CHEMICAL BIOLOGY
Membrane Protein Structure, Techniques to Study
Gavin King and Ann M. Dixon, University of Warwick, Coventry, United Kingdom
doi: 10.1002/9780470048672.wecb653
The biological importance of membrane proteins has been recognized worldwide for many years, but historically these proteins have proved difficult to characterize structurally because of a variety of experimental challenges. Recently, technological advances across several disciplines have prompted considerable progress in three-dimensional structure determination of membrane proteins. This review describes the state-of-the-art methods that have successfully produced high-resolution membrane protein structures to date. Most notably, X-ray crystallography will be discussed, as this technique has made by far the largest contribution to our current knowledge of membrane protein structure. This is followed by discussion of nuclear magnetic resonance spectroscopy and cryo-electron microscopy, both techniques that have also been successful in producing high-resolution structures for membrane proteins, albeit to a lesser degree than X-ray crystallography. Finally, we will discuss atomic force microscopy. Although this technique cannot be used for atomic level structural determination, it offers distinct advantages for investigation of membrane protein oligomerization, dynamics, and large-scale conformational changes. Recent notable membrane protein structures are included throughout to illustrate progress in the field as well as the strengths and weaknesses of each method.
Structural, biophysical, and biochemical studies of membrane proteins have revealed the importance of this class of proteins in fundamental biological processes such as the import and export of nutrients and waste into and out of cells, cell division, and signaling, to name a few. With approximately one third of sequenced genomes encoding integral membrane and membrane-associated proteins, and two thirds of all drugs in development targeting membrane proteins, their importance is now well accepted. However, three-dimensional (3-D) structures of the majority of known membrane proteins still elude us. Of the 47,000 protein structures deposited in the Protein Data Bank (PDB), only about 1% are for membrane proteins. Solving the structures of these proteins has proved to be highly challenging, and it still represents the leading edge of protein structural biology. The slow progress is caused, in part, by difficulties in protein production and purification (especially for eukaryotic membrane proteins) and in part by experimental difficulties encountered because of their large size and the requirement for detergent/lipid solubilization. Despite these obstacles, significant advancement has been made over the past decade. Careful production of membrane proteins and major breakthroughs in instrumentation have yielded stunning results for several classes of membrane proteins, including channels, pores, and receptors. The resulting structural data have given us an insight into the architecture of both simple and complex membrane proteins as well as their function (for an excellent summary of known structures, see http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html). In this review, we will cover the four most successful and commonly used experimental methods for determination of membrane protein structures, namely X-ray crystallography, nuclear magnetic resonance spectroscopy, electron microscopy, and atomic force microscopy, with particular emphasis on recent advances. We note that molecular dynamics simulations have also contributed significantly to our current understanding of membrane protein structure, and closely accompany the above listed methods; however a description of these computational methods is beyond the scope of this review.
Membrane Protein Structures by X-Ray Crystallography
X-ray crystallography is by far the most successful method for solving membrane protein structures, having provided approximately 80% of the membrane protein structures currently held in the PDB. The first membrane protein structure solved by X-ray crystallography (XRC) was that of the photoreaction center from Rhodopseudomonas viridis, originally solved at 3 A resolution in 1985 (1). Since that time, XRC has been applied to a wide variety of membrane protein families. This technique typically produces 3-D structural data with resolution ranging from 1.5 to 3.5 A, making it the highest resolution technique currently available for structure determination.
XRC determines the precise arrangement of atoms within a crystal by analyzing the scattering (diffraction) of X-rays by electrons to produce an electron density map. The rate-limiting step in structure determination by XRC is the production of well-ordered 3-D crystals, which has proved to be highly challenging as illustrated by the scarcity of membrane protein structures. Obtaining sufficient quantities of highly purified protein for numerous crystallization trials is often difficult, as membrane proteins can be toxic to heterologous hosts. Recent advances in automation and miniaturization have greatly reduced the quantity required, currently enabling investigators to test up to 100,000 conditions in parallel per day using less than 300 mg of protein (2). Despite these advances, however, crystallization remains a “trial-and-error” process, involving numerous variables and exacerbated for membrane proteins by the need for solubilizing agents such as detergents, which can greatly destabilize protein folds and shield crystal contacts.
The diffraction size and quality of the crystals greatly impacts the resolution of XRC and, therefore, is of utmost importance. The X-ray source also plays a key role in the quality of XRC data. Traditional X-ray sources can suffer from poor spatial coherence and low beam strength. For these reasons, synchrotron sources are now commonly used to collect data. A synchrotron source produces high-energy X-rays that more effectively penetrate the crystal to interact with atoms in the protein. The X-ray beam from a synchrotron source is more coherent, has a higher concentration, and can be accurately focused on very small targets.
3-D crystallization of membrane proteins
The first, and often most difficult, step in XRC is the production of 3-D crystals of purified protein. It is important to emphasize the importance of protein purity for successful crystallization because, although it is true that a nonhomogenous mixture may crystallize, it has been noted by some researchers that difficulties in crystallizing a protein may be negated by further purification. Crystallization of membrane proteins is a complicated process, and various approaches have been used, including vapor diffusion, microdialysis, batch crystallization and the recently developed lipid-phase methods. Detailed information on membrane protein crystallization can be found in several excellent reviews in the “Further Reading” section. The general principle of protein crystallization involves the supersaturation of a protein solution. The addition of precipitating agents such as salts, organic solvents, or polymers triggers the crystallization process.
Detergents play a vital role in the crystallization of membrane proteins. However, as mentioned, they can also have a destabilizing effect on the protein. For successful crystallization, the detergent micelles must be accommodated in the crystal lattice with minimum effect on the formation of crystal contacts. The choice of detergent and its associated properties (polarity of head group, aliphatic chain length, size) must be explored for each protein (3). Generally, charged detergents should be avoided because of the risk of repulsion between protein-detergent complexes. The length of aliphatic chains should be balanced between the need to cover the protein’s hydrophobic surfaces and the need to have as low a detergent volume as possible to maximize protein-protein contacts. The phase behavior of detergents and lipids is also a pertinent consideration. Phase separation from a micellar phase (detergent-rich) toward a nonmiscible phase (detergent-poor) can occur at high-detergent and precipitant concentrations. These phase boundaries have been exploited to enhance crystallization of membrane proteins (4). The use of additives such as small amphiphiles (e.g., heptane-triol, LDAO) can also enhance crystallization by reducing the volume of detergent in the protein-detergent complex, leading to increased numbers of crystal contacts (5). However, the overriding problem with the use of detergents is the increase in the number of variables that need to be optimized in crystallization trials, which is already considerable. Coupled with the use of additives, this presents a huge number of possibilities. The absence of a general set of rules to guide detergent/additive choice is currently a significant problem for crystallization, and it increases the timescale for each structure.
A significant landmark in membrane protein crystallization has been the development of lipidic cubic phase technology (6). This technically challenging approach is capable of producing crystals that diffract beyond 2 A while providing an environment resembling that of a natural membrane. Lipidic cubic phases are gel-like materials into which the protein is embedded. Crystallization is then initiated by the addition of precipitants. If successful, the resulting crystals contain ordered layers of protein-lipid sheets, with contacts formed within and between the sheets. Lipidic cubic phases have been successfully used to crystallize several membrane proteins, including archeal seven transmembrane proteins (6, 7). Co-crystallization with antibody fragments (Fv fragments of Fab domains) has also been used to enhance membrane protein crystallization (8, 9). The fragments form bridges between protein-detergent complexes, thus increasing the number of protein-protein contacts. Using this method, crystallization was achieved for the cytochrome bc1 complex (8), the KcsA potassium channel (10), and more recently the human β-2 adrenergic receptor (Fig. 1a) (11-13). A caveat to this approach is that antibody production must be tailored to the protein under study, which can be expensive and time consuming.
The process of crystallization is still largely a process of trial and error, but in an attempt to formulate general rules, the cumulative experiences of X-ray crystallographers are being compiled into online databases such as the Marseille Protein Crystallization Database (14) and the Biological Macromolecule Crystallization Database (15). These and similar databases will provide useful starting points in crystallization trials and may greatly accelerate the process.
Collection of X-ray diffraction data
A detailed examination of the crystallographic method is beyond the scope of this article, sources of more detailed information can be found in the “Further Reading” section. The general method of XRC involves five steps (2): 1) crystallization of purified protein (discussed above); 2) measurement of crystal diffraction; 3) phase determination; 4) phase and electron density calculations; 5) model building. Once crystals are formed, they are exposed to X-rays and diffraction data are collected. Exposure to X-rays can damage the crystals, and this can be reduced by freezing the crystal under a nitrogen stream at 100 K. This approach is not always favorable for membrane proteins as crystal contacts can be quite weak and, therefore, highly sensitive to temperature. Cooling can further destabilize the crystal by causing the detergents to undergo phase transitions that perturb crystal contacts. For these reasons, cooling may be limited to 4 °C, which often proves sufficient to collect diffraction data. Smaller crystals are more amenable to freezing but often yield poor quality data, being best suited for use with synchrotron beams (16). After collection of diffraction data, phase determination is commonly carried out for soluble proteins using the method of molecular replacement. This method is less useful for membrane proteins because of the lack of known structures. Another method for phase determination is the “heavy-atom” method, which involves soaking crystals in heavy-atom solutions. Unfortunately, this method is also not as effective for membrane proteins because of the reduced binding of heavy atoms, which is a direct result of the decrease in hydrophilic surfaces, lack of accessibility, and nonspecific interactions of hydrophobic heavy-atom compounds with detergents. An alternative approach to the phase problem involves multi-wavelength anomalous diffraction phasing using selenomethionine (17). Diffraction and phasing data are used to create an electron-density map and to build a preliminary model. The model is further refined by multiple rounds of energy minimization and manual manipulations. Finally, simulated annealing is performed to produce the final structure of the protein.
Recent notable structures
Human β-2 adrenergic receptor
In 2007, this structure was solved to a resolution of 2.4 A (11) (Fig. 1a), and it represents the first structure of a ligand-activated G-protein-coupled receptor. Crystallization was achieved using the antibody cocrystallization approach. This work revealed structural features not seen before in this protein family, including cholesterol-mediated intermolecular associations and an extracellular loop containing a helix.
Sodium-potassium pump
The structure of the sodium-potassium pump was solved in 2007 to a resolution of 3.5 A (Fig. 1b), in complex with two bound rubidium ions (13). Structures were also solved for other members of the P-type ATPase family (18, 19) around the same time revealing the structural similarity between family members as well as the conformational changes that occur during function.
Acid-sensing ion channel
This high-resolution structure (1.9 A) solved in 2007 was the first structure of an acid-sensing ion channel (19). It belongs to the degenerin/epithelial sodium channel family of ion channels, which play essential roles in diverse biological processes such as mechanotransduction and ion homeostasis. The structure (Fig. 1c) revealed the trimeric state of the protein channel as well as a possible mechanism for activation, involving long-range conformational changes triggered by proton binding.
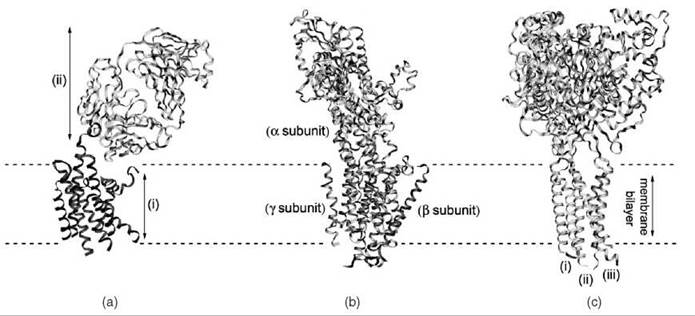
Figure 1. Ribbon representations of three membrane protein structures determined using X-ray crystallography. (a) The human β-2 adrenergic receptor was crystallized using the technique of antibody cocrystallization (11) (PDB ID: 2R4R). The structures of the receptor (i) and the antibodies (ii) are shown (b) The structure of the sodium-potassium pump (12) (PDB ID: 3B8E) is shown, indicating the three subunits of the protein. (c) The structure of the acid-sensing ion channel 1 at low pH (13) (PDB ID: 2QTS), which is composed of three chains (i, ii, and iii) that assemble to form a trimer.
Membrane Protein Nuclear Magnetic Resonance Spectroscopy
Solution-state nuclear magnetic resonance (NMR) spectroscopy has also proved very useful in membrane protein structure determination, particularly for smaller proteins. Like XRC, solution NMR requires the use of high concentrations of protein as well as solubilizing agents like detergents, and therefore, it requires a similar level of optimization. However, when optimal conditions are achieved, NMR provides high-resolution structural data and has the added advantage of providing information on function, dynamics, topology, and interactions of the protein with the membrane, ligands, and other proteins.
NMR determines the arrangement of atoms in a protein by exploiting the quantum magnetic property that certain atomic nuclei will align their magnetic moment with an external magnetic field (B0). Perturbation of B0 by a second perpendicular magnetic field leads to a response of the atomic nuclei that is detected as the NMR signal. NMR signals provide information on both short- and long-range interatomic distances that are used with simulated annealing methods to determine the 3-D structure of a protein. The lifetime of the NMR signal is related to the size of the protein, with signals for large, slowly tumbling proteins relaxing very rapidly. This relaxation leads to severe broadening of the signals, and it places a practical limit on the size of proteins that can be analyzed. Thus far, solution-state NMR has yielded the global fold of an 82-kDa soluble protein (20); however, for membrane proteins, the practical limit is around 40 kDa, making it most useful for proteins or individual domains. NMR studies of several membrane proteins have been carried out in the solid-state (in lipid bilayers), and this will be discussed briefly in the next section. However, a larger number of membrane proteins has been studied in solution, and this will be the main focus here.
Solid-state NMR
Solid-state NMR will only be considered in brief because of the limited number of novel membrane protein structures produced. For comprehensive reviews of this technique, the reader is referred to the “Further Reading” section. Around 17 structures (mainly small peptides) solved by solid-state NMR have been deposited in the PDB, with the first being gramicidin A in 1997 (21) and the most recent being the backbone structure of Influenza A M2 proton channel transmembrane domain (22). Solid-state NMR differs from the solution-state in that the molecule under investigation is static or slowly tumbling, resulting in broad NMR peaks with low resolution and sensitivity. This fundamental problem has been tackled by development of MAS (magic-angle spinning) (23) and REDOR (rotational echo double resonance) (24) experiments. Advantageously, solid-state NMR methods enable membrane proteins to be studied in lipid bilayers resembling native membranes; a disadvantage is that proton detection is challenging, with most studies focusing on isotopically labeled samples.
Membrane protein samples for solution-state NMR
Three-dimensional structure determination by NMR requires complete assignment of spectral peaks (or resonances), and the success of this process is highly dependent on sample preparation. Similarly to XRC, obtaining purified protein is a rate-limiting step in NMR structure determination. Resolution and sensitivity can be increased through isotopic labeling of proteins with 15N and 13C, either by expression in labeled media or by incorporation of labels during synthesis. Once a labeled protein has been obtained, solution conditions (e.g., temperature, pH, and ionic strength) must be optimized for NMR. For membrane proteins, this also involves selection of a solubilizing agent, which can solubilize the protein at a sufficiently high concentration for NMR (typically ~1 mM), produce high-resolution spectra, and maintain the protein in its native conformation. Detailed protocols for protein and sample preparation can be found in the “Further Reading” section.
Detergents are frequently used to solubilize membrane proteins for solution-state NMR. Rapidly tumbling protein-micelle complexes are formed (Fig. 2a) (25-27) that are capable of producing narrow linewidths and high-resolution spectra while maintaining the quaternary structure of the protein. A detergent screen is often required to find the optimum detergent for a particular protein, and extensive studies of detergents suitable for NMR have been performed (28). Although no detergent is generally applicable, in numerous cases, dodecylphosphocholine (DPC) and lyso-palmitoyl phospatidylglycerol (LPPG) have produced high-quality solution spectra, providing a strong starting point in detergent screens (28). The use of detergents is currently limited to proteins of <40 kDa (protein-micelle complexes of <80 kDa). Alternatives to detergents have also been employed with moderate success. Organic solvent mixtures have proved useful (29) and offer the added advantage that proteins tumble more rapidly producing higher quality spectra. However, these mixtures are considered poor membrane mimetics, casting doubt on the relevance of resulting structures. Bicelles (Fig. 2b), or lipidic disk-shaped particles that more closely resemble bilayers, have also been useful in studies of small transmembrane peptides and can be used in both solution and solid-state NMR (30). Further solubilizing agents include lipopeptides and amphipols (Fig. 2d), which have been used in initial studies of OmpA, PagP, and DAGK (31-33), whereas nanodisks (Fig. 2e) (34) and lipid cubic phases (Fig. 2f) (35) are recent developments that may prove useful in the future.

Figure 2. Membrane mimetics used in NMR studies of membrane proteins. (a) Small, spherical detergent micelles composed of amphiphillic detergent monomers. Dodecylphosphocholine (DPC) micelles were employed to determine the structure of OmpA (25) (PDB ID: 1G90) (b) Bicelles, composed of lipid/detergent mixtures, have been used recently to solve the structure of the Bnip3 transmembrane domain dimer (26) (PDB ID: 2J5D) (c) Synthetic lipid bilayers closely approximate the natural environment of membrane proteins and have been used with solid-state NMR to determine the structure of the fd bacteriophage pVIII coat protein (27) (PDB ID: 1MZT). (d) Amphipols are amphiphillic polymers consisting of a hydrophilic backbone onto which numerous hydrophobic chains are attached that can coat the hydrophobic region of membrane proteins. (e) Nanodisks are self-assembling structures composed of a phospholipid bilayer disk encircled by an engineered membrane scaffold protein. (f) Lipid cubic phases are primarily used in X-ray crystallography studies but recently have shown promise for use in NMR studies as well. (g) NMR structure of the phospholamban pentamer (PDB ID: 1ZLL), looking down on the central cavity. (h) Side view of the phospholamban pentamer shown in (g) overlaid with a refined version of the structure published in 2006 (PDB ID: 2HYN).
NMR data collection and structure determination
Once a suitable membrane protein sample is prepared, spectral assignments are made. Assignment is another rate-limiting step in NMR structure determination, and it has greatly benefited from the development of the TROSY (transverse relaxation optimized spectroscopy) triple resonance experiments, which improve spectral resolution and sensitivity (36). Although TROSY experiments will not be covered in detail here, briefly these experiments reduce relaxation of NMR signals at high magnetic field strengths, thus increasing the molecular mass of proteins that can be studied to ~50kDa. Additional benefits can be realized by using TROSY in conjunction with a selectively deuterated protein and deuterium decoupling, acting to further reduce broadening of signals (37). TROSY experiments have been used successfully to assign the resonances of both β-barrel (25, 38) and a-helical membrane proteins (39).
After completion of the spectral assignment, NMR-derived short- and long-range distances can be determined by measurement of nuclear Overhauser enhancements (or NOEs). An NOE is an interaction between a pair of atoms <5.0 A apart, and the intensity of the NOE can be related to the distance (r) separating the pair. The use of NOEs can be problematic for helical proteins as they tend to have few long-range NOEs. The use of methyl protonation can increase the number of NOEs; however, because of the poor chemical shift dispersion of methyl groups in helical membrane proteins, this approach is generally ineffective. An alternative is the use of residual dipolar couplings (RDCs) to obtain long-range distances. RDCs are derived from the difference in coupling constants in an aligned and unaligned state, providing information on the orientation of internuclear vectors relative to the external magnetic field (40). Several methods have been developed to produce weak alignment of membrane proteins, thus enabling the measurement of RDCs. Polyacrylamide gels can be used for small-to-medium proteins to produce an anisotropic environment where protein orientations are limited, resulting in weak alignment (40). DNA nanotubes are also a promising alignment method with the added advantage that they are resistant to detergent and do not reduce achievable protein concentrations (41). Alignment can also be achieved by incorporation of paramagnetic lanthanide ions into diamagnetic proteins (42). Once aligned samples are prepared, RDCs are accurately measured using a set of TROSY-based experiments (43) Importantly for helical proteins, a plot of backbone amide RDCs versus residue number produces a wave pattern with a periodicity of 3.6 residues per cycle for helical regions, allowing detection of helical secondary structure and the tilt of the helix relative to the magnetic field (44). The use of RDCs for structure refinement has been demonstrated for several membrane proteins, including OmpA (45), Vpu (46), and MerF (47).
Finally, NMR-derived distance information as well as information about dihedral angles (obtained from chemical shifts) is incorporated into structure calculations performed using molecular dynamics and simulated annealing programs such as CNS (48) and XPLOR-NIH (49) to calculate the protein structure.
Recent notable structures
Mistic
Despite its uncertain status as a membrane protein, the structure of Mistic from Bacillus subtilis determined in detergent micelles highlights a promising approach to solving the 3-D structure of multispanning helical membrane proteins by solution-state NMR (50). Backbone and side-chain assignment was achieved by partial deuteration and full 13C/15N labeling of the protein. The conformation of its four α-helices was determined using 13C chemical shift-derived angle restraints, NOEs, and hydrogen bond restraints. Importantly, 487 long-range restraints from 5 paramagnetic spin labels and 29 long-range NOEs allowed the fold of the protein to be determined.
Outer membrane protein a (OmpA)
In 2001, the structure of the transmembrane domain of OmpA (Fig. 2a) was solved in DPC micelles, and it represented the largest membrane protein structure (19 kDa) to have been solved by NMR at that time (25). TROSY triple-resonance experiments enabled a large number of the residues to be assigned for the deuterated protein. Relatively limited structure restraints (NOEs, dihedral angles, interstrand hydrogen bonds) revealed an eight-stranded β barrel structure for the transmembrane domain of this ion channel that was very similar to the existing X-ray structure.
Phospholamban pentamer
Phospholamban is a homopentameric membrane protein involved in muscle contraction through regulation of the calcium pump in cardiac muscle cells. The structure of the unphosphorylated protein solved in DPC micelles reveals a symmetric pentamer of phospholamban monomers (Fig. 2g) stabilized by leucine/isoleucine zipper motifs along the transmembrane domains (51). Notably, another structure was produced for phospholamban (Fig. 2h) that used a variant of the traditional simulated annealing and molecular dynamics protocol that reduced the chances of entrapment in local minima (52).
Membrane Protein Structures by Electron Microscopy
Electron microscopy (EM) has also made major contributions to the field of membrane protein structure determination, and could potentially lead the field before long. This technique provides major advantages in that the structural features of membrane proteins can be measured in their native membranes or reconstituted lipid bilayers, and not in detergents as required by (and which often limits the success of) XRC and solution NMR techniques. Although electron microscopy has historically produced lower resolution structures than XRC and NMR, recent improvements in instrumentation and sample preparation have produced data with resolution that rivals even the best X-ray structures (53).
The first reported 3-D model of a membrane protein was obtained using EM. This ground-breaking work was reported by Henderson and Unwin in 1975, who analyzed two-dimensional (2-D) crystals of bacteriorhodopsin within the purple membrane to produce a density map with 7A resolution and a structural model of the protein (54). This data provided our first view of the architecture of a membrane protein, revealing key features such as membrane spanning a-helices. Fifteen years and many instrumental and methodological improvements later, a model of bacteriorhodopsin at atomic resolution was reported, marking the first structure solved by electron microscopy (55). Since that time, structures of important membrane protein classes, including pores (53, 56), receptors (57-59), channels (60, 61), transporters (62), and enzymes (63), have been solved using EM.
EM can provide structural information for a protein at a variety of different resolutions depending on the microscope used, the method of staining, and the condition of the sample. State-of-the-art electron microscopes can provide resolution of up to a few angstroms for good quality samples. In order to obtain high-resolution structural information from EM, one important factor is the electron source. Electrons emitted from a heated metal cathode suffer from poor spatial coherence and yield only a small amount of signal at high resolution. An alternative electron source that is rapidly gaining widespread use, the field emission gun (FEG), produces a much more coherent electron beam by limited heating in combination with an electric field. Certainly most of the current high-resolution EM structures have been acquired using an EM equipped with an FEG.
Another important factor in obtaining high-resolution data is the minimization of radiation damage to the sample. Radiation damage results from exposure of the protein sample to the electron beam as it is being imaged, and it greatly limits the resolution of the data. This loss of resolution is one of the primary reasons that XRC has had more success in obtaining structural data for proteins sensitive to radiation. One method that greatly reduces radiation damage and represents a major milestone in the field is electron cryomicroscopy (cryo-EM), in which samples are prepared in a frozen hydrated state and imaging takes place at liquid nitrogen or liquid helium temperatures under high vacuum. Cryo-EM is now the method of choice for structure determination by EM, and it is used almost exclusively in recent reports. Additional improvements in the resolution of cryo-EM data are achieved by imaging samples present in 2-D crystals, a subject that will be covered in more detail in the next section.
Preparation of samples for cryo-EM analyses
In order to produce 3-D reconstructions of membrane protein structures effectively, cryo-EM is most successful for samples present as single particles or in 2-D crystals (as opposed to the 3-D crystals used in XRC). Single-particle cryo-EM is used primarily for large proteins and complexes that do not form crystals (64). In this approach, images are obtained for particles fixed to a carbon-film surface and coated in a layer of heavy metal salts, a process called negative staining that acts to protect the sample from beam damage and improve image contrast, or particles suspended within a layer of vitreous ice (cryo-EM). These images are then averaged after alignment of the various particle orientations.
Although single-particle EM has been most successfully and routinely used for soluble proteins, structural information for several membrane proteins including the ryanodine receptor ion channel (58), the L-type Ca2+ channel (61), the voltage-gated K+ channel (60), and the inosotol (1,4,5)-triphosphate receptor (65) has also been obtained using this method. However, single-particle EM has thus far resulted in low-resolution structural information, typically yielding data with resolutions in the range. Another disadvantage of single-particle cryo-EM is the lower limit for molecular weight, which is currently approximately 250 kDa. This limit results from the need to distinguish between individual particles and molecules in the surrounding medium, and it is unlikely to change in the future (66).
The highest resolution cryo-EM data has thus far been obtained for proteins present in 2-D crystals (a technique known as electron crystallography). This technique measures the structural features of membrane proteins reconstituted into 2-D crystals in the presence of lipid bilayers. In contrast to the 3-D crystals used in XRC, in which proteins are solubilized in detergent micelles that can disrupt crystal formation and reduce crystal quality, formation of 2-D crystals forces the membrane proteins to pack within a lipid bilayer, thus restoring their native environment. Another advantage of 2-D crystallization is that it requires very small amounts of protein (as opposed to NMR, which requires milligram quantities).
As with the growth of 3-D crystals, the growth of 2-D crystals is often the most difficult step in cryo-EM analysis of a membrane protein. Two-dimensional crystallization of membrane proteins can be achieved by mixing the protein with detergent and lipids, and then slowly decreasing the detergent concentration (e.g., by dialysis). Two-dimensional crystals can also be formed by vesicle fusion, addition of crystallizing agents, addition of detergents to reduce the lipid:protein ratio, or adsorption onto lipid monolayers (for a review of 2-D crystallization techniques, see the “Further Reading” section). Because the fundamental mechanisms of 2-D crystal formation are thus far very poorly understood, extensive screening of suitable lipids and lipid:protein ratios is required (67) to obtain a high-quality crystal. However, in some cases, membrane proteins that have proved very resistant to 3-D crystal formation have readily formed 2-D crystals in lipid membranes.
Two types of 2-D crystal have been analyzed by cryo-EM: sheet-like (planar) crystals and tubular crystals. Planar crystals are tilted in the electron beam in order to obtain different projection maps of the crystal at a variety of angles. Maps collected at different angles are then averaged together to produce a 3-D reconstruction (68). However, the planar crystal cannot be tilted through all angles in the microscope (for example, a sample rotated through 90° would then be parallel to the electron beam) and typically can only be rotated through 70-75°. The resulting resolution is, therefore, spatially heterogeneous and appears to have what is known as a “missing cone” of data. Alternatively, membrane proteins can also crystallize into tubular crystals where, in a single crystal “tube” one can potentially see all orientations of a protein without the need to tilt the crystal. Tubular crystals overcome the “missing cone” of information and provide resolution that is equal in all directions (59).
Collection of cryo-EM data
High-resolution cryo-EM data can be collected in two forms: as electron images (69) or as electron diffraction patterns. Cryo-EM images contain information on both amplitude and phase, which can be analyzed after Fourier transformation. The quality of the amplitude data can be improved if combined with electron diffraction data, which contains only amplitude information. In this way, EM overcomes one of the main difficulties in XRC. In XRC, only diffraction patterns are obtained. X-rays cannot be used to form an image of the crystal; therefore, the phase information is lost. In contrast, electron microscopes contain electron lenses that can capture phase information.
Structure determination and other applications of cryo-EM
A 3-D structure is determined by fitting an atomic model to a 3-D density map at a given resolution. This map is determined by analyzing EM images and electron diffraction patterns. Because the membrane protein is embedded within a lipid bilayer, 2-D crystals more closely resemble its native environment indeed, the protein Aquaporin 1 (AQP1) embedded in a 2-D crystal was shown to maintain its biological activity (70). Because they are more likely to adopt their native fold in a lipid bilayer, 2-D crystals are thought to provide more biologically relevant structural information about protein-protein and protein-lipid interactions. This is illustrated by the 3-D structure of the transporter protein EmrE, which has been solved by both XRC (71) and cryo-EM (62). Comparison of the two structures reveals important differences between the structure of EmrE in a 3-D crystal (formed in detergent) and its structure in a 2-D crystal (embedded in a bilayer) that result from the presence of lipids.
Apart from the determination of a static structure, which provides a mere snapshot of the protein at a particular moment in time, cryo-EM of 2-D crystals also allows us to study conformational changes of a protein over time, thus yielding critical insight into function. In these time-resolved studies, conformational changes are induced in the protein (by changing pH, adding ligand, etc.) and then trapped by freezing the crystal. Freezing can take place at several time points after induction of a conformational change to produce a series of structures that describe a reaction pathway. Time-resolved studies have been performed to investigate the photocycle of bacteriorhodopsin (72), the gate-opening mechanism of the acetylcholine receptor (57), and more recently, the pH-dependent mechanism for a Na+/H+ antiporter (73).
Recent notable structures
Aquaporin-0
The structure of this water-selective membrane pore protein (Fig. 3a and 3b) represents the highest resolution structure obtained from electron crystallography to date (53). Data were obtained for Aquaporin-0 in double-layered 2-D crystals, and its staggering 1.9-A resolution clearly reveals water molecules within the pore. The data also reveal associated lipids, allowing key protein-lipid interactions to be modeled.
Aquaporin-4
This 3.6-A structure of Aquaporin-4 (Fig. 3c) was determined by electron crystallography of double-layered 2-D crystals (56). Features in the structure show that Aquaporin-4 can form membrane junctions, and they suggest for the first time its role in cell adhesion. This structure is of additional interest in that it is the first structure of a multispanning mammalian membrane protein obtained by purely recombinant methods.
Glutathione transferase-1
At 3.2 A, the resolution of this recent structure also rivals that obtained in XRC (63). This structure provides a strong insight into the function of the protein, and it reveals key differences between the glutathione binding site of this membrane-spanning enzyme and that of its soluble counterparts.
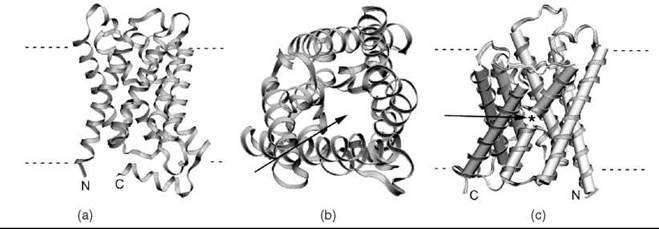
Figure 3. Ribbon representations of three membrane protein structures determined using electron microscopy. (a) The structure of Aquaporin 0 AQP0 (53) (PDB ID: 2B6O), the highest resolution structure of a membrane protein to be solved by cryo-EM to date. (b) A view down the pore of AQP0 (arrow), which allows water molecules to pass across the membrane. (c) The rat AQP4 (56) (PDB ID: 2D57) structure, highlighting the gap (arrow and *) between two helices that align to span the membrane.
Imaging Membrane Proteins by AFM
The 2-D crystals of membrane proteins discussed in the above section can also be analyzed using atomic force microscopy (AFM) (74). Although it is a low-resolution technique restricted to surface contouring, and as such cannot provide atomic level structural information, AFM has developed into a powerful tool for investigation of oligomerization, dynamics, and large-scale conformational changes. AFM also provides several advantages over EM. As mentioned, high-resolution EM analyses require coating of the sample, low temperatures, and high vacuum. In contrast, AFM images (or topographs) can be collected under physiological conditions allowing investigation of structure and conformational changes involved in function (e.g., gating of channels). AFM is also the only imaging technique that can collect data on liquid samples, and it is therefore able to provide, in addition to images of 2-D crystals, images of membrane bilayers in an aqueous environment.
AFM was first used to analyze a membrane protein in 1990, when images were collected on hydrated purple membranes (75). In these images, the global arrangement of bacteriorhodopsin molecules was observed for the first time at near-physiological conditions (75). The resolution of AFM images has improved steadily over the past 20 years, and now the technique is regularly used to observe individual proteins and macromolecular assemblies in both 2-D crystals (74-78) and in native membranes (75, 81, 82). From AFM images, information has been obtained for a wide variety of membrane proteins, including Class A G-protein-coupled receptors (81), pumps (78, 83), channels (76), enzymes (84), and pores (80). Larger protein complexes, such as the light harvesting complexes I and II, have also been studied in both 2-D crystals (79) and in their native membranes (82).
Currently, the resolution of AFM is lower than that of NMR, XRD, or cryo-EM. The highest resolution AFM data thus far has been collected on samples in 2-D crystals, producing structural data with resolutions between 9 and 14 A (77-80). In general terms, AFM works by raster scanning (scanning line by line) a very sharp tip attached to a flexible cantilever over a sample in order to produce a contour map of its surface. In order to maximize resolution, vertical fluctuations of the tip must be minimized. This is achieved using a self-regulating feedback system (or servo system), which keeps the cantilever deflection approximately constant by making small adjustments in the vertical displacement of the sample. A 2-D crystal provides an ideal sample for AFM as it is hard and flat, with the protein embedded in a densely packed array, thus restricting any lateral or vertical movement of the protein. Another prerequisite for high-resolution data is minimal interaction between the tip and the sample, and this is greatly influenced by the size and the geometry (or sharpness) of the tip. The tip geometry is most commonly an inverted pyramid. Depending on its sharpness, as the tip moves across a surface, structures may interact with both the end of the tip as well as the sides of the tip. Any interactions with the sides of the tip will cause a broadening of the signal, thus reducing the lateral resolution of the image. This effect, also known as tip convolution, is most noticeable on surfaces with considerable height differences. Most of the tips used on high-resolution instruments are now commercially fabricated, with the best tips having a radius of curvature of >5 nm.
Structure determination and other applications of AFM
As mentioned, individual membrane proteins and complexes in 2-D crystals and in densely packed, noncrystalline arrays can be imaged by AFM at sub-nanometer resolution (76-80, 84). This makes AFM ideally suited to provide information on the conformation and oligomeric state of membrane proteins in their native membranes. One very nice example of this is the study of rhodopsin in its native membrane (81). AFM was used to image native disk membranes in aqueous solutions, and revealed the organization of rhodopsin into dimers and higher oligomers. By imaging membranes in aqueous solutions, AFM also allows us to observe conformational changes of biomolecules as they function.
Because deflections of the cantilever can be detected in the 10-50 pN range, making single-molecule force measurements possible, AFM can also be used to study protein folding. In these studies, the AFM tip is displaced toward the sample until a protein is attached by contact adhesion, the tip is then retracted. Force versus distance curves are recorded before and after adhesion in order to determine the forces involved in unfolding or “unzipping” the protein. In addition to the force versus distance curves, images can also be acquired before and after an unfolding event to observe the effects on the surrounding environment (85).
Recent structural studies
KirBac3.1
AFM is a powerful method of providing information on larger scale structure, especially when combined with higher resolution structural data. This fact is beautifully illustrated in the investigation of the KirBac3.1 potassium channel (76). AFM imaging of KirBac3.1 embedded in a lipid bilayer has revealed the tetrameric assembly of the protein (Fig. 4a and 4c) as well as large conformational changes upon ligand binding (Fig. 4b and 4d), thus providing insight into the gating mechanism of this channel.

Figure 4. High-resolution AFM images of the surface of the KirBac3.1 potassium channel in a 2-D crystal in the presence of EDTA (a) or Mg2+ (b) allow us to watch the channel open and close. The channel is in its open state in the absence of Mg2+, and that can be seen clearly from the image shown in (c), which reveals a central cavity ~30 A across. (d) This cavity is no longer visible in the image of KirBac3.1 in its closed state, which is a result of adding Mg2+. (Reprinted from Reference 76 with permission from Elsevier.)
F0F1-ATP synthase
The F0F1-ATP synthase is responsible for synthesizing ATP in many organisms. This enzyme is composed of two rotary motors (F0 and F1) connected by a central stem, and the stoichiometry of these two motors is of critical importance to energy conversion. F0F1-ATP synthases are very large, making them unsuitable for study by NMR or X-ray crystallography. However, AFM is ideally suited to such applications and has been used to determine the oligomeric states of the F0 and F1 motors in several species of bacteria and plants to shed new light on how the enzymes function (84, 86, 87).
Summary
The aim of this review has been to summarize the key techniques currently used for determination of membrane protein structures. Thus far, XRC has produced the largest number of membrane protein structures, and it has achieved the highest resolution of any other technique. However, this technique still requires a high-quality crystal, which is currently the rate-limiting step in the process. Steady progress in our understanding of crystal growth and the development of new crystallization methods show great potential for future studies. NMR has proved to be a valuable technique for the study of small membrane proteins and individual domains of larger proteins. In addition to 3-D structural information, NMR can also provide information on dynamics and ligand binding. NMR also has the advantage that it does not require crystals, which are difficult and time-consuming to produce, and instead uses a large range of membrane mimetics and solubilizing agents. The major limitations of NMR remain the upper limit on protein size and difficulties in resolving resonances for assignment purposes. Continued developments in instrumentation, experimental methods, and improved membrane mimetics will greatly advance the field. EM has also made a large impact on our knowledge of membrane proteins and promises to equal or surpass the more static structural methods of XRC and NMR. Because of recent new developments in technology, cryo-EM has caught up with XRC and NMR in terms of resolution and is well positioned to lead the field in the future. Both EM and AFM offer the unique advantage that membrane proteins can be studied in synthetic lipid bilayers as well as their native membranes, producing more biologically relevant structural data. Furthermore, these methods allow study of conformational changes over time, in response to such events as ligand binding or unfolding. However, the most exciting prospect for future studies of membrane proteins comes when data from all these methods are used in concert, alongside molecular dynamics simulations, to describe the structures and functions of these essential proteins.
References
1. Deisenhofer J, Epp O, Miki K, Huber R, Michel H. Structure of the protein subunits in the photosynthetic reaction centre of Rhodospeudomonas viridis at 3 A resolution. Nature 1985; 618-624.
2. Lundstrom K. Structural genomics for membrane proteins. Cell. Mol. Life Sci. 2006; 63:2597-2607.
3. Wiener MC. The development of membrane protein crystallization screens based upon detergent solution properties. Biophys. J. 2002; 82:29a.
4. Arnold T, Linke D. Phase separation in the isolation and purification of membrane proteins. BioTechniques 2007; 43:427-430.
5. Wiener MC. Existing and emergent roles for surfactants in the three-dimensional crystallization of integral membrane proteins. Curr. Opin. Coll. Int. Sci. 2001; 6:412-419.
6. Landau EM, Rosenbusch JP. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:14532-14535.
7. Gordeliy VI, Schlesinger R, Efremov R, Buldt G, Heberle J. Crystallization in lipidic cubic phases: a case study with bacteriorhodopsin. Methods Molec. Biol. 2003; 228:305-316.
8. Hunte C, Koepke J, Lange C, Rossmanith T, Michel H. Structure at 2.3 angstrom resolution of the cytochrome bc(1) complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure 2000; 8:669-684.
9. Zhou YF, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 angstrom resolution. Nature 2001; 414:43-48.
10. Doyle DA, Cabral JM, Pfuetzner RA, Kuo AL, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998; 280:69-77.
11. Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007; 318:1258-1265.
12. Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 2007; 449:316-323.
13. Morth JP, Pedersen BP, Toustrup-Jensen MS, Sorensen TL, Petersen J, Andersen JP, Vilsen B, Nissen P. Crystal structure of the sodium-potassium pump. Nature 2007; 450:1043-1049.
14. Charles M, Veesler S, Bonnete F. MPCD: a new interactive on-line crystallization data bank for screening strategies. Acta Cryst. D-Biol. Cryst. 2006; 62:1311-1318.
15. Gilliland GL, Tung M, Blakeslee DM, Ladner JE. Biological macromolecule crystallization database, version-3.0 - New features, data and the NASA archive for protein crystal-growth data. Acta Cryst. D-Biol. Cryst. 1994; 50:408-413.
16. Dauter Z. Efficient use of synchrotron radiation for macromolecular diffraction data collection. Prog. Biophys. Mol. Biol. 2005; 89:153-172.
17. Hendrickson WA, Horton JR, Lemaster DM. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD) - a vehicle for direct determination of 3-dimensional structure. EMBO J. 1990; 9:1665-1672.
18. Olesen C, Picard M, Winther AM, Gyrup C, Morth JP, Oxvig C, Moller JV, Nissen P. The structural basis of calcium transport by the calcium pump. Nature. 2007; 450:957-959.
19. Pedersen BP, Buch-Pedersen MJ, Morth JP, Palmgren MG, Nissen P. Crystal structure of the plasma membrane proton pump. Nature 2007; 450:1111-1114.
20. Tugarinov V, Choy W-Y, Orekhov VY, Kay LE. Solution NMR-derived global fold of a monomeric 82-kDa enzyme. Proc Natl Acad Sci U.S.A. 2005; 102:622-627.
21. Ketchem RR, Lee KC, Huo S, Cross TA. Macromolecular structural elucidation with solid-state NMR-derived orientational constraints. J. Biomol. NMR. 1996; 8:1-14.
22. Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from Influenza A virus. Biophys. J. 2007; 92:4335-4343.
23. Fu R, Cross TA. Solid-state nuclear magnetic resonance investigation of protein and polypeptide structure. Annu. Rev. Biophys. Biomol. Struct. 1999; 28:235-268.
24. Tycko R. Biomolecular solid state NMR: advances in structural methodology and applications to peptide and protein fibrils. Annu. Rev. Phys. Chem. 2001; 52:575-606.
25. Arora A, Abildgaard F, Bushweller JH, Tamm LK. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat. Struct. Biol. 2001; 8:334-338.
26. Bocharov EV, Pustovalova YE, Pavlov KV, Volynsky PE, Goncharuk MV, Ermolyuk YS, Karpunin DV, Schulga AA, Kirpichnikov MP, Efremov RG, Maslennikov IV, Arseniev AS. Unique dimeric structure of BNip3 transmembrane domain suggests membrane permeabilization as a cell death trigger. J. Biol. Chem. 2007; 282:16256-16266.
27. Marassi FM, Opella SJ. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci. 2003; 12:403-411.
28. Krueger-Koplin RD, Sorgen PL, Krueger-Koplin ST, Rivera-Torres IO, Cahill SM, Hicks DB, Grinius L, Krulwich TA, Girvin ME. An evaluation of detergents for NMR structural studies of membrane proteins. J. Biomol. NMR. 2004; 28:43-57.
29. Fillingame RH, Dmitriev OY. Structural model of the transmembrane F0 rotary sector of H+-transporting ATP synthase derived by solution NMR and intersubunit cross-linking in situ. Biochim. Biophys. Acta 2002; 1565:232-245.
30. Prosser RS, Evanics F, Kitevski JL, Al-Abdul-Wahid MS. Current applications of bicelles in NMR studies of membrane-associated amphiphiles and proteins. Biochemistry 2006; 45:8453-8465.
31. Gorzelle BM, Hoffman AK, Keyes MH, Gray DN, Ray DG, Sanders CR. Amphipols can support the activity of a membrane enzyme. J. Am. Chem. Soc. 2002; 124:11594-11595.
32. McGregor CL, Chen L, Pomroy NC, Hwang P, Go S, Chakrabartty A, and Prive, GG. Lipopeptide detergents designed for the structural study of membrane proteins. Nat. Biotechnol. 2003; 21:171-176.
33. Zoonens M, Catoire LJ, Giusti F, Popot JL. NMR study of a membrane protein in detergent-free aqueous solution. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:8893-8898.
34. Nath A, Atkins WM, Sligar SG. Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry 2007; 46:2059-2069.
35. Boyle-Roden E, Hoefer N, Dey KK, Grandinetti PJ, and Caffrey M. High resolution 1H NMR of a lipid cubic phase using a solution NMR probe. J. Magn. Reson. 2007; 189:13-19.
36. Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:12366-12371.
37. Grzesiek S, Anglister J, Ren H, Bax A. C-13 line narrowing by H2 decoupling in H2/C13/N15-enriched proteins - Application to triple-resonance 4D J-connectivity of sequential amides. J. Am. Chem. Soc. 1993; 115:4369-4370.
38. Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CR, Prive GG, Bishop RE, Kay LE. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:13560-13565.
39. Oxenoid K, Kim HJ, Jacob J, Sonnichsen FD, Sanders CR. NMR assignments for a helical 40 kDa membrane protein. J. Am. Chem. Soc. 2004; 126:5048-5049.
40. Bax A. Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci. 2003; 12:1-16.
41. Douglas SM, Chou JJ, Shih WM. DNA-nanotube-induced alignment of membrane proteins for NMR structure determination. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:6644-6648.
42. Kamen DE, Cahill SM, Girvin ME. Multiple alignment of membrane proteins for measuring residual dipolar couplings using lanthanide ions bound to a small metal chelator. J. Am. Chem. Soc. 2007; 129:1846-1847.
43. Liang B, Bushweller JH, Tamm LK. Site-directed parallel spinlabeling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy. J Am Chem Soc. 2006; 128:4389-4397.
44. Mesleh MF, Opella SJ. Dipolar waves as NMR maps of helices in proteins. J. Magn. Reson. 2003; 163:288-299.
45. Cierpicki T, Liang BY, Tamm LK, Bushweller JH. Increasing the accuracy of solution NMR structures of membrane proteins by application of residual dipolar couplings. High-resolution structure of outer membrane protein A. J. Am. Chem. Soc. 2006; 128:6947-6951.
46. Park SH, Mrse AA, Nevzorov AA, Mesleh MF, Oblatt-Montal M, Montal M, Opella SJ. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein “u” (Vpu) from HIV-1. J. Mol. Biol. 2003; 333:409-424.
47. Howell SC, Mesleh MF, Opella SJ. NMR structure determination of a membrane protein with two transmembrane helices in micelles: MerF of the bacterial mercury detoxification system. Biochemistry 2005; 44:5196-5206.
48. Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice, LM, Simonson, T, and Warren, GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Cryst. D-Biol. Cryst. 1998; 54:905-921.
49. Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003; 160:65-73.
50. Roosild TP, Greenwald J, Vega M, Castronovo S, Riek R, Choe S. NMR structure of Mistic, a membrane-integrating protein for membrane protein expression. Science 2005; 307:1317-1321.
51. Oxenoid K, Chou JJ. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:10870-10875.
52. Potluri S, Yan AK, Chou JJ, Donald BR, Bailey-Kellogg C. Structure determination of symmetric homo-oligomers by a complete search of symmetry configuration space, using NMR restraints and van der Waals packing. Proteins 2006; 65:203-219.
53. Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two dimensional crystals of aquaporin-0. Nature 2005; 438:633-638.
54. Henderson R, Unwin PN. Three dimensional model of purple membrane obtained by electron microscopy. Nature 1975; 257:28-32.
55. Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 1990; 213:899-929.
56. Hiroaki Y, Kazutoshi T, Kamegawa A, Gyobo N, Nishikawa K, Suzuki H, Walz T, Sasaki S, Mitsuoka K, Kimura K, Mizoguchi A, Fujiyoshi Y. Implications of the aquaporin-4 structure on array formation and cell adhesion. J. Mol. Biol. 2006; 355:628-639.
57. Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature 2003; 423:949-955.
58. Samso M, Shen X, Allen PD. Structural characterization of the RyR1-FKBP12 interaction. J. Mol. Biol. 2006; 356:917-927.
59. Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 2005; 346:967-989.
60. Orlova EV, Papakosta M, Booy FP, van Heel M, Dolly JO. Voltage-gated K+ channel from mammalian brain: 3D structure at 18A of the complete (alpha)4(beta)4 complex. J. Mol. Biol. 2003; 326:1005-1012.
61. Serysheva II, Ludtke SJ, Baker MR, Chiu W, Hamilton SL. Structure of the voltage-gated L-type Ca2+ channel by electron cryomicroscopy. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:10370-10375.
62. Ubarretxena-Belandia I, Baldwin JM, Schuldiner S, Tate CG. Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. EMBO J. 2003; 22:6175-6181.
63. Holm PJ, Bhakat P, Jegerschold C, Gyobu N, Mitsuoka K, Fujiyoshi Y, Morgenstern R, Hebert H. Structural basis for detoxification and oxidative stress protection in membranes. J. Mol. Biol. 2006; 360:934-945.
64. Henderson, R. Realizing the potential of electron cryo-microscopy. Rev. Biophys. 2004; 37:3-13.
65. Jiang QX, Thrower EC, Chester DW, Ehrlich BE, Sigworth FJ Three-dimensional structure of the type 1 inositol 1,4,5-trisphosphate receptor at 24 angstrom resolution. EMBO J. 2002; 21: 3575-3581.
66. Lacapere JJ, Pebay-Peyroula E, Neumann JM, Etchebest C. Determining membrane protein structures: still a challenge! Trends Biochem. Sci. 2007; 32:259-270.
67. Lacapere JJ, Stokes DL, Olofsson A, Rigaud JL. Two-dimensional crystallization of Ca-ATPase by detergent removal. Biophys. J. 1998; 75:1319-1329.
68. Amos LA, Henderson R, Unwin PN. Three-dimensional structure determination by electron microscopy of two-dimensional crystals. Prog. Biophys. Mol. Biol. 1982; 39:183-231.
69. Baldwin JM, Henderson R, Beckman E, Zemlin F. Images of purple membrane at 2.8 A resolution obtained by cryo-electron microscopy. J. Mol. Biol. 1988; 202:585-591.
70. Walz T, Smith BL, Zeidel ML, Engel A, Agre P. Biologically active two-dimensional crystals of aquaporin CHIP. J. Biol. Chem. 1994; 269:1583-1586.
71. Ma C, Chang G. Structure of the multidrug resistance efflux transporter EmrE from Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:2852-2857.
72. Subramaniam S, Lindahl M, Bullough P, Faruqi AR, Tittor J, Oesterhelt D, Brown L, Lanyi J, Henderson R. Protein conformatinal changes inthe bacteriorhodopsin photocycle. J. Mol. Biol. 1999; 287:145-161.
73. Vinothkumar KR, Smits SH, Kuhlbrandt W. pH-induced structural change in a sodium/proton antiporter from Methanococcus jannaschii. EMBO J. 2005; 24:2720-2729.
74. Binnig G, Quate CF, Gerber C. Atomic force microscope. Phys. Rev. Letters. 1986; 56:930-933.
75. Butt H-J, Downing KH, Hansma PK. Imaging the membrane protein bacteriorhodopsin with the atomic force microscope. Biophys. J. 1990; 58:1473-1480.
76. Jaroslawski S, Zadek B, Ashcroft F, Venien-Bryan C, Scheuring S. Direct visualization of KirBac3.1 potassium channel gating by atomic force microscopy. J. Mol. Biol. 2007; 374:500-505.
77. Muller DJ, Fotiadis D, Scheuring S, Muller SA, Engel A. Electrostatically balanced subnanometer imaging of biological specimens by atomic force microscopy. Biophys. J. 1999; 76:1101-1111.
78. Persike N, Pfeiffer M, Guckenberger R, Radmacher M, Fritz M. Direct observation of different surface structures on high-resolution images of native halorhodopsin. J. Mol. Biol. 2001; 310:773-780.
79. Scheuring S, Seguin J, Marco S, Levy D, Breyton C, Robert B, Rigaud JL. AFM characterization of tilt and intrinsic flexibility of Rhodobacter sphaeroides light harvesting complex 2 (LH2). J. Mol. Biol. 2003; 325:569-580.
80. Walz T, Tittmann P, Fuchs KH, Muller DJ, Smith BL, Agre P, Gross H, Engel A. Surface topographies at subnanometer-resolution reveal asymmetry and sidedness of Aquaporin-1. J. Mol. Biol. 1996; 264:907-918.
81. Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. Rhodopsin dimers in native disc membranes. Nature 2003; 421:127-128.
82. Scheuring S, Sturgis JN. Chromatic adaptation of photosynthetic membranes. Science 2005; 309:484-487.
83. Sapra KT, Besir H, Oesterhelt D, Muller DJ. Characterizing molecular interactions in different Bacteriorhodopsin assemblies by single-molecule force spectroscopy. J. Mol. Biol. 2006; 355: 640-650.
84. Seelert H, Poetsch A, Dencher NA, Engel A, Stalhberg H, Muller DJ. Proton-powered turbine of a plant motor. Nature 2000; 405:418-419.
85. Oesterhelt F, Oesterhelt D, Pfeiffer M, Engel A, Gaub HE, Muller DJ. Unfolding pathways of individual bacteriorhodopsins. Science 2000; 288:143-146.
86. Pogoryelov D, Yu J, Meier T, Vonck J, Dimroth P, Muller DJ. The c15 ring of the Spirulina platensis F-ATP synthase: F1/F0 symmetry mismatch is not obligatory. EMBO Rep. 2005; 6:1040-1044.
87. Stahlberg H, Muller DJ, Suda K, Fotiadis D, Engel A, Matthey U, Meier T, Dimroth P. Bacterial (Na+)-ATP synthase has an undecameric rotor. EMBO Rep. 2001; 2:229-233.
Further Reading
Caffrey M. Membrane protein crystallization. J. Struct. Biol. 2003; 142:108-132.
De Angelis AA, Jones DH, Grant CV, Park SH, Mesleh MF, Opella SJ. NMR experiments on aligned samples of membrane proteins. Meth. Enzymol. 2005; 394:350-382.
Engel A. Robert Feulgen Lecture. Microscopic assessment of membrane protein structure and function. Histochem. Cell Biol. 2003; 120:93-102.
Fernandez C, Wider G. TROSY in NMR studies of the structure and function of large biological macromolecules. Current opinion in structural biology. 2003; 13:570-580.
Janovjak H, Kedrov A, Cisneros DA, Sapra KT, Struckmeier J, Muller DJ Imaging and detecting molecular interactions of single transmembrane proteins. Neurobiol. Aging 2006; 27:546-561.
McPherson A. Introduction to protein crystallization. Methods 2004; 34:254-265.
Mosser G. Two-dimensional crystallogenesis of transmembrane proteins. Micron. 2001; 32:517-540.
Muller DJ, Sapra KT, Scheuring S, Kedrov A, Frederix PL, Fotiadis D, Engel, A. Single-molecule studies of membrane proteins. Curr. Opin. Struct. Biol. 2006; 16:489-495.
Prive GG. Detergents for the stabilization and crystallization of membrane proteins. Methods 2007; 41:388-397.
Qian B, Raman S, Das R, Bradley P, McCoy AJ, Read RJ, Baker D. High-resolution structure prediction and the crystallographic phase problem. Nature 2007; 450:259-264.
Sanders CR, Sonnichsen F. Solution NMR of membrane proteins: practice and challenges. Magn. Reson. Chem. 2006; 44:S24-40.
Smyth MS, Martin JHJ. X-ray crystallography. J. Clin. Pathol.-Mol. Pathol. 2000; 53:8-14.
Tamm LK, Liang B. NMR of membrane proteins in solution. Prog. Nucl. Magn. Reson. Spectr. 2006; 201-210.
Watts A, Straus SK, Grage SL, Kamihira M, Lam YH, Zhao X. Membrane protein structure determination using solid-state NMR. Meth. Mol. Biol. 2004; 278:403-473.
Werten PJ, Remigy HW, de Groot BL, Fotiadis D, Philippsen A, Stahlberg H, Grubmuller H, Engel A. Progress in the analysis of membrane protein structure and function. FEBS Letts. 2002; 529: 65-72.
Wiener MC. A pedestrian guide to membrane protein crystallization. Methods 2004; 34:364-372.
See Also
Protein Expression, Systems for
Solubilize Membrane Proteins, Techniques to
Crystallization of Proteins, Overview of Applications in Chemical Biology
NMR for Proteins
Imaging Techniques for Proteins