, Mainolfi N. The intermolecular Pauson-Khand reaction. Angew. Chem. Int. Ed. 2005; 44:3022-3037.
131. Shibata T. Recent advances in the catalytic Pauson-Khand-type reaction. Adv. Synth. Catal. 2006; 348:2328-2336.
132. Chung CY. Transition metal alkyne complexes: the Pauson-Khand reaction. Coord. Chem. Rev. 1999; 188:297-341.
133. Brummond KM, Kent JL. Recent advances in the Pauson-Khand reaction and related (2+2+1) cycloadditions. Tetrahedron 2000; 56:3263-3283.
134. Nishimura T, Uemura S. Novel palladium catalytic systems for organic transformations. Synlett 2004; 201-216.
135. Sigman MS, Jensen DR. Ligand-modulated palladium-catalyzed aerobic alcohol oxidations. Acc. Chem. Res. 2006; 39:221-229.
136. Schultz MJ, Sigman MS. Recent advances in homogeneous transition metal-catalyzed aerobic alcohol oxidations. Tetrahedron 2006; 62:8227-8241.
137. Lane BS, Burgess K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 2003; 103:2457-2473.
138. Schroder M. Osmium tetraoxide cis hydroxylation of unsaturated substrates. Chem. Rev. 1980; 80:187-213.
139. Kobayashi S, Sugiura M. Immobilization of osmium catalysts for asymmetric dihydroxylation of olefins. Adv. Synth. Catal. 2006; 348:1496-1504.
140. Tsuji J. Synthetic applications of the palladium-catalyzed oxidation of olefins to ketones. Synthesis 1984; 369-383.
141. Gorin, D. J, Toste F. D. Relativistic effects in homogeneous gold catalysts. Nature 2007; 446:395-403.
142. Knowles, W. S. Asymmetric Hydrogenations (Nobel Lecture). Angew. Chem., Int. Ed. 2002; 41:1998-2007.
143. Knowles, W. S. Asymmetric Hydrogenations - The Monsanto L-Dopa Process. pp. 23-38 in; Blaser, H.-U, Schmidt, E. eds. Asymmetric Catalysis on Industrial Scale. 2004. Wiley-VCH: Weinheim.
144. Farina V, Reeves JT, Senanayake CH, Song JJ. Asymmetric synthesis of active pharmaceutical ingredients. Chem. Rev. 2006; 106:2734-2793.
145. Jacobsen EN, Pfalz A, Yamamoto H. Comprehensive Asymmetric Catalysis. 2004. Springer, New York.
146. Kolodiazhnyi OI. Multiple stereoselectivity and its application in organic synthesis. Tetrahedron 2003; 59:5953-6018.
147. Guo H-C, Ma J-A. Catalytic asymmetric tandem transformations triggered by conjugate additions. Angew. Chem., Int. Ed. 2006; 45:354-366.
148. Keller E, Feringa BL. Catalytic enantioselective annulations via 1,4-addition-aldol cyclization of functionalized organozinc reagents. J. Am. Chem. Soc. 1999; 121:1104-1105.
149. Au-Yeung TT-L, Chan S-S, Chan ASC. Partially hydrogenated 1,1'-binaphthyl as ligand scaffold in metal-catalyzed asymmetric synthesis. Adv. Synth. Catal. 2003; 345:537-555.
150. Lennon IC, Pilkington CJ. The application of asymmetric hydrogenation for the manufacture of pharmaceutical intermediates: the need for catalyst diversity. Synthesis 2003; 1639-1642.
151. Biirgi T, Baiker A. Heterogenoeus enantioselective hydrogenation over cinchona alkaloid modified platinum: mechanistic insights into a complex reaction. Acc. Chem. Res. 2004; 37:909-917.
152. Tang W, Zhang, X. New Chiral Phosphorus Ligands for Enantioselective Hydrogenation. Chem. Rev. 2003; 103:3029-3069.
153. Laneman SA. Transition metal catalyzed hydrogenations, isomerizations, and other reactions. In: Handbook of Chiral Chemicals, 2nd ed. Ager DJ, ed. 2006. CRC Press, Boca Raton, FL. pp. 185-247.
154. Knowles WS. Asymmetric hydrogenation. Acc. Chem. Res. 1983; 16:106-112.
155. Noyori R, Hashiguchi S, Iwasawa Y. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 1997; 30:97-102.
156. Noyori R, Okhuma T. Asymmetric catalysis by architectural and functional molecular engineering: practical chemo- and stereoselective hydrogenation of ketones. Angew. Chem., Int. Ed. 2001; 40:40-73.
157. Cui X, Burgess K. Catalytic homogeneous asymmetric hydrogenations of largely unfuctionalized alkenes. Chem. Rev. 2005; 105:3272-3296.
158. Corey EJ, Helal CJ. Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: a new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew. Chem. Int. Ed. 1998; 37:1987-2012.
159. Roesky PW, Mueller TE. Enantioselective catalytic hydroamination of alkenes. Angew. Chem. Int. Ed. 2003; 42:2708-2710.
160. Riant O, Mostefai N. Recent advances in the asymmetric hydrosilylation of ketones, imines and electrophilic double bonds. Synthesis 2004; 2943-2958.
161. Braum M, Meier T. New developments in stereoselective palladium-catalyzed allylic alkylations of preformed enolates. Synlett 2006; 661-676.
162. Walsh PJ. Titanium-catalyzed enantioselective additions of alkyl groups to aldehydes: mechanistic studies and new concepts in asymmetric catalysis. Acc. Chem. Res. 2003; 36:739-749.
163. Denmark SE, Fu J. Catalytic enantioselective addition of allylic organometallic reagents to aldehydes and ketones. Chem. Rev. 2003; 103:2763-2793.
164. Betancort JM, Garcia C, Walsh PJ. Development of the first practical catalyst for the asymmetric addition of alkyl- and arylzinc reagents to ketones. Synlett 2004; 749-760.
165. Groger H. Catalytic enantioselective Strecker reactions and analogous syntheses. Chem. Rev. 2003; 103:2795-2827.
166. Brunel J-M, Holmes IP. Chemically catalyzed asymmetric cyano-hydron syntheses. Angew. Chem. Int. Ed. 2004; 43:2752-2778.
167. Chen F-X, Feng X. Synthesis of racemic tertiary cycanohydrins. Synlett 2005; 892-899.
168. Achard TRJ, Clutterbuck LA, North M. Asymmetric catalysis of carbon-carbon bond-forming reactions using metal (salen) complexes. Synlett 2005; 1828-1847.
169. Dai L.-X, Tu T, You S.-L, Deng W.-P, Hou X. L. Asymmetric catalysis with chiral ferrocene ligands. Acc. Chem. Res. 2003; 36:659-667.
170. Trost BM, Crawley ML. Asymmetric transition-metal-catalyzed allylic alkyations: applications in total synthesis. Chem. Rev. 2003; 103:2921-2943.
171. Belda O, Moberg C. Molybdenum-catalyzed asymmetric allylic alkylations. Acc. Chem. Res. 2004; 37:159-167.
172. Trost BM, Van Vranken DL. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 1996; 96:395-422.
173. Mlynarski J. Direct asymmetric aldol-Tischenko reaction. Eur. J. Org. Chem. 2006; 4779-4786.
174. Christoffers J, Baro A. Construction of quaternary stereocenters: new perspectives through enantioselective Michael reactions. Angew. Chem., Int. Ed. 2003; 42:1688-1690.
175. Hayashi T, Yamasaki K. Rhodium-catalyzed asymmetric 1,4-addition and its related asymmetric reaction. Chem. Rev. 2003; 103:2829-2844.
176. Christoffers J, Koripelly G, Rosiak A, Roessle M. Recent advances in metal-catalyzed asymmetric conjugate additions. Synthesis 2007; 1279-1300.
177. Oestreich M. Strategies for catalytic asymmetric electrophilic a halogenation of carbonyl compounds. Angew. Chem. Int. Ed. 2005; 44:2324-2327.
178. Noyori R. Asymmetric catalysis: science and opportunitites (Nobel lecture). Angew. Chem. Int. Ed. 2002; 41:2008-2022.
179. Kumar P, Naidu V, Gupta P. Application of hydrolytic kinetic resolution (HKR) in the synthesis of bioactive compounds. Tetrahedron 2007; 63:2745-2785.
180. Jacobsen EN. Asymmetric catalysis of epoxide ring-opening reactions. Acc. Chem. Res. 2000; 33:421-431.
181. Punniyamurthy T, Velusamy S, Iqbal J. Recent advances in transition metal catalyzed oxidation of organic substrates with molecular oxygen. Chem. Rev. 2005; 105:2329-2363.
182. McGarrigle EM, Gilheany DG. Chromium- and manganese-salen promoted epoxidation of alkenes. Chem. Rev. 2005; 105:1563-1602.
183. Xia QH, Ge HQ, Ye CP, Liu ZM, Su KX. Advances in homogeneous and heterogeneous catalytic asymmetric epoxidation. Chem. Rev. 2005; 105:1603-1662.
184. Rose E, Andrioletti B, Zrig S, Quelquejeu-Etheve, M. Enan- tioselective epoxidation of olefins with chiral metalloporphyrin catalysts. Chem. Soc. Rev. 2005; 34:573-583.
185. Katsuki T. Catalytic asymmetric oxidations using optically active (salen) manganese(III) complexes as catalysts. Coord. Chem. Rev. 1995; 140:189-214.
186. Frohn M, Shi Y. Chiral ketone-cataolyzed asymmetric epoxidation of olefins. Synthesis 2000; 1979-2000.
187. Katsuki T. Chiral metallosalen complexes: structures and catalyst tuning for asymmetric epoxidation and cyclopropanation. Adv. Synth. Catal. 2002; 344:131-147.
188. Lauret C, Roberts SM. Asymmetric epoxidation of α,β-un-saturated ketones catalyzed by poly(amino acids). Aldrichim. Acta 2002; 35:47-51.
189. Muller P, Fruit C. Enantioselective catalytic aziridinations and asymmetric nitrene insertions into CH bonds. Chem. Rev. 2003; 103:2905-2919.
190. Ramon DJ, Yus M. In the arena of enantioselective synthesis, titanium complexes wear the laurel wreath. Chem. Rev. 2006; 106:2126-2208.
191. Katsuki T, Martin VS. Asymmetric epoxidation of allylic alcohols: the Katsuki-Sharpless epoxidation reaction. Org. Reactions 1996; 48:1.
192. Sharpless K. B. Searching for new reactivity (Nobel lecture). Angew. Chem. Int. Ed. 2002; 41:2024-2032.
193. Fernandez I, Khiar N. Recent developments in the synthesis and utilization of chiral sulfoxides. Chem. Rev. 2003; 103:3651-3705.
194. Zaitsev AB, Adolfsson H. Recent developments in asymmetric dihydroxylations. Synthesis 2006; 1725-1756.
195. Kolb HC, VanNieuwenhze MS, Sharpless KB. Catalytic asymmetric dihydroxylation. Chem. Rev. 1994; 94:2483-2547.
196. Muniz K. Imido-osmium(VIII) compounds in organic synthesis I. Aminohydroxylation and diamination reactions. Chem. Soc. Rev. 2004; 33:166-174.
197. O’Brien P. Sharpless asymmetric aminohydroxylation: scope, limitations, and use in synthesis. Angew. Chem. Int. Ed. 1999; 38:326-329.
198. Bodkin JA, McLeod MD. The Sharpless asymmetric aminohydroxylation. J. Chem. Soc., Perkin Trans. I 2002; 2733-2746.
199. Davies HML, Beckwith REJ. Catalytic enantioselective C-H activation by means of metal-carbenoid-induced C-H insertion. Chem. Rev. 2003; 103:2861-2903.
200. Davies HML, Loe 0. Intermolecular C-H insertions of donor/ acceptor-substituted rhodium carbenoids: a practical solution for catalytic enantioselective C-H activation. Synthesis 2004; 2595-2608.
201. Dounay AB, Overman LE. The asymmetric intramolecular Heck reaction in natural product total synthesis. Chem. Rev. 2003; 103:2945-2963.
202. Shibasaki M, Vogl EM, Ohshima T. Asymmetric Heck reaction. Adv. Synth. Catal. 2004; 346:1533-1552.
203. Ager DJ, Laneman SA. Reduction of 1,3-dicarbonyl systems with ruthenium-biarylphosphine catalysts. Tetrahedron: Asymmetry 1997; 8:3327-3355.
204. Federsel H-J, Larsson M. An innovative asymmetric sulfide oxidation: the process development history behind the new antiulcer agent esomeprazole. In: Asymmetric Catalysis on Industrial Scale. Blaser H-U, Schmidt E, eds. 2004. Wiley-VCH, Weinheim, Germany. pp. 413-436.
205. Dai L-X. Chiral metal-organic assemblies—a new approach to immobilizing homogeneous asymmetric catalysts. Angew. Chem. Int. Ed. 2004; 43:5726-5729.
206. Corma A, Garcia H. Silica-bound homogeneous catalysts as recoverable and reuseable catalysts in organic synthesis. Adv. Synth. Catal. 2006; 348:1391-1412.
207. Heitbaum M, Glorius F, Escher I. Asymmetric heterogeneous catalysis. Angew. Chem., Int. Ed. 2006; 45:4732-4762.
208. Baleizao C, Garcia H. Chiral salen complexes: an overview to recoverable and reuseable homogeneous and heterogeneous catalysts. Chem. Rev. 2006; 106:3987-4043.
209. Dalko PI, Moisan L. Enantioselective organocatalysis. Angew. Chem. Int. Ed. 2001; 40:3726-3748.
210. Schreiner PR. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003; 32:289-296.
211. Dalko PI, Moisan L. In the golden age of organocatalysis. Angew. Chem. Int. Ed. 2004; 43:5138-5175.
212. Bolm C, Rantanen T, Schiffers I, Zani L. Protonated chiral catalysts: versatile tools for asymmetric synthesis. Angew. Chem. Int. Ed. 2005; 44:1758-1763.
213. Taylor MS, Jacobsen EN. Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem., Int. Ed. 2006; 45:1520-1543.
214. Lelais G, MacMillan DWC. Modern strategies in organic catalysis: the advent and development of iminium activation. Aldrichim. Acta 2006; 39:79-87.
215. Seayad J, List B. Asymmetric organocatalysis. Org. Biomol. Chem. 2005; 3:719-724.
216. Gaunt MJ, Johansson CCC, McNally A, Vo NT. Enantioselective organocatalysis. Drug Disc. Today 2007; 12:8-27.
217. Jarvo ER, Miller SJ. Amino acids and peptides as asymmetric organocatalysts. Tetrahedron 2002; 58:2481-2495.
218. List B. Proline-catalyzed asymmetric reactions. Tetrahedron 2002; 58:5573-5590.
219. Guillena G, Ramon DJ, Yus M. Organocatalytic enantioselective multicomponent reactions (OEMCRs). Tetrahedron: Asymmetry 2007; 18:693-700.
220. Shi Y. Organocataltic asymmetric epoxidation of olefins by chiral ketones. Acc. Chem. Res. 2004; 37:488-496.
221. Yang D. Ketone-catalyzed asymmetric epoxidation reactions. Acc. Chem. Res. 2004; 37:497-505.
222. Aggarwal VK, Winn CL. Catalytic, asymmetric sulfur ylide-mediated epoxidation of carbonyl compounds: scope, selectivity, and applications in synthesis. Acc. Chem. Res. 2004; 37:611-620.
223. Li A-H, Dai L-X, Aggarwal VK. Asymmetric ylide reactions: epoxidation, cyclopropanation, aziridination, olefination, and rearrangement. Chem. Rev. 1997; 97:2341-2372.
224. Ooi T, Maruoka K. Recent advances in asymmetric phase-transfer catalysis. Angew. Chem. Int. Ed. 2007; 46:4222-4266.
225. Kacprzak K, Gawronski J. Cinchona alkaloids and their derivatives: versatile catalysts and ligands in asymmetric synthesis. Synthesis 2001; 961-998.
226. Maruoka K, Ooi T. Enantioselective amino acid synthesis by chiral phase-transfer catalysts. Chem. Rev. 2003; 103:3013-3028.
227. O’Donnell MJ. The enantioselective synthesis of α-amino acids by phase-transfer catalysis with achiral Schiff base esters. Acc. Chem. Res. 2004; 37:506-517.
228. Lygo B, Andrews BI. Asymmetric phase-transfer catalysis utilizing chiral quaternary ammonium salts: asymmetric alkylation of glycine imines. Acc. Chem. Res. 2004; 37:518-525.
229. O’Donnell MJ, Wu SD, Huffman JC. A new active catalyst species for enantioselective alkylation by phase-transfer catalysis. Tetrahedron 1994; 50:4507-4518.
230. O’Donnell MJ. The preparation of optically active α-amino acids from the benzophenone imines of glycine derivatives. Aldrichim. Acta 2001; 34:3-15.
231. Palomo C, Oiarbide M, Garcia JM. Current progress in the asymmetric aldol addition reaction. Chem. Soc. Rev. 2004; 34:3-15.
232. Ooi T, Maruoka K. Asymmetrix organocatalysis of structurally well-defined chiral quaternary ammonium fluorides. Acc. Chem. Res. 2004; 37:526-533.
233. List B. Enamine catalysis is a powerful strategy for the catalytic generation and use of carbanion equivalents. Acc. Chem. Res. 2004; 37:548-557.
234. Allemann C, Gordillo R, Clemente FR, Cheong PH-Y, Houk KN. Theory of asymmetric organocatalysis of aldol and related reactions: rationalizations and predictions. Acc. Chem. Res. 2004; 37:558-569.
235. Saito S, Yamamoto H. Design of acid-base catalysis for the asymmetric direct aldol reaction. Acc. Chem. Res. 2004; 37:570-579.
236. Sorensen EJ, Sammis GM. A dash of proline makes things sweet. Science 2004; 305:1725-1726.
237. Northrup AB, MacMillan DWC. Two-step synthesis of carbohydrates by selective aldol reactions. Science 2004; 305:11752-11755.
238. Guillena G, Najera C, Ramon DJ. Enantioselective direct aldol reaction I. The blossoming of modern organocatalysis. Tetrahedron: Asymmetry 2007; 18:2249-2293.
239. Pihko PM. Activation of carbonyl compounds by double hydrogen bonding: an emerging tool in asymmetric catalysis. Angew. Chem. Int. Ed. 2004; 43:2062-2064.
240. List B. Asymmetric aminocatalysis. Synlett 2001; 1675-1686.
241. Notz W, Tanaka F, Barbas III CF. Enamine-based organocatalysis with proline and diamines: the development of direct catalytic asymmetric aldol, Mannich, Michael, and Diels-Alder reactions. Acc. Chem. Res. 2004; 37:580-591.
242. Tsogoeva SB. Recent advances in asymmetric organocatalytic 1,4-conjugate additions. Eur. J. Org. Chem. 2007; 1701-1716.
243. France S, Guerin DJ, Miller SJ, Leckta T. Nucleophilic chiral amines as catalysts in asymmetric synthesis. Chem. Rev. 2003; 103:2985-3012.
244. Basavaiah D, Rao PD, Hyma RS. The Baylis-Hillman reaction: a novel carbon-carbon bond forming reaction. Tetrahedron 1996; 52:8001-8062.
245. Ciganek E. The catalyzed α-hydroxylation and α-aminoalkylation of activated olefins (The Morita-Baylis-Hillman reaction). Org. Reactions 1997; 51:201.
246. Fu GC. Asymmetric catalysis with “Planar-chiral” derivatives of 4-(Dimethylamino) pyridine. Acc. Chem. Res. 2004; 37:542-547.
247. Miller, S. J. In search of peptide-based catalysts for asymmetric organic synthesis. Acc. Chem. Res. 2004; 37:601-610.
248. Tian S-K, Chen Y, Hang J, Tang L, McDaid P, Deng L. Asymmetric organic catalysis with modified cinchona alkaloids. Acc. Chem. Res. 2004; 37:621-631.
249. Marion N, Diez-Gonzalez S, Nolan SP. N-Heterocyclic carbenes as organocatalysts. Angew. Chem. Int. Ed. 2007; 46:2988-3000.
250. Merino P, Tejero T. Organocatalyzed asymmetric a-aminoxylation of aldehydes and ketones—an efficient access to enantiomerically pure α-hydroxycarbonyl compounds, diols, and even amino alcohols. Angew. Chem. Int. Ed. 2004; 43:2995-2997.
251. Adolfsson H. Organocatalytic hydride transfers: a new concept in asymmetric hydrogenations. Angew. Chem. Int. Ed. 2005; 44:3340-3342.
252. Enders D, Balensiefer T. Nucleophilic carbenes in asymmetric organocatalysis. Acc. Chem. Res. 2004; 37:534-541.
253. Cozzi F. Immobilization of organic catalysts: when, why and how. Adv. Synth. Catal. 2006; 348:1367-1390.
254. Wasilke J-C, Obrey SJ, Baker RT, Bazan GC. Concurrent tandem catalysis. Chem. Rev. 2005; 105:1001-1020.
255. Huerta FF, Minidis ABE, Backvall J-E. Racemization in asymmetic synthesis. Dynamic kinetic resolution and related processes in enzyme and metal catalysis. Chem. Soc. Rev. 2001; 30: 321-331.
256. Pamies O, Backvall J-E. Combination of enzymes and metal catalysts a powerful approach in asymmetric catalysis. Chem. Rev. 2003; 103:3247-3261.
257. Verzijl GKM, de Vries JG, Broxterman QB. Removal of the acyl donor residue allows the use of simple alkyl esters as acyl donors for the dynamic kinetic resolution of secondary alcohols. Tetrahedron: Asymmetry 2005; 16:1603-1610.
258. Constable DJC, Dunn PJ, Hayler JD, Humphrey GR, Leazer Jr JL, Linderman RJ, Lorenz K, Manley J, Pearlman BA, Wells A, Zaks A, Zhang TY. Key green chemistry research areas—a perspective from pharmaceutical manufacturers. Green Chem. 2007; 9:411-420.
Further Reading
Ager DJ, ed. Handbook of Chiral Chemicals. 2nd edition. 2006. CRC Press, Boca Raton, FL.
Blaser H-U, Schmidt E, eds. Asymmetric Catalysis on Industrial Scale. 2004. Wiley-VCH, Weinheim, Germany.
Collins AN, Sheldrake GN, Crosby J, eds. Chirality in Industry: The Commercial Manufacture and Applications of Optically Active Compounds. 1992. John Wiley & Sons, Chichester, UK.II: Developments in the Commercial manufacture and Applications of Optically Active Compounds. 1997. John Wiley & Sons, Chichester, UK.
Collins AN, Sheldrake GN, Crosby J, eds. Chirality in Industry
de Meijere A, Diederich F, eds. Metal-Catalyzed Cross-Coupling Reactions, 2nd edition. 2004. Wiley-VCH: Weinheim, Germany.
de Vries JG, Elsevier CJ, eds. The Handbook of Homogeneous Hydrogenation. 2007. Wiley VCH, Weinheim, Germany.
Drauz K, Waldman H, eds. Enzyme Catalysis in Organic Synthesis: A Comprehensive Handbook. 1995. VCH, New York.
Jacobsen EN, Pfalz A, Yamamoto H. Comprehensive Asymmetric Catalysis. 2004. Springer, New York.
Patel RN, ed. Biocatalysts in the Pharmaceutical and Biotechnology Industries. 2007. CRC Press, Boca Raton, FL.
Sheldon RA. ChiroTechnology: Industrial Synthesis of Optically Active Compounds. 1993. Marcel Dekker Inc., New York.
Wong CH, Whitesides GM. Enzymes in Organic Synthesis. 1994. Pergamon Press, Oxford, UK.
See Also
Enzyme Catalysis, Chemical Strategies for