CHEMICAL BIOLOGY
Platinum Anticancer Drugs, Chemical Biology of
Jenny Zhang, School of Chemistry, The University of Sydney
Renee Whan, The Electron Microscopy Unit, The University of Sydney
Trevor Hambley, School of Chemistry, The University of Sydney
doi: 10.1002/9780470048672.wecb657
In the past, studies into the distribution of platinum anti-cancer agents inside cancer cells were experimentally challenging because of a lack of readily available and sensitive platinum mapping techniques. Recent advancements in the area of platinum mapping in cells have meant that the biologic fate of cisplatin and its derivatives can be studied in several new ways. In particular, electron microscopy, fluorescence microscopy, and synchrotron radiation induced X-ray emission have made significant contributions in the study of cisplatin in vitro. The applications of these three techniques and the insights they provide into the intracellular distributions and interactions platinum complexes are reviewed here. The strengths and weaknesses of each technique and their potentials for additional use in this field of research have been evaluated.
Introduction
Platinum-based anticancer drugs including cisplatin, carboplatin, and oxaliplatin are the most widely used and effective chemotherapeutic agents in the current arsenal (1, 2). They are believed to exert their action by binding to nuclear DNA that leads ultimately to the affected cell undergoing apoptosis (3). To reach the nucleus, the complex must traverse the cell membrane, the cytoplasm, and the nuclear membrane to avoid the detoxification mechanisms of the cell along the way. Less than 1% of the platinum that enters the cell successfully makes this journey and, consequently, the great bulk of the platinum suffers other fates (4, 5). Determining how the cell deals with platinum is important for understanding and for overcoming unwanted toxicities and inherent and acquired resistance, but is experimentally challenging
Biologic Background
Although the cytotoxicity of cisplatin (and its derivatives) is accepted widely to be a result of its interactions with DNA, much is still unknown about the interactions of cisplatin on a cellular level. To begin with, the intracellular uptake of cisplatin is still the subject of much debate. Some studies have suggested that cisplatin diffuses into cells passively, as uptake has been observed to be not saturable, not dependent on pH, and not inhibited by coadministration of structural analogs (6, 7). Other studies have argued that some component of cisplatin uptake is carrier mediated, as cisplatin accumulation is modulated by ATP, membrane potential, ion concentrations, and several transportomes, which includes the copper influx transporter CTR1 and the ATP dependent copper efflux transporter ATP7B (8-15). Indeed, it has been observed that cisplatin accumulation is reduced in many cisplatin-resistant cell lines (16-18). Hence, the mode of platinum influx and efflux in cells plays an important part in drug resistance, and it needs to be understood better.
Another area that requires additional understanding is the role of cytoplasmic organelles in the fate of platinum drugs. Platinum has been shown to be localized within the Golgi apparatus, endoplasmic reticulum, mitochondria, and lysosomes, the cytosol, and the nucleus. However, only the consequences of nuclear localization have been defined satisfactorily (19-21).
It has been observed that the Golgi apparatus and the endoplasmic reticulum change in morphology after cisplatin treatment. One explanation for this change is that protein translation has been inhibited after cisplatin localization in these organelles (22, 23). Another suggestion is that these organelles contain enzymes and proteins that are linked directly to mechanisms of cisplatin induced resistance, including glutathione-S-transferase and metallotheins (21, 24, 25). Hence, the changes observed in these organelles may be caused by the participation of these enzymes in the deactivation of cisplatin.
It is well documented that the aquated form of cisplatin localizes within the mitochondria, and that the inhibition of respiration and spontaneous release of calcium ions into the cytoplasm accompanies this localization. Furthermore, it has been shown that the number of mitochondrial DNA-platinum adducts is greater than that of genomic DNA adducts. However, some studies have shown that the damage done to the mitochondria is subsequent to lysosomal and nuclear localization, which suggests that this organelle has only a secondary role in cisplatin-mediated cytotoxicity (23, 26-30).
The involvement of lysosomes in the action of cisplatin is also of interest. Cells that have been treated with cisplatin have an increased number and size of lysosomes, suggesting that they may play an important role in detoxification of the drug (8). In other studies, this organelle has been implicated in the mechanisms of resistance, with several alterations that have been identified in cisplatin-resistant cell lines. These alterations include a decreased number of the organelles, a more alkaline lumen, and altered recycling of the luminal contents between the endosomal and lysosomal compartments (31, 32). Interestingly, lysosomes have also been implicated as a potential site of induced cytotoxicity. Endogenous and/or exogenous stress can cause the membrane of the lysosome to permeabilize and release hydrolases. If this release is uncontrolled, it can lead to necrosis and selective release of two classes of hydrolases (sphingolipidases and cathepsins) that can trigger the so-called “extrinsic” pathway to apoptosis. Abnormalities in lysosomal cathepsins (found in many cancer types) can degrade the extracellular matrix of the cell and lead to tumor metastasis (33-37).
Thus, the mode of platinum entry into cells and its subsequent intracellular pathways and interactions with subcellular organelles are defined poorly. Addressing where and how platinum drugs are processed in cells could greatly help elucidate the cause of unwanted toxicities as well as the phenomena of drug-resistance, ultimately improving drug design. In the past, techniques such as cell fractionation, atomic absorption spectroscopy, and light and electron microscopy were employed to study the distribution of cisplatin in vitro (8, 19, 21, 30). A lack of readily available, accurate, and sensitive platinum-mapping techniques limited the research in this area. In the past decade, new and improved techniques have emerged for the detection of platinum in cells. Here, we review and evaluate recent developments in the study of the fate and distribution of platinum complexes in cancer cells, achieved through the use of three important imaging techniques: electron microscopy, fluorescence imaging, and synchrotron radiation induced X-ray emission.
Platinum Imaging Techniques
Transmission electron microscopy
Transmission electron microscopy (TEM) has been used extensively in biology for direct visualization of ultrastructural details and platinum deposits in cells. The underlying physics behind TEM is similar to that of ordinary light microscopy; however, the resolutions achieved by TEM can be some 400-fold greater than that of light microscopy (38). Briefly, the mechanics behind TEM involves an illuminating source, the electron gun, that sends a beam of electrons through a vacuum and onto the specimen of interest. An image is formed according to the differential absorption of electrons by various parts of the specimen; this image is amplified via a series of electromagnetic or electrostatic lenses and is projected onto a fluorescent screen. As all manufactured transmission electron microscopes guarantee a resolution of 0.344 nm, TEM is particularly useful in cellular biology, where resolutions of around 2 nm (the width of one third of a mitochondrial membrane) are needed (38, 39).
The preparation of biologic samples for TEM involves several steps. First, the sample is fixed, usually chemically, to preserve the structural integrity and the spatial information of the cells while simultaneously terminating any biologic activity. After fixation, the sample is buffered, dehydrated, and embedded in an epoxy resin, then sectioned into thin slices (that range from 80 to 800 nm) and stationed onto a grid, ready for examination by the electron microscope. For specific staining of cellular components, immunolabeling can be employed; the contrast between cellular components or the background can be heightened by staining with heavy metal salts, such as those of uranium, lead, and osmium, all of which have sufficient mass and electron density to scatter the electron beam (40).
The high resolution attainable by TEM has led to its use in biologic platinum studies in two ways; to reveal ultrastructural morphologic changes in drug treated biological samples and to map directly the platinum moiety in cells.
Several studies have used TEM to examine morphologic features of organ tissues, sarcoma, and tumour cells, before and after cisplatin treatment to gain insights into its toxicity and mode of action (41, 42). Among the more recent findings from these fine-structure studies was the observation that the morphology of rat kidney tissues changes after cisplatin treatment in vivo (8). It was observed that cisplatin induced a large increase in the number and the size of lysosomes in the cells of the kidney, which suggests that lysosomal sequestration is a mechanism used by the kidney for the removal of platinum and other heavy metals (8, 43). Other similar fine-structure studies revealed significant changes in mouse kidney morphology after cisplatin incubation, with observed features such as necrotic proximal tubules, damaged tubular basement membrane, and short irregular microvilli (44).
In in vitro studies, Yang et al. (26) found that the morphology of the mitochondria to be changed significantly after one and 4 hour incubations with 50 μM cisplatin on the head and neck squamous cell carcinoma cell line (HSNSCC). The mitochondria showed swelling, distortion of the cristae as well as the inner and outer membranes, whereas the nucleus and nucleoli remained unchanged. This finding led to the authors to challenge the notion that the site of action of cisplatin is solely with nuclear DNA, which suggests that cisplatin could act directly on the mitochondria to cause the release of cytochrome c trigger in the apoptotic cascade (26). In another study conducted by Meijera et al. (45) on human small cell lung carcinoma cancer cells (GLC4), a high nucleus to cytoplasm ratio was observed after 4 hour incubations with 333 μM cisplatin. Furthermore, similar cisplatin treatments on the cisplatin resistant sub-line, GLC-CDDP, were shown to cause swelling of the Golgi apparatus and an increase in the numbers of mitochondria (45).
Early attempts to map the distribution of the electron-rich platinum moieties inside cells employing TEM have met with some success, with platinum(II)-pyrimidine complexes being amongst the first platinum species to be mapped (46). In this study, it was observed that cells stained with platinum(II)- thymine/uracil complexes, prepared as 1% solutions, showed platinum staining in the chromatin, nucleolus, and ribosomes with high selectivity. However, it should be noted that the concentration present in a 1% platinum complex solution is much greater than those in normal experimental dosages. In a separate study by Khan and Sadler (47), gray electron dense areas were also observed to be localized primarily within the nucleus and nucleolus of human epithelial carcinoma (HeLa) cells after 4 hour incubations with high concentrations of cisplatin (200 μM). These gray areas were later confirmed by electron-probe X-ray analysis to contain platinum (47).
In later TEM studies by Ghadially et al. (48), a lower concentration of cisplatin (30 μM) was incubated in HeLa and human lymphoblastoid (RPMI 6410) cells for periods that ranged from 1 hour to 4 days. No intracellular platinum was detected in either cell line. However, similar incubations with platinum(II)-uracil resulted in development of lysosome-like bodies in the cytosol, which are referred to as “platinosomes,” that contain electron dense species identified by X-ray analysis as platinum (48). In a related study, similar concentrations of cisplatin were injected into rabbit knee joints and incubated for several days. Again, no platinum was detected in the intracellular or extracellular compartments of the synovial cells, whereas platinum was observed to accumulate only in the “platinosomes” after platinum-uracil incubation (49). This set of studies highlights the effect of incubation concentrations of the drug on the cellular distribution results, as well as, drawing attention to the uptake of some platinum(II) complexes in cytoplasmic organelles.
In more recent attempts to map the cellular distribution of cisplatin, Beretta et al. (50) incubated human ovarian cancer cells (A2780) with cisplatin concentrations of up to 100 μM for 30 minutes and observed electron dense spots, identified as large platinum deposits, distributed in the cell cytoplasm and nucleus. Additionally, it was observed that the platinum deposits made blunt contacts with the plasma membrane, which suggests that the cellular influx of cisplatin is through an endocytosis-independent manner that is consistent with the passive diffusion theory of cisplatin uptake (50).
Recent developments in TEM have resulted in the emergence of immunoelectron microscopy, in which antibodies labeled with gold particles are employed to detect certain intracellular antigens under the electron microscope. Using this new technique, intracellular platinum-intrastrand cross-links, formed in GLC4 cells after 4-hour cisplatin (0-333 μM) incubations, were identified by the employment of a GPt antibody stain. This stain binds to Pt-DNA adducts selectively and quantitative analysis could be performed by counting the gold labels per cm2 of area of interest on the images of three or more individual cells. It was found that the distribution of Pt-DNA adducts was the same for both GLC4 and GLC4-CDDP cell lines, with adducts detected in the nucleus (preferentially at loci with high-density chromatin), and in the mitochondria. DNA platination was found to be highest in apoptotic and dividing cells (45). It should be noted that again, high cisplatin incubation concentrations were required to acquire accurate quantitative Pt-adduct formation data.
Overall, TEM has proven to be a valuable tool for biologic platinum studies. It can provide qualitative information, with excellent resolution, regarding the intracellular sites of platinum interactions and accumulations. Quantitative information regarding certain structures, such as Pt-adducts, can now be acquired. Despite these advantages, limitations exist in the use of TEM as a mapping technique for platinum. TEM cannot attain visual information of living samples, and the process of fixing, dehydration, and resin embedding can significantly affect the distribution of the platinum species inside cells. Furthermore, TEM lacks the sensitivity to detect single platinum moieties accurately, mainly because of the damage caused to the ultrathin sample sections by the high-energy beams (51). Hence, TEM is limited to the detection of clusters or deposits of platinum metals; sparse distributions of platinum remain undetected. Additionally, the cisplatin incubation concentrations required for the detection of platinum deposits and Pt-adducts in cells are much greater than normal experimental dosages. This finding could produce misleading results in relation to the intracellular distribution of platinum.
Fluorescence imaging
Currently, fluorescence microscopy is one of the most useful techniques for the examination of drug localization in fixed and live cells. However, to use this technique, the drug must be fluorescent, or labeled with a fluorescent tag.
Two forms of fluorescence microscopy are observed in the literature: wide-field epifluorescence microscopy and laser scanning confocal microscopy. Briefly, the mechanics behind both techniques involves irradiation of the sample with high-energy photons, which excite appropriate fluorophores to high-energy states. After excitation, the fluorophores undergo relaxation in the forms of nonradiative transitions between excited states, and this energy can be lost as heat to the solvent. Fluorescence results as the fluorophore relaxes even more back to its ground state that emits a photon with a longer wavelength than the original excitation photon. In both techniques, this process is performed on a light microscope platform. The two fluorescent techniques differ in how the resultant fluorescence is collected and initially excited. In wide-field epifluorescence microscopy, the sample is illuminated using a high-energy light source, such as a Hg or Xe lamp, and the emitting photons are collected and amplified using a charged coupled device camera. The resulting images can be in “red color;” however, this technique collects emitted fluorescence across a large depth of field, including light outside the focal plane, which limits axial resolution to ~1 |lm. In confocal microscopy, the sample is excited by scanning with a laser, and the emitting photons are collected and amplified using photon multiplier tubes. The resulting images are in “gray scale;” however, the additional employment of a pinhole positioned before the detector eliminates out of focus light, which enables resolutions of around 0.1-0.2 μm in the XY plane. This method allows the sample to be “optically sectioned” without physical sectioning, which enables three-dimensional images to be created (52, 53).
Several advantages exist in using fluorescence microscopy in drug distribution studies. Unlike TEM, fluorescence microscopy is a noninvasive technique that can acquire three-dimensional images of living cells in real time with minimal preparation. Additionally, the spatial distribution of fluorescence within cells can be detected with high sensitivity and signal specificity; hence, both qualitative and quantitative data can be acquired at relatively low drug incubation concentrations. Furthermore, intracellular organelles can be labeled with fluorescent organelle specific stains to identify the sites of drug accumulation in co-localization experiments (52, 54).
Recently, platinum agents have been labeled with various fluorescent tags for fluorescence microscopy studies. The earliest work of this kind was conducted by Molenaar et al. (55), who tagged a cisplatin derivative with a carboxyfluoroscein diacetate (CFDA) moiety. The CFDA ligand is a nonfluorescent derivative of fluorescein and the CFDA-Pt complex becomes fluorescent following cellular uptake and acetate hydrolysis. In this study, human osteosarcoma cells (U2-OS) were incubated for 30 minutes in 10 μM CFDA-Pt. Using wide-field epifluorescence microscopy, the complex was observed to distribute initially throughout the entire cell, and then accumulate in the nucleus to form fine granular patterns at 3 hours after incubation. After 8 and 72-hour incubations with the complex, localization within the Golgi apparatus was identified through colocalization experiments employing a Golgi fluorescent probe. After 24 hours, no significant fluorescence was observed in the nuclei of the cells, which suggests that the fluorophore had undergone efflux from the organelle. The fluorescence distribution of the free CFDA fluorophore proved to be very different from the complex, which suggests that the platinum complex is not mimicking the behavior of the fluorophore (55). A later study by the same group (56) found that the treatment of biologically active dinuclear platinum complexes labeled with the same CFDA moiety in U2-OS cell lines shows very similar localization to the mononuclear CFDA-Pt complex (56). Therefore, fluorescence microscopy has allowed for the intracellular pathway of CFDA tagged platinum complexes to be deduced and subsequently, identified the Golgi as having a significant role in the cellular processing of these types of complexes.
In a similar study, Safaei et al (57) studied the distribution of a fluorescein-labeled cisplatin analog (FDDP) using epifluorescent microscopy in fixed human ovarian carcinoma cells (2008). This study showed the complex was localized within the cytoplasm after a 5 minute incubation with 2 μM F-DDP. After 10 to 30 minutes, fluorescence was observed in the nuclear regions. Like the Pt-CFDA complex, F-DDP also displayed fine granular patterns in the nucleus, and it was distributed in discrete vesicular structures, not localized diffusely throughout the cytoplasm. Staining with organelle-specific markers revealed that F-DDP was sequestered into the lysosomes, Golgi, and secretory compartments of the cells. The roles of these subcellular organelles in the processing of the complex were investigated by the use of various pathway inhibitors. These biologic tools in conjunction with fluorescence microscopy revealed that the secretory pathways, involving the Golgi and lysosomes played an important role in intracellular trafficking of these tagged complexes, particularly with regard to drug efflux (57).
In another fluorescence microscopy study, the trafficking pathways of a fluorescently tagged Alexa Fluor-platinum complex in a cisplatin-sensitive human KB epidermoid carcinoma cell line (KB-3-1) and its cisplatin-resistant subline (KB-CP-r) were compared. During the first 30 minutes of the incubation, the fluorescence from the complex moved from the membrane and into the Golgi apparatus; after 2 hours, the complex was distributed throughout the cell, including nuclear compartments. Internalization of the fluorescent complexes was very different in the cisplatin resistant subline, where the overall accumulation of the complex was lower, as determined by decrease of cellular fluorescence by flow cytometry. Only punctate cytoplasmic staining was observed after incubations, with little fluorescence in the cell nucleus. The altered distribution suggests that cellular resistance could be caused by modified trafficking, possibly involving the failure of cell membrane proteins that bind cisplatin (58).
From these studies, it can be observed that fluorescence microscopy can indeed be a powerful technique for studies into the biologic fate of platinum anticancer agents. It not only provides qualitative and quantitative distribution data, but also offers the option of real-life time imaging, which reveals cellular processes in action. Despite the usefulness of fluorescence imaging in distribution studies, inherent disadvantages exist with this technique. For example, it is difficult to ascertain whether the fluorescence observed is from the fluorophore-platinum complex or the fluorophore alone (should cleavage occur in the biologic environment). In addition, the attachment of the fluorophore creates a more lipophilic compound, and entry into the cell and the distribution will differ from the properties of the platinum complex alone, which affects such factors as cellular uptake (59, 60). Furthermore, a highly conjugated, aromatic fluorophore may act as an intercalator, which generates different reaction profiles with DNA. Hence, caution should be used when evaluating the distributions of fluorescence in cells.
The disadvantage associated with altered distribution because of the fluorescence tag does not apply when the drugs concerned are intrinsically fluorescent platinum species, such as those of the platinum-anthraquinone complexes. Platinum-anthraquinone complexes were designed to increase the DNA affinity of the platinum moiety by its chemical linkage to a bio-carrier, in this case an intrinsically fluorescent DNA intercalator (61, 62). In a series of time-lapse fluorescence microscopy studies conducted by Reedijk et al. (63, 64), the cellular distribution of a group of dinuclear cationic platinum-anthraquinone complexes was observed in live parental A2780 cells. At the low incubation concentration of 3 μM, the complexes were observed to enter the cells slowly, with fluorescence predominantly being localized within the cytosol after 2 hours. After 24 hours, the complexes were localized in the lysosomes whereas the free ligand was found to be additionally localized in the nucleus. In contrast, studies using the same complexes in the cisplatin resistant A2780 subline show that the complexes were encapsulated by the lysosomes at the beginning of their incubation, which suggests that lysosomal sequestration plays an important role in the resistance profile of A2780 cells. However, additional DNA titration studies found that the fluorescence of the anthraquinone was quenched in the presence of DNA; hence, any nuclear localization of the drug may not have been detected (63, 64).
Additional studies using the same complexes in both cisplatin sensitive and resistant forms of U2-OS cells also showed differences in the cellular processing of the drugs; both the free ligands and platinum complexes were rapidly found to accumulate in the nucleus, after uptake into the Golgi, which was hypothesized to be involved in transport of the platinum complexes out of the cells (56). In this set of studies, fluorescence imaging helped to illustrate the differences in the intracellular interactions of dinuclear platinum-anthraquinone complexes in different cell lines and in the different resistance profiles of A2780 and U2-OS resistant sub lines.
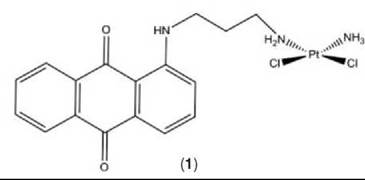
In a separate fluorescence microscopy study, the distribution of a mononuclear neutral platinum-anthraquinone complex, Pt-1 C3 (1), was examined in human colon adenocarcinoma cells (DLD-1) and A2780 cells (see Fig. 1). In both cell lines, fluorescence was observed to be predominantly localized within lysosomes after 1-, 4-, and 24-hour incubations with the complex, with no colocalization with mitochondrial and nucleic acid fluorescent probes. These results were in conflict with the high cytotoxicity profile observed for the platinum complex and cellular uptake studies that showed high platinum content within the nucleus after 4 hour incubations (65). Several possible reasons exist for the lack of fluorescence detected in the nucleus. The amount of DNA binding may be too low to detect by con- focal techniques. Alternatively, the platinum cytotoxin could be cleaved from the fluorescent anthraquinone part of the complex and is still entering the nucleus. Last, quenching of the molecules fluorescence could have occurred through reaction with an endogenous molecule such as DNA. Hence, although the bio-distribution of these intrinsically fluorescent drugs will not be altered because fluorescence tagging is unnecessary, limitations still exist in the application of fluorescence imaging in the mapping of these intrinsically fluorescent platinum complexes in cells.
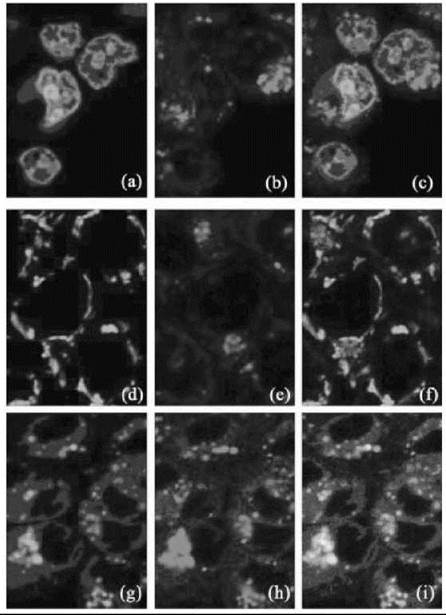
Figure 1. ''Going down: Nucleic acid probe, mitochondrial probe, lysosomal probe. Going across: Probe, drug, merge of probe and drug. Co-localisation images of cells incubated in a) SYTO-21 nucleic acid stain, b) Pt1C3 for 2-4 hours c) both nucleic acid stain and Pt1C3; d) MitoTracker Green mitochondrial stain, e) Pt1C3 for 2-4 hours, f) both mitochondrial stain and Pt1C3; and g) LysoTracker Green lysosomal stain, h) Pt1C3 for 2-4 hours, i) both lysosomal stain and Pt1C3. Figure reproduced with permission from Reference [65] with kind permission from ScienceDirect, Elsevier.''
A recently embraced technique in the monitoring of platinum in cells is that of synchrotron radiation induced X-ray emission (SRIXE). SRIXE is essentially synchrotron-based X-ray fluorescence microscopy and allows for the accurate two-dimensional analysis of elemental distribution within biologic samples (provided that the element is heavier than fluorine). The high intensity of X-rays derived from third generation synchrotron sources gives rise to the high sensitivity of platinum detection, with detection possible down to 5-10 ppm, and the high spatial resolution of about 0.15 x 0.15 μm2 (66-68). At this level of elemental sensitivity, SRIXE has advantages over electron microscopy because the detection of platinum is possible at much lower drug concentrations. Furthermore, the location of the unmodified platinum complex can be monitored directly using SRIXE, and it is not merely inferred indirectly by the imaging of chemically modified analogs of the drug as done in fluorescence microscopy studies. Additionally, it is not necessary to fix or to section samples for SRIXE because of the highly penetrating nature of hard X-rays (photons with energy > 1 keV), and the samples can be studied relatively close to their natural hydrated state (with some cryogenic treatments) (69).
Briefly, the principle behind SRIXE involves the focusing of a beam of hard X-rays from a synchrotron source to a submicron area section on a sample. When absorbed by core electrons of the elements in the sample, these photons result in ejection of these electrons. Electrons from a higher energy orbital move down to fill the vacancies left, which results in the release of photons with energy that is equivalent to the difference in the binding energies of the two shells. This X-ray fluorescence is detected and converted into an X-ray spectrum, conveying characteristic peaks (see Fig. 2) (66). The identity of the elements can be deduced from the energies of the peaks, and the integration of the peaks correlates to the quantities of the elements present. By this method, many elements can be identified concurrently, which allows maps of the distribution of these elementals to be generated at submicron resolution (66-68).

Figure 2. Schematic diagram illustrating of the components in the X-ray fluorescence microscope apparatus. Figure reproduced with permission from Reference 68 with kind permission from ScienceDirect, Elsevier.
So far, studies that use SRIXE to map the distribution of platinum inside cells have been limited, and all studies have been conducted at low resolution. The first of these studies was conducted by Ilinski et al. (68), who used SRIXE to compare the uptake of platinum(II) complexes in the cisplatin-sensitive 2008 cell line and its cisplatin-resistant subline. In this study, incident X-ray energy of 11.7 keV (just above the platinum 2p3/2 absorption edge) was used, and La fluorescence lines were collected with resolutions of 1 x 0.2 μm2. It was observed that the cisplatin accumulation in the cisplatin-resistant subline was half that of the cisplatin-sensitive cell line after 24-hour incubation with 10 μM cisplatin. In contrast, Pt103, a cisplatin derivative that displayed high activity against cisplatin-resistant tumors, showed a higher accumulation after the same incubation period. This suggests that increase of Pt103 cellular uptake was responsible for its bypass of cisplatin resistance mechanisms in cisplatin-resistant 2008 cells. However, the accuracy of this experiment was compromised because of the overlap of the fluorescence peaks between Pt La and Zn Kβ when 11.7 keV is used as the incident energy (68). Regardless, the potential of SRIXE to map the distribution of platinum quantitatively within individual cells was demonstrated.
Following this, Hall et al. (60, 70) used SRIXE to examine elemental distribution within A2780 cells that had been treated with cisplatin and platinum(IV) complexes. This study employed incident energy of 13.4 keV (above the 2p1/2 absorption edge), and emissions from both Ka and La fluorescence were collected at a resolution of 0.3 x 0.3 μm2. It was observed that cisplatin and the platinum(IV) analogs accumulated in the nuclei of the cells after 24-hour incubations with 20 μM solutions (note that zinc elemental maps were used to identify the cell nucleus - see Fig. 3). This finding suggests that most platinum(IV) species had been reduced, and it was confirmed qualitatively by collecting micro-XANES spectra at points of high platinum concentration within the cells. The observation was also confirmed quantitatively by obtaining XANES spectra of bulk cell samples and quantifying the percentage of remaining platinum(IV) (60, 70). Hence, this study confirmed that SRIXE can be used for qualitative and quantitative studies of platinum accumulation inside cells, and it showed that the coupling of SRIXE with XANES affords a powerful technique in which both the distribution and oxidation state of a drug within a biologic system can be determined.

Figure 3. Elemental maps of Cl, K, Ca, Cu, Zn and Pt, respectively, obtained from an A2780 cell treated with cisplatin for 24 hours. Figure reproduced with permission from Reference 60 with kind permission from ScienceDirect, Elsevier.
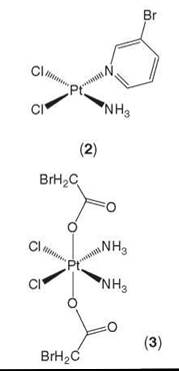
In later studies, Hall et al. (71) employed the same method as above to study the intracellular distribution of platinum complexes that carry bromine labels (2, 3) or anthraquinones (1). Again, after 24 hours, all complexes were found to be localized within the nucleus, with little platinum being in the cytosol. In the case of the platinum(II)-3-bromopyridine complex (2), the bromine was also observed to colocalize with the platinum. This finding is consistent with the amine moieties that remain bound to platinum(II) complexes inside cells. In another case of the platinum(IV)-bromoacetate complex (3), the distribution of the bromine was much more diffuse when compared with that of the platinum, which is consistent with the reduction of the platinum(IV) complex. However, whether the reduction occurred before or after cell entry is still unclear (see Fig. 4) (71). Hence, by this method of labeling, the bio-transformation of platinum(IV) agents can be monitored via SRIXE.

Figure 4. SRIXE maps showing the distribution of Br (left column) and Pt (right column) in A2780 whole cancer cells treated with cis-(PtCl2(3-Brpyr)(NH3)) (top row) or cis, trans, c/s-(PtCl2(OAcBr)2(NH3)2) (bottom row). Figure reproduced with permission from Reference 71 with kind permission from Springer Science and Business Media.
SRIXE has also been recently employed to investigate cis-platin resistance. Shimura et al. (72) has measured the quantitative difference in cisplatin accumulation between lung carcinoma cells (PC-9) and its cisplatin-resistant subline. Using an incident energy of 15 keV and a resolution of 1.5 x 0.75 μm, it was found that at 12 hour incubations using 1 μM cisplatin, the intensity of platinum in the resistant subline was 2.6 fold less than observed for the sensitive cell line (72). By using the same method as Ilinski et al. (67), Chen et al. (73) mapped the distribution of cisplatin in intrinsically cisplatin-resistant human melanoma cells (MNT-1) and in human epidermoid carcinoma cells (KB-3-1). This study found that cisplatin was sequestered into the melanosomes, and nuclear accumulation was reduced.
Although SRIXE is relatively new to cellular platinum studies, it has proven to be an excellent and reliable technique for the detection of platinum drugs in cells. Currently, success of elemental distribution studies using SRIXE is limited by the comparatively poor resolution. In the above studies, organelles, such as mitochondria, Golgi, endoplasmic reticulum, and lysosomes were not resolved because many of these subcellular organelles and structures are smaller than 0.1 μm (52). Hence, SRIXE in platinum studies currently lacks the high resolution found in TEM and fluorescence microscopy studies.
Conclusion and Future Applications
Advancements in imaging techniques, as illustrated above, has facilitated the significant progress made by studies of the intracellular distribution of platinum anticancer agents, providing deeper insight into the cellular fate of these drugs. All techniques mentioned have the potential to make even more valuable contributions biologic studies of platinum complexes. In terms of imaging, TEM provides the highest level of two-dimensional morphologic spatial resolutions. Additional developments into immunoelectron microscopy may allow for the intracellular mapping of more platinum-protein structures.
Fluorescence microscopy uniquely allows for real time, three-dimensional tracking of drugs in live cells at low incubation concentrations. When employed in conjunction with tools such as pathway inhibitors, this technique can elucidate drug trafficking pathways and reveal the roles of intracellular components in the drug action.
SRIXE is a powerful technique that can map the distribution of platinum with very high sensitivity and at low incubation concentrations of the unmodified platinum drug. With future improvements to its resolving power, this technique has the potential to obtain unprecedented information on the distribution of both endogenous and exogenous elements at a sub-cellular level.
In addition to the advancement of the mentioned techniques, the future of the imaging of cellular platinum may also benefit from alternative techniques, such as that of secondary-ion mass spectrometry and autoradiography. However, these techniques have yet to be used in investigations of the in vitro distributions of platinum complexes.
References
1. Weiss RB, Christian MC. New cisplatin analogues in development. Drugs 1993; 46:360-377.
2. Kelland LR, Sharp SY, O’Neill CF, Raynaud FI, Beale PJ, Judson IR. Mini-review: discovery and development of platinum complexes designed to circumvent cisplatin resistance. J. Inorg. Biochem. 1999; 77:111-115.
3. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 2003; 22:7265-7279.
4. Tilby MJ, Johnson C, Knox RJ, Cordell J, Roberts JJ, Dean CJ. Sensitive detection of DNA modifications induced by cisplatin and carboplatin in vitro and in vivo using a monoclonal antibody. Cancer Res. 1991; 51:123-129.
5. Akaboshi M, Kawai K, Maki H, Akuta K, Ujeno Y, Miyahara T. The number of platinum atoms binding to DNA, RNA and protein molecules of HeLa cells treated with cisplatin at its mean lethal concentration. Japanese J. Cancer Res. 1992; 83:522-526.
6. Binks SP, Dobrota M. Kinetics and mechanism of uptake of platinum-based pharmaceuticals by the rat small intestine. Biochem. Pharmacol. 1990; 40:1329-1336.
7. Gately DP, Howell SB. Cellular accumulation of the anticancer agent cisplatin: A review. Brit. J. Cancer 1993; 67:1171-1176.
8. Aggarwal SK. A histochemical approach to the mechanism of action of cisplatin and its analogs. J. Histochem. Cytochem. 1993; 41:1053-1073.
9. Safaei R, Holzer AK, Katano K, Samimi G, Howell SB. The role of copper transporters in the development of resistance to Pt drugs. J. Inorgan. Biochem. 2004; 98:1607-1613.
10. Canada RG, Andrews PA, Mack KM, Haider, A. The effects of terbium on the accumulation of cisplatin in human ovarian cancer cells. Biochim. Biophys. Acta, Mol. Cell Res. 1995; 1267:25-30.
11. Andrews P, Albright K. 1991. Role of membrane ion transport in cisplatin accumulation. Paper presented at the Platinum and other metal coordiantion compounds in cancer chemotherapy.
12. Christen RD, Jekunen AP, Jones JA, Thiebaut F, Shalinsky DR, Howell SB. In vitro modulation of cisplatin accumulation in human ovarian carcinoma cells by pharmacologic alteration of microtubules. J. Clin. Invest. 1993; 92:431-440.
13. Andrews PA, Mann SC, Huynh HH, Albright KD. Role of the sodium, potassium-adenosine triphosphatase in the accumulation of cis-diamminedichloroplatinum(II) in human ovarian carcinoma cells. Cancer Res. 1991; 51:3677-3681.
14. Holzer AK, Katano K, Klomp LWJ, Howell SB. Cisplatin rapidly down-regulates its own influx transporter hCTR1 in cultured human ovarian carcinoma cells. Clin. Cancer Res. 2004; 10:6744-6749.
15. Samimi G, Safaei R, Katano K, Holzer AK, Rochdi M, Tomioka M, Goodman M and Howell SB. Increased expression of the copper efflux transporter ATP7A mediates resistance to cis-platin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin. Cancer Res. 2004; 10:4661-4669.
16. Kalayda GV, Jansen BAJ, Molenaar C, Wielaard P, Tanke HJ, Reedijk J. Dinuclear platinum complexes with N,N’-bis(amino-alkyl)-1,4-diaminoanthraquinones as linking ligands. Part II. Cellular processing in A2780 cisplatin-resistant human ovarian carcinoma cells: new insights into the mechanism of resistance. J. Biol. Inorganic Chem. 2004; 9:414-422.
17. Pereira-Maia E, Garnier-Suillerot A. Impaired hydrolysis of cis-platin derivatives to aquated species prevents energy-dependent uptake in GLC4 cells resistant to cisplatin. J. Biol. Inorganic Chem. 2003; 8:626-634.
18. Loh SY, Mistry P, Kelland LR, Abel G, Harrap KR. Reduced drug accumulation as a major mechanism of acquired resistance to cisplatin in a human ovarian carcinoma cell line: circumvention studies using novel platinum (II) and (IV) ammine/amine complexes. Brit. J. Cancer 1992; 66:1109-1115.
19. Lindauer E, Holler E. Cellular distribution and cellular reactivity of platinum(II) complexes. Biochem. Pharmacol. 1996; 52:7-14.
20. Dietel M. What’s new in cytostatic drug resistance and pathology. Pathol. Res. Pract. 1991; 187:892-905.
21. Sharma RP, Edwards IR. cis-Platinum: subcellular distribution and binding to cytosolic ligands. Biochem. Pharmacol. 1983; 32:2665-2669.
22. Leibbrandt MEI, Wolfgang GHI, Metz AL, Ozobia AA, Haskins JR. Critical subcellular targets of cisplatin and related platinum analogs in rat renal proximal tubule cells. Kidney Inter- nat. 1995; 48:761-770.
23. Cece R, Petruccioli MG, Cavaletti G, Barajon I, Tredici G. An ultrastructural study of neuronal changes in dorsal root ganglia (DRG) of rats after chronic cisplatin administrations. Histol. Histopathol. 1995; 10:837-845.
24. Andrews PA, Schiefer MA, Murphy MP, Howell SB. Enhanced potentiation of cisplatin cytotoxicity in human ovarian carcinoma cells by prolonged glutathione depletion. Chem.-Biol. Interact. 1988; 65:51-58.
25. Hamilton TC, Winker MA, Louie KG, Batist G, Behrens BC, Tsuruo T, Grotzinger KR, McKoy WM, Young RC, Ozols RF. Augmentation of adriamycin, melphalan, and cisplatin cytotoxicity in drug-resistant and -sensitive human ovarian carcinoma cell lines by buthionine sulfoximine mediated glutathione depletion. Biochem. Pharmacol. 1985; 34:2583-2586.
26. Yang Z, Schumaker LM, Egorin MJ, Zuhowski EG, Guo Z, Cullen KJ. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin. Cancer Res. 2006; 12:5817-5825.
27. Gemba M, Nakatani E, Teramoto M, Nakano S. Effect of cisplatin on calcium uptake by rat kidney cortical mitochondria. Toxicol. Lett. 1987; 38:291-297.
28. Berry JP, Brille P, LeRoy AF, Gouveia Y, Ribaud P, Galle P, Mathe G. Experimental ultrastructural and x-ray microanalysis study of cisplatin in the rat: intracellular localization of platinum. Cancer Treat. Rep. 1982; 66:1529-1533.
29. Giurgiovich AJ, Diwan BA, Olivero OA, Anderson LM, Rice JM, Poirier MC. Elevated mitochondrial cisplatin-DNA adduct levels in rat tissues after transplacental cisplatin exposure. Carcinogenesis 1997; 18:93-96.
30. Andrews PA, Albright KD. Mitochondrial defects in cis-diamminedichloroplatinum(II)-resistant human ovarian carcinoma cells. Cancer Res. 1992; 52:1895-1901.
31. Chauhan SS, Liang XJ, Su AW, Pai-Panandiker A, Shen DW, Hanover JA, Gottesman MM. Reduced endocytosis and altered lysosome function in cisplatin-resistant cell lines. Brit. J. Cancer 2003; 88:1327-1334.
32. Safaei R, Katano K, Samimi G, Holzer A, Naerdemann W, Howell SB. 2004. Cisplatin resistance is associated with reduced lyssomal structures and markers in ovarian cancer cells. Paper presented at the American Association for Cancer Research.
33. Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer. 2005; 5:886-897.
34. Castino R, Demoz M, Isidoro C. Destination \“lysosome\”:a target organelle for tumour cell killing? J. Molec. Recog. 2003; 16:337-348.
35. Artal-Sanz M, Samara C, Syntichaki P, Tavernarakis N. Lysosomal biogenesis and function is critical for necrotic cell death in Caenorhabditis elegans. J. Cell Biol. 2006; 173:231-239.
36. Werneburg N, Guicciardi M, Bronk S, Gores G. Tumour necrosis factor-alpha-associated lysosomal permeabilization is cathepsin B dependent. Am. J. Physiol. Gastrointest. Liver Physiol. 2002; 283:G947-G956.
37. Muenchen HJ and Aggarwal SK. Enhanced immune system activation after treatment with novel antineoplastic platinum agents. Anticancer Res. 1998; 18:2631-2636.
38. Rochow TG, Tucker PA. Transmission Electron Microscopy and Electron Diffraction. 2nd ed. 1994. Plenum Press, New York. pp. 265-294.
39. Dykstra MJ, Reuss LE. Transmission Electron Microscopy. 2nd ed. 2003. Kluwer Academic/Plenum Publishers, New York. pp. 287-321.
40. Dykstra MJ, Reuss LE. Specimen Preparation for Electron Microscopy. 2003. Kluwer Academic/Plenum Publishers, New York. pp. 1-122.
41. Sodhi A, Aggarwal SK. Effects of cis-dichlorodiammine platinum (II) in the regression of sarcoma 180: a fine structural study. J. Natl. Cancer Inst. 1974; 53:85-101.
42. Aggarwal SK, Sodhi A. Cytotoxic effects of cis-dichlorodiammine platinum(II) on the mammalian cells in vitro: a fine structure study. Cytobiolgie 1973; 7:366-374.
43. Miller CM, Aggarwal SK. Cispaltin-induced changes in the adrenal of the rat. Proc. Electron Microsc. Soc. Am. 1992; 50:652-653.
44. Portilla D, Li S, Nagothu KK, Megyesi J, Kaissling B, Schnackenberg L, Safirstein RL, Beger RD. Metabolomic study of cisplatin-induced nephrotoxicity. Kidney Int. 2006; 69:2194-2204.
45. Meijera C, van Luyn MJA, Nienhuis EF, Blom N, Mulder NH, de Vries EGE. Ultrastructural morphology and localisation of cisplatin-induced platinum-DNA adducts in a cisplatin-sensitive and -resistant human small cell lung cancer cell line using electron microscopy. Biochem. Pharmacol. 2001; 61:573-578.
46. Aggarwal SK, Wagner RW, McAllister PK, Rosenberg B. Cell-surface-associated nucleic acid in tumorigenic cells made visible with platinum-pyrimidine complexes by electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 1975; 72:928-932.
47. Khan MUA, Sadler PJ. Distribution of a platinum anti-tumor drug in HeLa cells by analytical electron microscopy. Chem. Biol. Interact. 1978; 21:227-232.
48. Ghadially FN, Lock CJL, Yang-Steppuhn SE, Lalonde JMA. Platinosomes produced in cultured cells by platinum coordination complexes. J. Submicrosc. Cytol. 1981; 13:223-230.
49. Ghadially FN, Lock CJL, Lalonde JMA, Ghadially R. Platinosomes produced in synovial membrane by platinum coordination complexes. Virchows Arch., B, Cell Pathol. 1981; 35:123-131.
50. Beretta GL, Righetti SC, Lombardi L, Zunino F, Perego P. Electron microscopy analysis of early localization of cisplatin in ovarian carcinoma cells. Ultrastruct. Pathol. 2002; 26:331-334.
51. Ruben GC. Ultrathin (1nm) vertically shadowed platinum-carbon replicas for imaging individual molecules in freeze-etched biological DNA and material science metal and plastic specimens. J. Electron. Microsc. Tech. 1989; 13:335-354.
52. Hibbs AR. Confocal Microscopy for Biologies. 2004. Kluwer Academic/Plenum Publishers, New York. pp. 1-29.
53. Cox G. Optical Imaging Techniques in Cell Biology. 29-37 2007. CRC Press, Taylor and Francis Group, Boca raton.
54. Gumbleton M, Stephens DJ. Coming out of the dark: the evolving role of fluorescence imaging in drug delivery research. Adv. Drug Deliv. Rev. 2005; 57:5-15.
55. Molenaar C, Teuben JM, Heetebrij RJ, Tanke HJ, Reedijk J. New insights in the cellular processing of platinum antitumor compounds, using fluorophore-labeled platinum complexes and digital fluorescence microscopy. J. Biol. Inorg. Chem. 2000; 5:655-665.
56. Kalayda GV, Jansen BAJ, Wielaard P, Tanke HJ, Reedijk J. Dinuclear platinum anticancer complexes with fluorescent N,N’-bis (aminoalkyl)-1,4-diaminoanthraquinones: cellular processing in two cisplatin-resistant cell lines reflects the differences in their resistance profiles. J. Biol. Inorg. Chem. 2005; 10:305-315.
57. Safaei R, Katano K, Larson BJ, Samimi G, Holzer AK, Naerdemann W, Tomioka M, Goodman M, Howell SB. Intracellular localization and trafficking of fluorescein-labeled cisplatin in human ovarian carcinoma cells. Clin. Cancer Res. 2005; 11:756-767.
58. Liang XJ, Shen DW, Chen KG, Wincovitch SM, Garfield SH, Gottesman MM. Trafficking and localization of platinum complexes in cisplatin-resistant cell lines monitored by fluorescence- labeled platinum. J. Cell. Physiol. 2005; 202:635-641.
59. Kalayda GV, Zhang G, Abraham T, Tanke HJ, Reedijk J. Application of fluorescence microscopy for investigation of cellular distribution of dinuclear platinum anticancer drugs. J. Med. Chem. 2005; 48:5191-5202.
60. Hall MD, Dillon CT, Zhang M, Beale P, Cai Z, Lai B, Stampfl APJ, Hambley TW. The cellular distribution and oxidation state of platinum(II) and platinum(IV) antitumour complexes in cancer cells. J. Biol. Inorg. Chem. 2003; 8:726-732.
61. Gibson D, Binyamin I, Haj M, Ringel I, Ramu A, Katzhendler J. Anthraquinone intercalators as carrier molecules for second-generation platinum anticancer drugs. Eur. J. Med. Chem. 1997; 32:823-831.
62. Gibson D, Gean KF, Katzhendle J, Ben-Shoshan R, Ramu A, Ringel I. Preparation, characterization, and anticancer activity of a series of cis-PtCl2 complexes linked to anthraquinone intercalators. J. Med. Chem. 1991; 34:414-420.
63. Jansen BAJ, Wielaard P, Kalayda GV, Ferrari M, Molenaar C, Tanke HJ, Brouwer J, Reedijk J. Dinuclear platinum complexes with N,N’-bis(aminoalkyl)-1,4-diaminoanthraquinones as linking ligands. Part I. Synthesis, cytotoxicity, and cellular studies in gA2780 human ovarian carcinoma cells. J. Biol. Inorg. Chem. 2004; 9:403-413.
64. Kalayda GV, Jansen BAJ, Molenaar C, Wielaard P, Tanke HJ, Reedijk J. Dinuclear platinum complexes with N,N’-bis (aminoalkyl)-1,4-diaminoanthraquinones as linking ligands. Part II. Cellular processing in A2780 cisplatin-resistant human ovarian carcinoma cells: new insights into the mechanism of resistance. J. Biol. Inorg. Chem. 2004; 9:414-422.
65. Alderden RA, Mellor HR, Modok S, Hambley TW, Callaghan R. Cytotoxic efficacy of an anthraquinone linked platinum anticancer drug. Biochem. Pharmacol. 2006; 71:1136-1145.
66. Sparkes CJ. X-Ray Fluorescence Microprobe for Chemical Analysis. 1980. Plenum Press, New York. pp. 459-509.
67. Ilinski P, Lai B, Cai Z, Yun W, Legnini D, Talarico T, Cholewa M, Webster LK, Deacon GB, Rainone S, Phillips DR, Stampfl APJ. The direct mapping of the uptake of platinum anticancer agents in individual human ovarian adenocarcinoma cells using a hard x-ray microprobe. Cancer Res. 2003; 63:1776-1779.
68. Fahrni CJ. Biological applications of X-ray fluorescence microscopy: exploring the subcellular topography and speciation of transition metals. Curr. Opin. Chem. Biol. 2007; 11:121-127.
69. Paunesku T, Vogt S, Maser J, Lai B. X-ray fluorescence microprobe imaging in biology and medicine. J. Cell. Biochem. 2006; 99:1489-1502.
70. Hall MD, Foran GJ, Zhang M, Beale PJ, Hambley TW. XANES Determination of the Platinum Oxidation State Distribution in Cancer Cells Treated with Platinum(IV) Anticancer Agents. J. Am. Chem. Soc. 2003; 125:7524-7525.
71. Hall MD, Alderden RA, Zhang M, Beale PJ, Cai Z, Lai B, Stampfl APJ, Hambley TW. The fate of platinum(II) and platinum(IV) anti-cancer agents in cancer cells and tumors. J. Struct. Biol. 2006; 155:38-44.
72. Shimura M, Saito A, Matsuyama S, Sakuma T, Terui Y, Ueno K, Yumoto H, Yamauchi K, Yamamura K, Mimura H, et al. Element array by scanning X-ray fluorescence microscopy after cis-diamminedichloro-platinum(II) treatment. Cancer Res. 2005; 65:4998-5002.
73. Chen KG, Valencia JC, Lai B, Zhang G, Paterson JK, Rouzaud F, Berens W, Wincovitch SM, Garfield SH, Leapman RD, Hearing VJ, Gottesman MM. Melanosomal sequestration of cytotoxic drugs contributes to the intractability of malignant melanomas. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:9903-9907.