CHEMICAL BIOLOGY
Naturally Occurring Peptide Hormones and Neurotransmitters, Synthesis of
Zhihua Liu and Victor J. Hruby, Department of Chemistry, The University of Arizona, Tucson, Arizona
doi: 10.1002/9780470048672.wecb658
How to synthesize biologically active polypeptides chemically has long been an important challenge for many fields in chemistry and biology. In the past, the development of solution-phase and solid-phase synthesis techniques has made it possible to synthesize large molecules like peptides with molecular weights up to 10,000. The purpose of this review is to provide an overview of how to prepare natural bioactive peptides synthetically. Because of space limitations, this review concentrates on peptide hormones and neurotransmitters.
Abbreviations: Standard abbreviations as recommended by IUPAC-1UB Commission are used in this review (1). Other abbreviations include: Acm, acetamidomethyl; Bn, benzyl; Boc: t-butyloxycarbonyl; Bom, benzyloxymethyl; BHA, benzhydrylamine; Cbz, benzyloxycarbonyl; cHx, cyclohexyl; DCC, N,N-dicyclohexylcarbodiimide; DCM, dichloromethane; DIC, N,N-diisopropylcarbodiimide; DMF, N,N-dimethylformamide; DMSO, dimethyl sulphoxide; For, formyl; Fmoc, 9-fluorenylmethyloxycarbonyl; HMP, 4-hydroxymethylphenoxyacetyl; HOAt, 1-hydroxy-7-azabenzotriazole; HOBt, 1-hydroxybenzotriazole-benzotriazole; MBHA, 4-methylbenzhydrylamine; Mtr, 4-methoxy-2,3,6-trimethylphenylsulphonyl; Np, nitrophenyl; NSu, N-succinimide; ONsu, N-hydroxysuccinimide; Trt, triphenylmethyl; Pam, 4-(hydroxymethyl) phenylacetamidomethyl; PAL, 5-[4-(9-fluorenylmethyloxycarbonyl)aminomethyl-3,5,-dimethoxyphenoxy]valeric acid; Pcp, pentachlorophenyl; PEG, polyethylene glycol; Pha, phenoxyacetyl; Pmc, 2,2,5,7,8-pentamethyl-chroman-6-sulphonyl; Tcp, trichlorophenyl; TFA, trifluoroacetic acid; TFAA, trifluoroacetic anhydride; Tos, p-toluenesulfonyl.
Peptide hormones and neurotransmitters are among the most important classes of natural products in living systems, including in humans. They modulate and regulate most major biological functions and behaviors that are necessary for the maintenance of life and health, including feeding behavior, reproduction, response to stress, pain, cardiovascular function, kidney function, energy homeostasis, aggression, maternal behavior, learning behavior, and many others. In addition, these hormones often are intimately involved in virtually all major diseases, including hypertension and other cardiovascular dysfunctions, diabetes, obesity, cancer, CNS diseases, and many others. The recognition
that polypeptides played such a central role in health and disease is a relatively recent scientific development. The discovery of insulin as the principle modulator (along with glucagon—a later discovery) of glucose levels in the blood and the determination of the structure and total synthesis of the peptide hormone and neurotransmitter oxytocin, which controls uterine contraction, milk ejection, and later maternal behavior; socialization; and aspects of sexual behavior are milestones not only in peptide and protein chemistry, biology, and medicine, but also in modern drug discovery and design. Especially in the case of oxytocin immediately after its structure determination and total synthesis by du Vigneaud and co-workers in the early 1950s, he and many others began to modify the “functional groups” and structure of oxytocin to determine those features important to biological activity. Of central importance in this regard was the development of increasingly efficient synthetic methods (if you cannot make it you cannot study it) and analytical methods for the preparation and purification of polypeptides. In this review, we will outline synthetic methods that have been developed for the synthesis of polypeptide hormones and neurotransmitters, and in the process we also point out the key analytical procedures that have been used in the synthesis and purification of these important biological natural products. See Table 1 for an overview of this article and its contents.
Table 1. Peptides discussed in this article
|
Name |
Target |
Structure features |
Major biological activities |
Synthesis techniques |
|
Atrial natriuretic peptide (ANP)
Cholecystokinin (CCK)
Gastrin
Glucagon-like peptide 1 (GLP1)
Glucose-dependent insulinotropic polypeptide (GIP) Motilin Neuropeptide Tyrosine (NPY) Peptide Tyrosine Tyrosine (PYY) Secretin Somatostatin
Substance P
Vasoactive intestinal peptide (VIP)
Corticotropin-releasing factor (CRF or CRH) Gonadotropin-releasing factor (GnRF or GnRH)
Growth hormone releasing factor (GRF or GRH) Thyrotropinreleasing factor
Angiotensin II Amylin
Glucagon Insulin
Pancreatic polypeptide
Adrenocorticotropin (ACTH) Melanocyte-stimulating hormones (MSH)
Oxytocin
Vasopressin
Relaxin
Calcitonin
Calcitonin gene-related peptide (CGRP)
Dynorphin
Endorphin
Enkephalin
Leumorphin
Neurokinin A & B Eledoisin, Physalaemin and Kassinin Gastrin releasing peptide (GRP) Neurotensin
Bradykinin |
ANP-R1 to R3 receptors in the peripheral system CCK-A receptor, CCK-B receptor
CCK-B receptor
GLP-1 receptor
GIP receptor
Motilin receptor NP Y1 to Y5 receptors PYY receptor Secretin receptor Somatostatin 1 to 5 receptors
Neurokinin 1 receptor
VPAC1 and VPAC2 receptors
CRH-R1 and CRH-R2 receptors
GnRH I and II receptor
GRH receptor
TRF receptor
Angiotensin II AT1 and AT2 receptor Amylin receptor
Glucagon receptor Insulin receptor
Pancreatic polypeptide receptor, NP Y4 receptor Melanocortin 2 receptor Melanocortin 1 to 5 receptors
Oxytocin receptor
Arginine vasopressin receptor 1A, 1B, 2 Relaxin/insulin-like family peptide receptor 1 to 4
Calcitonin receptor
CGRP1 and 2 receptors
k-opioid receptor
μ,δ-opioid receptors
μ,δ-opioid receptors
k-opioid receptor
Neurokinin 1 to 3 receptors Neurokinin 1 to 3 receptors Gastrin-releasing peptide receptor Neurotensin 1, 2 receptors, Sortilin 1 receptor Bradykinin 1, 2 receptors |
28-residue peptide with a disulfide bridge 33-residue peptide amide with a Tyr sulfonic acid 14- and 17-residue peptide amide with a Glu rich region 30- and 31-residue peptide amide differed by a C-terminal Gly 42-residue peptide
22-residue peptide 36-residue peptide amide 36-residue peptide amide 27-residue peptide amide 14- and 28-residue peptide with a disulfide bridge 11-residue peptide amide
28-residue peptide amide, highly basic 41-residue peptide amide
10-residue peptide amide
40-44-residue peptide amide
3-residue peptide amide
8-residue peptide 37-residue peptide amide with one disulfide bridge 29-residue peptide Contains 21-residue A chain with an intra disulfide bridge and 30-residue B chain, linked by two disulfide bridges 36-residue peptide amide
39-residue peptide 12-22-residue peptide or peptide amide, with His-Phe-Arg-Trp as the major pharmacophore 9-residue peptide amide with a disulfide bridge 9-residue peptide amide with a disulfide bridge Contains 24-residue A chain with an intra disulfide bridge and 29-residue B chain, linked by two disulfide bridges 32-residue peptide amide with a disulfide bridge 37-residue peptide amide with a disulfide bridge 13- or 17-residue peptides
16- to 31-residue peptides
5-residue peptides different at the C –termili sequence 29-residue peptide
10-residue peptide amide 10- or 11-residue peptide amide 27-residue peptide amide 13-residue peptide
9-residue peptide |
Modulate blood pressure
Modulate digestion system
Stimulate gastric acid secretion
Stimulate insulin secretion
Inhibit gastric acid and stimulate insulin secretion Stimulate migrating motor complex Increase food intake Inhibit food digestion Regulate pH of duodenal contents Inhibit gut peptides and growth hormone Involved in pain, vomitting, cellular growth, mood regulation Inhibit gastric acid and stimulate water and electrolytes secretion Involved in stress response
Stimulate the release of luteinizing hormone and follicle-stimulating hormone Stimulates growth hormone secretion
Stimulates thyroid- stimulating hormone and prolactin secretion Increase blood pressure Regulate blood glucose level
Increase blood glucose level Lower blood glucose level
Inhibit pancreatic secretion
Stimulate the cortex of adrenal gland Darken skin and hair
Facilitate birth and breastfeeding
Regulate the retention of water
Facilitate birth
Lower blood calcium and phosphate levels Lower blood pressure
Endogenous k-opioid receptor agonist Have analgesic effect
Have analgesic effect
Endogenous k-opioid receptor agonist Similar activities as substance P Similar activities as substance P Stimulate gastrin release Lower blood pressure, increase blood glucagon, glucose levels Lower blood pressure, pain related |
Solid-phase Na-Boc chemistry
Solution-phase and solid-phase Na–Boc chemistry Solution-phase and solid-phase Na–Fmoc chemistry Solid-phase Na–Fmoc chemistry
Solution-phase chemistry
Solid-phase Na-Boc chemistry Solid-phase Na-Boc chemistry Solid-phase Na-Fmoc chemistry Solid-phase Na-Boc chemistry Solid-phase Na-Boc chemistry
Solid-phase Na-Boc chemistry
Solid-phase Na-Boc chemistry
Solid-phase Na-Boc chemistry
Solid-phase Na-Boc chemistry
Solid-phase Na-Boc chemistry
Solid-phase Na-Boc chemistry
Solid-phase Na-Boc chemistry Solid-phase Na-Boc chemistry
Solid-phase Na-Boc chemistry Solid-phase Na-Boc chemistry
Solid-phase Na-Boc chemistry
Solid-phase Na-Boc chemistry Solid-phase Na-Boc chemistry
Solid-phase Na–Boc chemistry
Solid-phase Na-Boc chemistry
Solid-phase Na-Fmoc and Na–Boc chemistry
Solid-phase Na-Fmoc chemistry
Solid-phase Na-Boc chemistry
Solid-phase Na–Fmoc chemistry
Solid-phase Na-Fmoc and Na-Boc chemistry Enzymetic synthesis
Solid-phase Na-Boc chemistry
Solid-phase Na-Boc chemistry Solid-phase Na-Boc chemistry Solid-phase Na–Boc chemistry
Solid-phase Na–Boc chemistry |
Atrial natriuretic peptide (ANP)
ANP is a 28-amino-acid peptide first discovered by de Bold et al. (2). It is released from heart atrial myocytes in response to a local arterial wall stretch. ANP acts on outer adrenal cells to decrease aldosterone production and blood pressure, increase salt and water excretion, and transudate plasma water to the interstitium (3).
The sequence of ANP is as follows: Ser-Leu-Arg-Arg-Ser-Ser-c(Cys-Phe-Gly-Gly-Arg-Ile-Asp-Arg-Ile-Gly-Ala-Gln-Ser-Gly-Leu-Gly-Cys)-Asn-Ser-Phe-Arg-Tyr-OH.
The structure of ANP shows a disulfide ring at position 7 to 23, which is shared by the other two natriuretic peptides (brain natriuretic peptide and C-type natriuretic peptide). ANP has been prepared synthetically via solid-phase peptide synthesis using the Na-Boc strategy. von Geldern et al. (4) used a Biosearch Model 9500 automated synthesizer (Biosearch Technologies, Novato, CA) to assemble the amino acid sequence on a Merrifield resin preloaded with Na-Boc-Tyr. A standard protocol for solid-phase synthesis (DCC/HOBt as coupling reagents; TFA for Na-Boc removal) and side-chain protections was used (refer to the sections on “Somatostatin synthesis” and “Angiotensin II synthesis” for details). The peptide was cleaved from the resin, and all protecting groups were deprotected with anhydrous HF that contained 10% anisole. The crude peptide was extracted with acetic acid, and the disulfide bond was formed by oxidation with iodine (0.01 N in ethanol; ~2 eq.) for 1 hour; excess iodine was removed by addition of aqueous sodium thiosulfate until the solution was colorless. The crude ANP was first purified on a column of Amberlyst XAD-16 nonionic adsorbent, and finally by high-performance liquid chromatography (HPLC) using a C-18 column, obtaining a more than 95% pure product.
Gut peptide hormones
Cholecystokinin (CCK)
CCK is a 33-amino-acid peptide hormone first isolated from porcine intestinal mucosa a long time ago (5). It exists in both the brain and the gastrointestinal systems, and it is responsible for central and pancreatic biological effects, including stimulating gallbladder contraction and bile flow and increasing the production of pancreatic digestive enzymes.
The sequence of porcine cholecystokinin-33(CCK-33) is as follows: Lys-Ala-Pro-Ser-Gly-Arg-Val-Ser-Met-Ile-Lys-Asn-Leu-Gln-Ser-Leu-Asp-Pro-Ser-His-Arg-Ile-Ser-Asp-Arg-Asp-Tyr(SO3H)-Met-Gly-Trp-Met-Asp-Phe-NH2.
Although discovered in the 1920s, the relatively long sequence and a tyrosine sulfonic acid residue make CCK a challenge for chemical synthesis compared with gastrins, which are related peptides that share identical C-termini. The first successful synthesis was not reported until Sakakibara’s group announced a solution-phase synthesis (6) in 1987, and in the same year, Penke’s group reported another successful synthesis by employing the solid-phase method (7). In the solution-phase study, the peptide was prepared via classic procedures. The Na-Boc proctected amino acids were coupled with the water-soluble carbodiimide (WSCI), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide, and HOBt (WSCI-HOBT method) (6). The coupling of protected peptides with L-amino acid residues at the C-terminus was carried out using WSCI together with 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine (HOOBT), which is known to minimize racemization of activated amino acid residues (WSCI-HOOBT method) (6). The side-chain protections were as follows: Asp was protected by a cHx group; Arg and His by Tos groups; Lys by a Cl-Cbz group; Tyr by a Br-Cbz group; Ser by a Pha group; Trp by a For group. The fully protected peptide thus obtained was deprotected by HF in the presence of anisole and Met. The Pha and For groups were not removed under these conditions. The Tyr residue was then sulphated with 40 equivalents of pyridinium acetylsulphate in TFA in an ice bath for 2 hours. After precipitation by adding water, the product was treated with 40 equivalents of 0.1 M NaOH in DMSO in an ice bath for 10 minutes to remove the remaining Pha and For groups. The crude product was purified by CM-cellulose chromatography and then by reversed-phase HPLC to produce a homogeneous preparation of the final product with only a 5% yield from the fully protected peptide. The scheme for the synthesis is shown in Fig. 1.
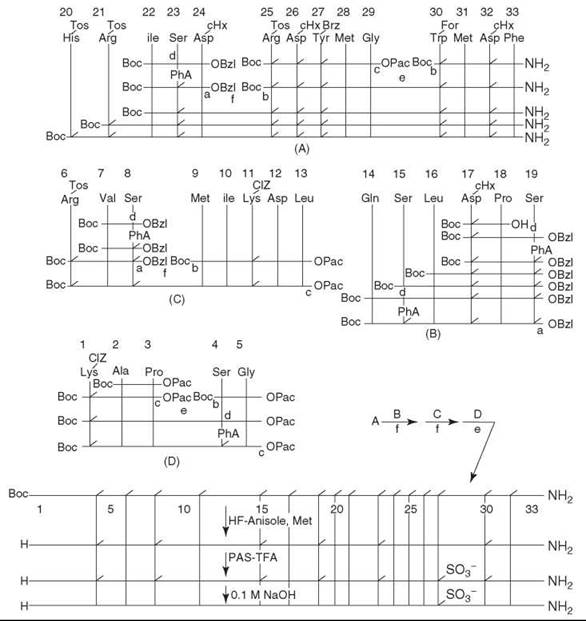
Figure 1. Scheme for the synthesis of porcine CCK-33. Peptide coupling was done by the WSCI-HOBT method unless otherwise indicated. (a) Catalytic hydrogenolysis. (b) TFA. (c) Zn-acetic acid. (d) phenoxyacetic anhydride. (e) WSCI-HOBY. (f) WSCI-HOOBT. OPac = phenacyl. Taken from Kurano et al. (6).
Penke’s group has reported two successful solid-phase syntheses of this peptide. The first study was using the Na-Boc chemistry published in 1987, with an overall yield of 1%. In 1991, they announced another synthesis using the Na-Fmoc strategy, and the protected Tyr(SO3H) was introduced into the peptide sequence during the chain assembly (8). A new solid support system, 4-succinylamido-2,2’,4’-trimethoxybenzhydrylamine (SAMBHA)-polystyrene resin, was also made for the synthesis to accommodate the acid-labile sulfate ester.
The anchor SAMBHA was prepared through a five-step synthesis (Fig. 2). First, 2-methoxy-4-acetamidobenzoic acid (0.03 mol) was dissolved at -20 °C in TFAA (15 mL) and reacted at 0 °C for 20 minutes. Then the mixture was dissolved in nitromethane (10 mL) and reacted with 2,4-dimethoxybenzene (0.035 mol) at room temperature for 25 minutes, followed by adding ice and ethylacetate and 10% sodium carbonate solution to neutralize the substance. The organic phase was washed three times with sodium carbonate solution and once with H2O and dried over sodium sulfate. After removing the solvents in vacuo, the oily residue was crystallized by triturating with ether, obtaining 2,2’,4’-trimethoxy-4-acetamidobenzophenone. The product was then dissolved in a mixture of 4N NaOH (30 mL) and methanol (30 mL) and was refluxed for 1 hour. Methanol was removed in vacuo, and 2,2’,4’-trimethoxy-4-aminobenzophenone was precipitated from the solution. After filtration and washing with H2O, the white crystals (0.01 mol) were dried, dissolved in pyridine (10 mol), and acylated with succinic anhydride (0.02 mol) by refluxing for 15 minutes. Pyridine was removed in vacuo; the oily residue was dissolved in a 1:1 mixture of ethyl acetate and 1 N HCl. The organic phase was separated, washed with H2O, dried over sodium sulfate, and evaporated to half volume. Ether was added, and the product 2,2’,4’-trimethoxy-4-succinylamidobenzophenone was precipitated as white crystals. The product (0.026 mol) was then dissolved in a mixture of pyridine (7.5 mL) and ethanol (50 mL), and it was refluxed for 45 minutes with hydroxylamine hydrochloride (0.067 mol). The solvent was then removed in vacuo, and the oily residue was triturated with 5% aqueous KHSO4 solution. The resulting solid was filtered, washed with H2O, and dried, obtaining 2,2',4'-trimethoxy-4-succinylamidobenzophenone oxime. The oxime (5 mmol) was suspended in glacial acetic acid (200 mL) and dissolved by warming. Then, 1-mL concentrated HCl and 0.2-g freshly prepared Pd-Catalyst were added, and the oxime was hydrogenated under pressure (1 M Pascal) for 5 hours. After filtering the catalyst and removing the solvents in vacuo, the oily residue was triturated with ether; the final product SAMBHA hydrochloride was crystallized as white solid. Four-mmol SAMBHA hydrochloride was then reacted overnight with Fmoc-OSu (4.4 mmol) in a mixture of H2O (5 mL) and dioxane (15 mL) in the presence of sodium bicarbonate (670 mg) to get Fmoc-SAMBHA, which was used to couple with aminomethyl polystyrene suspended in DCM in the presence of DCC in DMF. After 12 hours of reaction, the resin was washed with a 1:1 mixture of DMF and DCM, and the coupling reaction was repeated with Fmoc-SAMBHA and DCC. The Na-Fmoc group was cleaved by 10 minutes of treatment with 20% piperidine in DMF, and the SAMBHA resin was obtained with 0.33-mmol/g loading.
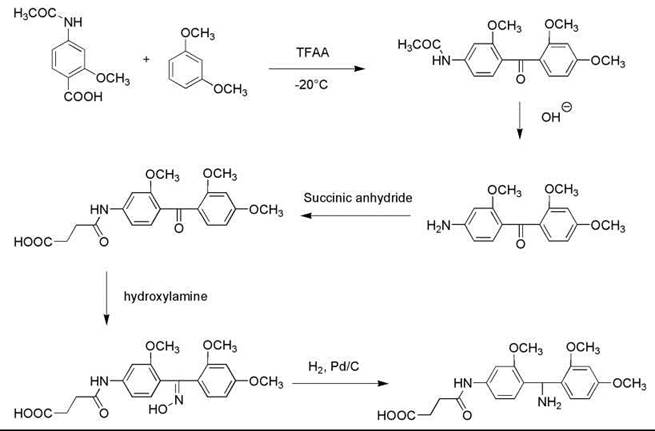
Figure 2. Synthesis of 4-succinylamido-2,2',4'-trimethoxybenzhydrylamine (SAMBHA).
Na-Fmoc-Tyr(SO3Na)OH was prepared by reacting tyrosine with chlorosulfonic acid in TFA at -20 °C for 5 minutes. Then Na-Fmoc group was added by reacting with Fmoc-Cl in a mixture of 10% sodium carbonate solution and dioxane.
The peptide was assembled using an Applied Biosystems 430A automatic peptide synthesizer (Applied Biosystems, Inc., Foster City, CA), and the SAMBHA-polystyrene resin was used as solid support. Amino acids were coupled as symmetric anhydrides using DCC in a 1:1 mixture of DCM-DMF or in pure DMF as coupling reagent, except that Na-Fmoc-Asp(Ot-Bu) and Na-Fmoc-Glu(Ot-Bu) were coupled as HOBt active esters and Na-Fmoc-Tyr(SO3Na) as a petafluorophenyl ester. The side-chain protections were as follows: Asp and Ser were protected by f-Bu groups; His by a Fmoc group; Lys by a Boc group; Arg by a Pmc group. Coupling was repeated if a ninhydrin test was positive. After completion of the synthesis, the peptide resin was washed with a 1:1 mixture of DMF-methanol that contained 0.4 g of NaOH for 1 minute, methanol for 2 minutes, and ether (4 x 2 minutes). Then the resin was dried. The protected pCCK-33 sulfate ester resin was washed with DCM (4 x 2 min) and was treated two times with 12 mL of 50% TFA solution in DCM that contained 5% ethanedithiol and 2% dimethyl sulfide (1 x 1min, 1 x 15 min, 1 x 1min). The TFA-containing solutions were pooled, and solvents were evaporated in vacuo. pCCK-33 was precipitated by adding peroxide-free ether.
Crude pCCK-33 was dissolved in the mixture of acetonitrile and 0.05 M (pH = 6.5) ammonium acetate buffer and was purified by prep-HPLC on a C18 column. The synthetic peptide was identified by using the native pCCK-33 standard. Pure fractions were pooled and lyophilized three times.
Gastrin
In 1905, Edkins discovered a hormone from the stomach and named it gastrin. Different forms and fragments of gastrin have been reported since then. The characteristic forms include a
17-residue amide called little gastrin and a 14-residue amide called minigastrin, which were known for their abilities of stimulating acid, pepsin, and pancreatic secretions in response to food (9).
The sequence of human little gastrin is as follows: pyroGlu-Gly-Pro-Trp-Leu-Glu-Glu-Glu-Glu-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2.
The sequence of human minigastrin is as follows: Trp-Leu-Glu-Glu-Glu-Glu-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2.
Gastrins and gastrin analogs have drawn great interest from chemists, and many total synthesis studies have been reported. Solution-phase synthesis was achieved by Anderson et al. in the 1960s (10). In 1980, Brown and coworkers reported a solid-phase synthesis of these two peptide amides (11). Methoxycarbonyl-functionalized polydimethylacrylamide resin was chosen as the solid support. It was converted into a primary amine by treating it with ethylenediamine. An internal reference-spacer amino acid was incorporated by couping with Na-Fmoc-norleucine anhydride and the cleavage of the Na-Fmoc group with 20% piperidine, followed by the addition of the reversible-linkage agent [2,4,5-trichlorophenyl 4-(hydroxymethyl)benzoate] in the presence of HOBt.
The peptide sequence was then assembled using initially Na-Boc-Phe anhydride in the presence of catalyst (dimethylaminopyridine) to establish the peptide-resin ester bond, followed by 16 cycles of Na-Fmoc amino-acid coupling. The symmetric anhydride method was used for coupling in all cases except for the last Gln, which was introduced as its p-nitrophenyl ester in the presence of HOBt. Deprotection of the Na-Boc groups was done by treating with HCl-AcOH, whereas the Na-Fmoc groups were removed by 20% piperidine in DMF. Gln was chosen as the N-terminal residue rather than pyroglutamic acid, which is present in little gastrin, so as to permit possible future extension into the prohormone series. Side-chain protections were as follows: Asp, Tyr, and Glu were protected by t-butyl groups. After completing the synthesis, side-chain t-butyl derivatives were removed by treatment with 90% aqueous TFA, and the free peptide was released from the resin by ammonolysis in saturated methanolic ammonia for 22 hours. Residual resin analysis indicated the removal of 91% of the peptide. At this stage, the product consisted of a mixture of the 17-residue N-terminal glutamine peptide amide and its cyclized pyroglutamyl analog (5:1). Cyclization was brought nearly to completion by treatment with 20% acetic acid (30 °C, 64 hours under Argon).
HPLC analysis showed the crude peptide has three major impurity peaks (Fig. 3a). The residual glutaminyl peptide (peak C) was removed by ion-exchange chromatography on DEAE cellulose DE52 (linear gradient of 0.01-1 M NH4OAc, pH 6.5); the impurities (peaks B and D) were removed by preparative HPLC on μ-Bondapak C-18 column using linear gradient of 18-45% MeCN in 0.01 M NH4OAc, pH 3.5, over 30 minutes, flow rate 2mL/min. The yield of purified [15-leucine] human little gastrin (Fig. 3b), which is identical by PHLC and TLC with an authentic sample, was 32%.
Minigastrin was obtained by stopping the peptide chain elongation after addition of the 14th residue; cleavage, deprotection, and HPLC purification were done similarly as mentioned above.
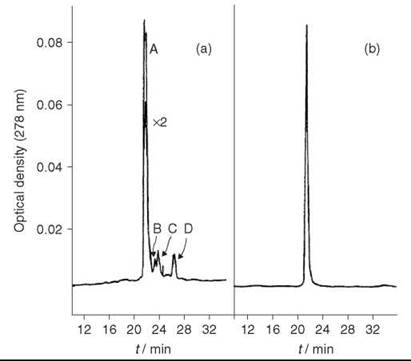
Figure 3. (a) HPLC analysis of crude little gastrin. (b) HPLC analysis of purified little gastrin. Taken from Brown et al. (11).
Glucagon-like peptide 1 (GLP1)
GLP1 is a peptide hormone produced by the posttranslation processing of proglucagon secreted from L-cells in the lower gut. It has two forms: GLP1(7-36), which is a 30-residue amide, and GLP1(7-37), which is a 31-residue glycine-extension peptide. It stimulates insulin secretion in response to the plasma glucose concentration change, and it is considered to have great potential in treating Type 2 diabetes.
The sequence of GLP-1(7-36) is as follows: His-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-NH2.
The sequence of GLP-1(7-37) is as follows: His-Ala-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Lys-Gly-Arg-Gly-NH2.
The total synthesis of GLP-1(7-36) was reported by Adelhorst et al. (12) in 1994 using the solid-phase peptide synthesis method. The peptide was synthesized on an ABI MED 422 multiple synthesizer. An Na-Fmoc strategy, which was modified according to Gausepohl et al. (13), was used starting from a Rink-resin. The side-chain protections were as follows: Asn, Gln, and His were proctected by Trt groups; Arg by a Pmc group; Asp, Glu, Tyr, Ser, and Thr by t-butyl groups; and Trp and Lys by Boc groups. The peptides were cleaved from the resin and deprotected by a treatment of TFA/triethylsilane/water (92.5:5:2.5) for 120 minutes and precipitated in t-butylmethyl ether and lyophilized from 10% acetic acid. The crude peptides were purified by reverse phase HPLC with Superpak Pep S C2/C18 column, obtaining a purity of >95%.
Glucose-dependent insulinotropic polypeptide (GIP)
Glucose-dependent insulinotropic polypeptide is originally known as gastric inhibitory polypeptide (GIP), which is a 42-residue peptide first isolated by Brown and Dryburgh (14). It is secreted from the duodenum and proximal jejunum in response to food. Two major physiological effects of GIP are inhibition of gastric acid secretion and stimulation of insulin release.
The sequence of porcine GIP is as follows: Tyr-Ala-Glu-Gly-Thr-Phe-Ile-Ser-Asp-Tyr-Ser-Ile-Ala-Met-Asp-Lys-Ile-Arg-Gln-Gln-Asp-Phe-Val-Asn-Trp-Leu-Leu-Ala-Gln-Lys-Gly-Lys-Lys-Ser-Asp-Trp-Lys-His-Asn-Ile-Thr-Gln-OH.
However, the first synthesized “GIP” (15) turned out not to be the real GIP, as the original sequence proposed by Brown and Dryburgh (14) was a 43-residue peptide. Ten years later, Jornvall et al. (16) revised the structure as one glutamine shorter. The first synthesis of the revised GIP structure was done by Mochizuki et al. (17) in 1981 by employing an azide-fragment condensation method in solution. The protected peptide fragments were prepared (Fig. 4), and the Trp residue in position 25 was introduced exceptionally by itself to the 26-42 or 26-43 peptide intermediate by the active ester method. The Cbz and Bn groups were removed by catalytic hydrogenation and the Boc and tBu groups by TFA treatment during the course of fragment condensation. After completing the synthesis, the crude protected peptide was treated with sodium in liquid ammonia to remove N-Cbz and N-Tos groups. The product was first purified by gel filtration on Sephadex G-25, followed by ion-exchange chromatography on CM-cellulose and HPLC with TSJ-Gel LS-420K C-18 column.
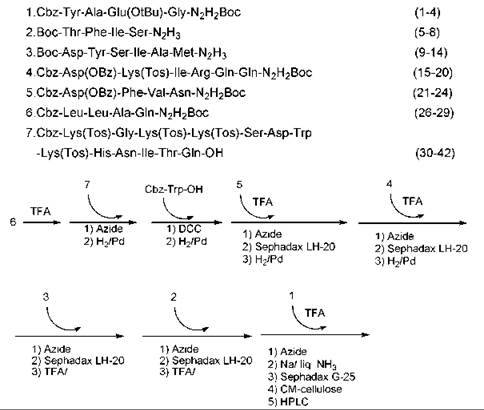
Figure 4. Protected fragments of peptide and synthesis scheme for GIP.
Motilin
Motilin is a 22-amino acid peptide first isolated from hog duodenal mucosa by Brown and colleagues in 1972 (18, 19). It was found to be released from endocrine cells of the duodenal-jejunal mucosa to control gastrointestinal muscles by controlling the migrating motor complex.
The sequence of motilin is as follows: Phe-Val-Pro-Ile-Phe-Thr-Tyr-Gly-Glu-Leu-Gln-Arg-Met-Gln-Glu-Lys-Glu-Arg-Asn-Lys-Gly-Gln-OH.
Ikota (20) reported the first solid-phase synthesis of motilin in 1980, and the overall yield was 1.8%. Coy et al. (21) improved the synthesis and got a 10% yield. Coy’s synthesis was carried out on a Beckman model 990 automatic peptide synthesizer using Na-Boc chemistry. Motilin was assembled stepwise on a 1% cross-linked BHA resin with Boc-α-benzyl-Glu as the first amino acid to be incorporated. In this manner, C-terminal Gln could be directly formed on HF cleavage of the final peptide from the resin. The detailed coupling schedule is shown in Table 2 (22). The symmetric anhydride method was used for coupling of all amino acids except for Asn and Gln, which were coupled as active HOBt esters, and DIC was used as the coupling reagent. The side-chain protections were as follows: Arg and His were protected by the Tos groups; Asp, Glu, Ser, and Thr by the Bn groups. Coupling reactions were monitored at each step by using the ninhydrin test. Couplings, which were incomplete after 1 hour, were recoupled by using the appropriate symmetric anhydride preformed at room temperature in DMF.
Table 2. Schedule of events for assembling the peptide BHA resin
|
Step |
Reagentor solvent and operations |
Time (min) |
|
1 |
DCM wash (x 3) |
1 |
|
2 |
33% TFA-DCM |
1 |
|
3 |
33% TFA-DCM |
25 |
|
4 |
DCM wash (x 3) |
1 |
|
5 |
EtOH wash (x 3) |
1 |
|
6 |
CHCl3 wash (x 3) |
1 |
|
7 |
10% Et3N-CHCl3 (x 2) |
5 |
|
8 |
CHCl3 wash (x 3) |
1 |
|
9 |
DCM wash (x 3) |
1 |
|
10 |
Na-Boc-amino acid (3 eq) + DIC (3 eq) in DCM* |
60 |
|
11 |
DCM wash (x 3) |
1 |
|
12 |
EtOH wash (x 3) |
1 |
*Asn and Gln were dissolved in DMF, Na-Boc-Arg(Tos) in 10% DMF-DCM. The extent of couplings was monitored by the ninhydrin test. Recouplings carried out by symmetric anhydride procedure in DMF if necessary.
After the completion of the solid-phase synthesis, 0.25 mmol of peptide-resin was treated with HF (30 mL) that contained 10% anisole for 30 minutes at 0 °C. After rapid removal of HF under a stream of nitrogen, free, deprotected peptide amide was precipitated by addition of ether, and then it was partially purified on Sephadex G-25. Additional purification was done on an octadecylsilyl-silica column (1.5 x 45 cm) by elution with a linear gradient formed from 200 mL each of 15 and 30% 1-propanol in 10% acetic acid at a flow rate of 2 mL/min. Examination of the fractions by TLC and HPLC located the peptide in the 3rd peak (Fig. 5). Fractions were pooled for maximum purity to give 70 mg (10%) of motilin.
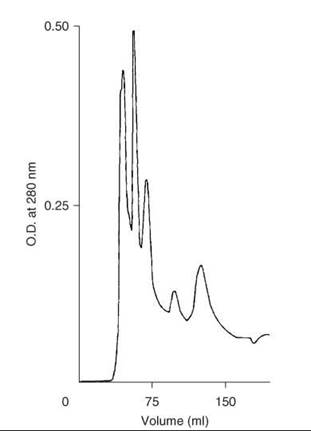
Figure 5. Purification of crude synthetic motilin on column of LRP-1 ODS silica. Taken from Coy et al. (21).
Neuropeptide Y (NPY)
NPY is a 36-amino acid peptide amide first isolated from porcine brain by Tatemoto et al. (23, 24) in 1982. It is found distributed in the central nervous system where it is involved in the control of blood pressure and appetite, and also in the peripheral nervous system, where it is a potent vasoconstrictor and presynaptic inhibitor of neurotransmission.
The sequence of porcine NPY is as follows: Tyr-Pro-Ser-Lys-Pro-Asp-Asn-Pro-Gly-Glu-Asp-Ala-Pro-Ala-Glu-Asp-Leu-Ala-Arg-Tyr-Tyr-Ser-Ala-Leu-Arg-His-Tyr-Ile-Asn-Leu-Ile-Thr-Arg-Gln-Arg-Tyr-NH2.
Krstenansky et al. (25) successfully prepared porcine NPY in 1987. It was synthesized by solid-phase techniques with an Applied Biosystems Inc. Model 430-A peptide synthesizer using MBHA resin. All residues were double coupled as the preformed symmetric anhydrides of the Na-Boc amino acid derivatives except for Asn and Gln, which were coupled with the DCC/HOBT methodology (refer to the section on “Corticotropin-releasing factor synthesis” for details). The side-chain protections were as follows: Arg and His were protected by Tos groups; Asp and Glu by cHx groups; Ser and Thr by Bn groups; Lys by a 2,6-Cl-Cbz group; Tyr by a 2-Br-Cbz group. After completing the synthesis, deprotection and the resin cleavage were performed by a treatment with HF that contained 5% anisole for 50 minutes at 0 °C. The HF was removed in vacuo at 0 °C, and the crude peptide was precipitated with ether and extracted with 30% acetic acid. The extract was lyophilized.
First-round purification was carried out on a 2.6 x 92-cm Sephadex G-15 column at 30 mL/h and 20 minutes per fraction (Fig. 6, top) (25). The fractions that contained the major peak detected by UV at 254 nm were combined and lyophilized. The residue was then purified for the second round by preparative HPLC on a 21.4 x 250 mm Rainin Dynamax C-18 column at 10 mL/min with 34% MeCN in 0.1% TFA. The main peak detected by the UV at 215 nm, and 280 nm was collected and lyophilized leaving 35 mg (3%) final product (Fig. 6, bottom).
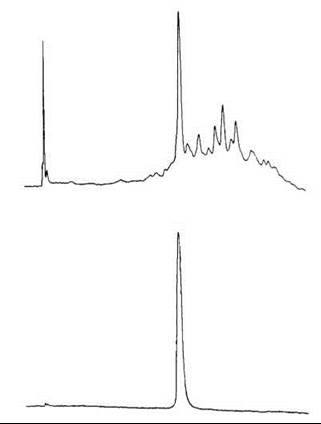
Figure 6. HPLC of synthetic porcine NPY. Top: after HF cleavage and Sephadex G-15 chromatography. Bottom: after preparative HPLC purification. Condition: Vydac 218TP54 (4.6 x 250 mm) C-18 column with a linear gradient of 25-50% acetonitrile in 0.1% TFA over 25 minutes at 2 mL/min. Taken from Krstenansky et al. (25).
Peptide tyrosine tyrosine (PYY)
PYY is a 36-amino-acid peptide originally isolated from porcine upper intestine by Tatemoto et al. (26, 27). It contains an N-terminal tyrosine and a C-terminal tyrosine amide and therefore is named peptide YY. It has a high degree of sequence homology (70%) with neuropeptide Y, and it strongly inhibits pancreatic exocrine secretion and jejunal and colonic motility as well as causes vasoconstriction.
The sequence of human PYY is as follows: Tyr-Pro-Ile-Lys-Pro-Glu-Ala-Pro-Gly-Glu-Asp-Ala-Ser-Pro-Glu-Glu-Leu-Asn-Arg-Tyr-Tyr-Ala-Ser-Leu-Arg-His-Tyr-Leu-Asn-Leu-Val-Thr-Arg-Gln-Arg-Tyr-NH2.
Tatemoto et al. (28) also reported a solid-phase synthesis of PYY. Human PYY was prepared manually using Na-Fmoc protection strategy on a 4-aminomethyl-3,5-dimethylphenoxyl resin (PAL resin). The coupling reaction was carried out using a five-fold excess of Na-Fmoc amino acid and DIC in DMF. The α-amino group of the growing peptide was deprotected with 20% piperidine in DMF for 10 minutes. The side-chain protections were as follows: Asp, Glu, Set, Thr, and Tyr were protected by t-Bu groups; Lys by a Boc group; His by a Trt group; Arg by a Mtr group. The peptide was deprotected and cleaved from the resin by a treatment with TFA/thioanisole/ethanedithiol/anisole (90/5/3/2) for 8 hours at room temperature. After filtration, TFA was removed in vacuo, and the peptide was precipitated by ether. The precipitate was redissolved in 0.1 M acetic acid and lyophilized. The crude peptide was purified using a reverse-phase HPLC column (MCI GEL ODS-1HU, 10 x 300 mm, Mitsubishi Kasei, Japan) with a linear gradient system of 0.1% TFA/water and 0.1% TFA/acetonitrile. The synthetic peptide obtained was found to coelute in HPLC with the natural peptide, and the results of amino acid and sequence analyses indicated that it was identical to human PYY.
Secretin
Secretin is a 27-residue peptide amide hormone produced by S cells of the duodenum. The primary effect of secretin is stimulating the release of bicarbonate from liver, pancreas, and duodenal tissues to inhibit gastrin-induced gastric acid release. It also enhances the effects of cholecystokinin and promotes normal growth and maintenance of the pancreas.
The sequence of secretin is as follows: His-Ser-Asp-Gly-Thr-Phe-Thr-Ser-Glu-Leu-Ser-Arg-Leu-Arg-Asp-Ser-Ala-Arg-Leu-Gln-Arg-Leu-Leu-Gln-Gly-Leu-Val-NH2.
Several classic syntheses of secretin have been reported using either fragment condensation or repetitive coupling of single residues (29); one solid-phase synthesis has also been reported (30). However, a peptide-stability problem arose, and it caused difficulty in developing an efficient solid-phase synthesis of the peptide. Coy and Gardner (31) developed a two-stage rapid purification method to avoid traditional long purification routes, and this approach solved potential degradation problem of the peptide.
Coy’s synthesis was carried out on a Beckman model 990 automatic peptide synthesizer (Beckman Instruments, Fullerton, CA) using Na-Boc chemistry. Secretin was assembled stepwise on a 1.0 mmol of 1% cross-linked BHA resin support using the synthesis schedule described in Table 2. The side-chain protections were as follows: Asp, Glu, Ser, and Thr were protected by Bn groups; Arg and His by Tos groups. Each amino acid was successively coupled in the presence of DIC and, in the case of Asn or Gln, 1 equivalent of HOBt was added. Coupling reactions were monitored at each step by using the ninhydrin test. Couplings that were incomplete within 1 hour were recoupled by using the appropriate symmetric anhydride preformed at room temperature in DMF.
After the completion of the solid-phase synthesis, the peptide resin was treated with HF that contained 10% anisole for 30 minutes at 0 °C. After rapid removal of HF under a stream of nitrogen, free, depretected peptide was precipitated by addition of ether, and it was washed and extracted into 2 M acetic acid. This solution was applied to a column (2.5 x 95 cm) of Sephadex G-25, which was eluted with 2 M acetic acid. Material that emerged just after the void volume as a major peak (254 nm) contained the major component confirmed by TLC. This material was injected onto a column (2.5 x 45 cm) of ODS silica LRP-1 (Whatman) (13-24 pm) and was eluted with a linear gradient of 15 and 35% 1-propanol in 0.1 M ammonium acetate (pH 4) at a flow rate of 5 mL/min and a pressure about 60psi. Fractions were examined by TLC and analytical HPLC, and the partially resolved peak emerging at 375 mL that contained the major component was found identical to a standard sample of secretin (Fig. 7). The peptide was lyophilized to give about 120 mg final product.
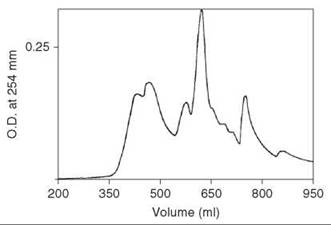
Figure 7. Chromatography of crude synthetic secretin eluted on a column (2.5 x 45 cm) of ODS silica LRP-1 (Whatman) (13-24 pm) with a linear gradient of 15 and 35% 1-propanol in 0.1 M ammonium acetate (pH 4) at a flow rate of 5 mL/min and pressure about 60 psi. Taken from Coy et al. (21).
Somatostatin
Somatostatin is a hormone that comprises two peptides, one built of 14 amino acids and the other of 28 amino acids. They are secreted not only by cells of the hypothalamus but also by delta cells of the stomach, intestine, and pancreas. They inhibit the release of numerous gut peptides, like CCK, gastrin, secretin, motilin, GIP, and they also inhibit insulin and glucagon secretion from the pancreas.
The sequence of hypothalamic somatostatin 14 is as follows: Ala-Gly-c(Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys)-OH.
The sequence of hypothalamic somatostatin 28 is as follows: Ser-Ala-Asn-Ser-Asn-Pro-Ala-Met-Ala-Pro-Arg-Glu-Arg-Lys-Ala-Gly-c(Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys)-OH.
Somatostatin 14 was isolated, characterized, and synthesized by Guillemin’s group in 1973 (32, 33). Several independent studies of the peptide’s synthesis were reported immediately after that (34-37). Among those studies, Rivier reported in detail the first solid-phase synthesis of the peptide in a highly purified form. The protected somatostatin tetradecapeptide was synthesized in a stepwise manner on chloromethylated resin prepared according to Stewart and Young (38). Na-Boc chemistry was used for the synthesis; all amino acids were coupled as symmetric anhydrides using DCC as the coupling reagent except for Asn, which was coupled as Np active ester. The side-chain protections were as follows: Thr and Ser were protected by Bn groups; Lys by a 2-Cl-Cbz group. Na-Boc-Cys(p-OMe-Bn) was used because it is easily removed by HF. Ala and Gly were introduced as a Cbz-protected dipeptide (Cbz-Ala-Gly-OH) to have a reliable internal standard to evaluate the amino-acid analyses. TFA was used for cleavage of the Na-Boc protecting groups, and 1,2-ethanedithiol was added for protection of the Trp residue from oxidation, which is a problem that has long been recognized in solid-phase synthesis. The completion of coupling was monitored by the ninhydrin test. The detailed schemes used for the synthesis are reported in Tables 3 and 4.
Table 3. Schedule for DCC Coupling in Solid-phase Synthesis of Somatostatin-14*
|
Step |
Reagents or solvents and operations |
Time (min) |
|
1 |
DCM wash, 80 mL (x 2) |
3 |
|
2 |
MeOH wash, 30 mL (x 2) |
3 |
|
3 |
DCM wash, 80 mL (x 3) |
3 |
|
4 |
50% TFA + 5% 1,2-ethanedithiol in DCM, 70 mL (x 2) |
10 |
|
5 |
DCM wash, 80 mL (x 2) |
3 |
|
6 |
Et3N 12.5% in DMF, 70 ml (x 2) |
5 |
|
7 |
MeOH wash, 40 mL (x 2) |
2 |
|
8 |
DCM wash, 80 mL (x 3) |
3 |
|
9 |
Boc-amino acid (10 mmol) in 30 mL of DMF (x 1) + DCC (10 mmol) in DMF |
30 |
|
10 |
MeOH wash, 40 mL (x 2) |
3 |
|
11 |
Et3N 12.5% in DMF, 70 ml (x 1) |
3 |
|
12 |
MeOH wash, 30 mL (x 2) |
3 |
|
13 |
DCM wash, 80 mL (x 2) |
3 |
* Aliquots taken for ninhydrin test: if negative, go back to step 1, if positive or slightly positive, go back to steps 9 → 13.
Table 4. Solid-phase synthesis of somatostatina schedule for Na-Boc-Asn-PNP coupling*
|
Step |
Reagents or solvents and operations |
Time (min) |
|
9 |
DMF wash, 60 mL (x 3) |
3 |
|
10 |
BocAsn-PNP (15 mmol) in 20 mL of DMF (x 1) |
800 |
|
11 |
MeOH wash, 30 mL (x 4) |
3 |
|
12 |
Et3N 12.5% in DMF, 30 ml (x 2) |
3 |
|
13 |
MeOH wash, 30 mL (x 2) |
3 |
|
14 |
DCM wash, 80 mL (x 3) |
3 |
*Same as lines 1-8 in Table (1) followed by changes shown. Aliquots taken for ninhydrin test: if negative, go back to step 1, if positive or slightly positive, go back to steps 9 → 13.
Cleavage and concomitant deprotection of the tetradecapeptide were achieved by HF in presence of anisole. After removing HF in vacuo and work-up, a white fluffy material that accounted for 60% of the calculated yield was obtained. A second HF treatment was applied to the resin, and 25% more of the peptide was obtained. The crude material was subjected to gel filtration. The main peak (Fig. 8, profile I) after lyophilization and routine handling was reapplied on the same column under the same conditions in an attempt to obtain one single symmetrical pattern (Fig. 8, profile II). A total yield of 28% in purified linear somatostatin was obtained.
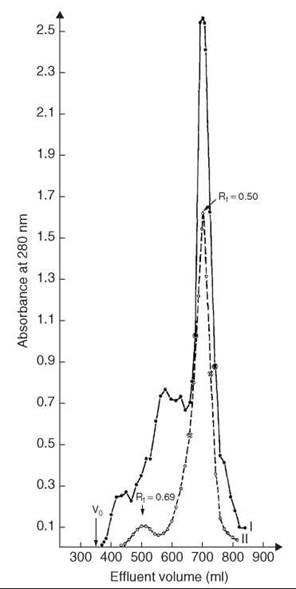
Figure 8. Gel filtration of crude and purified synthetic reduced somatostatin. Column: 2.5 x 200 cm Sephadex G-25F, 2 N AcOH, 10-2 M β-mercaptoethanol. V0 = hold-up volume: profile I(•) 1.25 g crude, yield 670-740 mL, 500 mg, 40%; profile II (bharathi) 500 mg from I, yield 660-728 mL, 350 mg, 28%. Taken from Rivier et al. (37).
Synthesis of somatostatin-28 has been reported by Nicolas et al. (39) in 1986. Na-Boc protected amino acids were assembled on chloromethyl/1% divinylbenzene resin by using solid-phase peptide synthesis techniques. The first amino acid was loaded by mixing the resin and Na-Boc-Cys(3,4-dimethylbenzyl)-OH in DMF for 18 hours at 50 °C in the presence of KI. The side-chain protections were as follows: Asp, Thr, Ser, and Glu were protected by Bn groups; Lys by a 2-Cl-Cbz group; Tyr by a 2-Br-Cbz group; Arp by a Tos group; Cys by a 3,4-dimethyl-Bn group; Trp by a For group. The coupling was done using the symmetric anhydride method according to the protocol shown in Table 5 (40), except Asn was first coupled for 15 hours as its p-nitrophenyl ester in DMF and then by the use of HOBt as reported (41). After the coupling was completed, the peptide-resin was subjected to steps 1-10, washed with EtOH, and dried. The cleavage of the peptide was done by treating the peptide-resin with liquid HF in the presence of anisole for 30 minutes at -20 °C and then 60 minutes at 0 °C. After removal of HF by a stream of nitrogen, the residue was stirred in EtOAc, and the solid material was filtered off. The filtrate was purified on a Sephadex G-25 column (2.6 cm x 100 cm) equilibrated and run with degassed 10% AcOH saturated with nitrogen. The main peak was lyophilized, which yielded 565 mg product. Deformylation and formation of the disulfide bond were carried out by dissolving 100 mg material in water (25 mL) that contained 75 pL hydrazine and taken to pH 11.5 with 1 M NaOH. After 2 minutes, the solution was diluted to 1000 mL with distilled water, adjusted to pH 8 with glacial acetic acid, and allowed to stand at 24 °C. After 22 hours, less than 1% free thiols remained as assessed by the Ellman’s method (42).
After lyophilization, the material was dissolved in 5 mL of 10% AcOH and filtered through a Sephadex G-50F column (2.5 cm x 91cm) in 10% AcOH. Isolation of material in the major peak gave 61 mg. This material was subjected to chromatography on CM-cellulose in a 1.5 cm x 35-cm column initially equilibrated with 10 mM NH4OAc, pH 4.2. A gradient was applied through a 200 mL constant volume mixing chamber that contained the initial buffer by introducing 0.45 M NH4OAc, pH 7. Material in the major peak was isolated to gave 16.5 mg (a 6% overall yield) of somatostatin-28.
Substance P
Substance P is an 11-amino-acid peptide amide hormone discovered by von Euler and Gaddum in 1931 (43) from certain tissue extracts, especially from intestinal plain muscles and brains of horses. It is involved in the transmission of pain impulses from peripheral receptors to the central nervous system. It is also involved in the vomit reflex, stimulates salivary secretions, and induces vasodilation. Antagonists seem to have antidepressant properties.
The sequence of substance P is as follows: Arg-Pro-Lys-Pro-Gln-Gln-Phe-Phe-Gly-Leu-Met-NH2.
This peptide was synthesized by Fisher et al. (44) in 1974 employing the solid-phase technique using Na-Boc chemistry. BHA-type resin was used for the synthesis on a Beckman Model 990 solid-phase peptide synthesizer. Dried BHA-resin hydrochloride was neutralized with triethylamine and stirred overnight with an excess of Na-Boc-Met and DCI in DCM. The remaining unreacted amino groups of the resin were acetylated with a mixture of Ac2O and triethylamine in DMF overnight. Amino-acid analysis of the resin, after hydrolysis in 6 N HCl-propionic acid (1:1), gave a value of 0.2 mM/g of Met. The side-chain protections were as follows: Lys was protected by a Cbz group; Arg by a Tos group. A 2.5-fold excess of each amino acid was used for coupling with a reaction time of 6 hours except for Gln, which was incorporated as a fivefold excess of its Np active ester with an 11-hour coupling time. Dry DCM was used as the coupling solvent except for Arg and Gln, which required purified DMF. Na-Boc deprotection was accomplished by 50% TFA in DCM with neutralization by 10% Et3N in DCM. The completion of coupling was monitored by the ninhydrin test, and recoupling was done when needed (refer to the sections on “Motilin synthesis” and “Secretin synthesis” for details). Cleavage of the peptide from the resin, with simultaneous removal of the protecting groups and formation of the carboxyl terminal amide, was affected with 20 mL of dry HF in the presence of 2mL of anisole for 1 hour at 0 °C. After removal of the excess HF in vacuo, the resin was washed with EtOAc to remove anisole, followed by 0.5 N HOAc to extract the peptide; 424 mg crude product was obtained and lyophilized.
Purification was done by gel filtration on a 102 x 2.5 cm column of Bio-Gel P2 eluted with 1.3% AcOH, with detection of the peptide peaks by UV at 256 nm. The main fraction (253 mg) was partitioned on a 100 x 1.5 cm column of Sephadex G-25 eluted with the system 0.1% AcOH-n-BuOH-Pyr (11:5:3) with detection of the peptide peaks by the Folin-Lowry procedure (45) at 700 nm, giving 90 mg (16.7%) of pure substance P.
Na-Fmoc synthesis of substance P also has been reported (46).
Table 5. Schedule for coupling in solid-phase synthesis of somatostatin-28
|
Step |
Reagents or solvents and operations |
Time (min) |
|
1 |
DCM wash, 15 mL (x 3) |
3 |
|
2 |
55% TFA-DCM wash, 15 mL (x 1) |
1 |
|
3 |
55% TFA-DCM wash, 15 mL (x 1) |
15 |
|
4 |
DCM wash, 15 mL (x 2) |
2 |
|
5 |
25% Dioxane-DCM wash, 15 mL (x 2) |
2 |
|
6 |
DCM wash, 15 mL (x 2) |
2 |
|
7 |
5% DIEA-DCM wash, 15 mL (x 2) |
2 |
|
8 |
DCM wash, 15 mL (x 2) |
2 |
|
9 |
5% DIEA-DCM wash, 15 mL (x 2) |
2 |
|
10 |
DCM wash, 15 mL (x 6) |
6 |
|
11 |
Symmetric anhydride of Boc-amino acid (1.5 mmole) in DCM 13 mL |
60* |
|
12 |
DCM wash, 15 mL (x 3) |
3 |
|
13 |
5% DIEA-DCM wash, 15 mL (x 2) |
2 |
|
14 |
DCM wash, 15 mL (x 6) |
6 |
|
15 |
Symmetric anhydride of Boc-amino acid (1.5 mmole) in DCM 13 mL |
60* |
|
16 |
DCM wash, 15 mL (x 3) |
3 |
|
17 |
33% EtOH-DCM wash, 15 mL (x 3) |
3 |
* In the case of Na-Boc-Val, -Leu, -Thr, 90 minutes of reaction time.
Vasoactive intestinal peptide (VIP)
VIP is a 28-amino-acid residue peptide amide isolated in 1970 by Said and Mutt in the course of the purification of secretin from porcine duodenum (47). It was characterized 2 years later as a highly basic peptide related to gut peptides. It inhibits acid and pepsin secretion, acts as a neurotransmitter in peripheral autonomic nervous system, and increases secretion of water and electrolytes from pancreas and gut.
The sequence of porcine VIP is as follows: His-Ser-Asp-Ala-Val-Phe-Thr-Asp-Asn-Tyr-Thr-Arg-Leu-Arg-Lys-Gln-Met-Ala-Val-Lys-Lys-Tyr-Leu-Asn-Ser-Ile-Leu-Asn-NH2.
Bodanszky reported the first synthesis of VIP using solution- phase peptide synthesis (48). Several other syntheses, both solid-phase and solution-phase, also have been reported (49-52). Among these syntheses, Fournier’s study is representative. Na-Boc solid-phase synthesis techniques were used. Peptide syntheses were carried out with a Vega peptide synthesizer (Vega Biotech., Tucson, AZ) in the automatic mode following operations as described (53) (similar to Tables 3 and 4) except that 1% D,L-methionine was used in TFA instead of 5% thioanisole. The first amino acid was loaded to the BHA resin (0.21 meq/g) via the DCC-HOBt procedure after neutralization of the resin with 25% triethylamine-DCM. The remaining amino acids were coupled as symmetric anhydrides and were prepared using a 6-molar excess of Na-Boc-amino acid and DCC. Nitrogen is bubbled through the suspension to eliminate air and moisture in the mixing reservoir. For Asn and Gln, couplings were achieved by the DCC-HOBt method. The side-chain protections were as follows: Ser, Thr, and Asp were protected by Bn groups; His by a Boc group; Tyr by a 2-Br-Cbz; Arg by a Tos group; Lys by a 2-Cl-Cbz group. Every coupling was monitored for completion using the ninhydrin test (refer to the sections on “Motilin synthesis” and “Secretin synthesis” for details). Excess amino groups were acetylated when the test showed slightly positive results (~1%). After the last amino acid was introduced, the cleavage and deprotection were executed in the Kel-F reaction vessels of a liquid HF apparatus. HF (8-10 mL/g) and a 5-molar excess D,L-methionine were added to the vessel that already contains anisole. The reaction proceeded for 30 minutes at -20 °C and for another 30 minutes at 0 °C. The HF was rapidly evaporated in vacuo, and the resin was washed with ether. The crude peptide was extracted with 30% acetic acid (~200 mL), and the solution was lyophilized.
The crude peptide was dissolved in a minimal volume of 0.005-M ammonium acetate buffer at pH 6.0, and then it was applied to a column of Whatman CM-23 ion exchange resin equilibrated with the same buffer. The peptide was eluted with a pH and ionic gradient of adding 0.20 M pH 7.5 ammonium acetate buffer to the initial one at a flow rate of 700 mL/h. The elution was monitored by UV at 280 nm. Fractions (12 mL) that corresponded to the major peak were collected and lyophilized twice to eliminate the volatile salt. A second chromatographic step on CM-23 resin was achieved with a pH and ionic gradient obtained by the addition of 0.12 M pH 7.5 to 0.005 M pH 5.9 at a flow rate of 700-1000 mL/h. In the third purification step, the semi-purified VIP was injected on a partition chromatography column packed with Sephadex G-50 resin and equilibrated with the solvent system 1-butanol, acetic acid, and 0.5%-pyridine (5:10:3). The flow rate was set to 20 mL/h, and 5-mL fractions were collected. Final purification (Fig. 9) was carried out on HPLC using a semipreparative Waters pBondapak C-18 column under isocratic conditions: methanol ammonium acetate 0.10 M, pH 5.2 (70:30). The flow rate was adjusted to 1.2 mL/min, and the peptide was detected at 246 nm by UV. Two-hundred fifty μL of 40 mg/mL peptide solution were injected. The main peak was collected and lyophilized, which gave an overall yield of 4%.
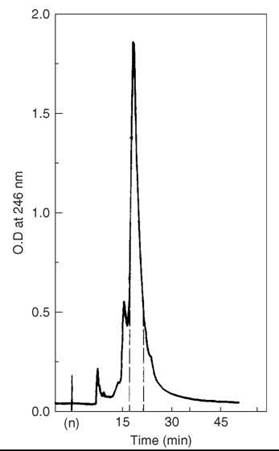
Figure 9. HPLC purification of partially purified VIP. Taken from Fournier et al. (52).
Corticotropin-releasing factor (CRF or CRH)
CRF is a 41-amino-acid peptide first isolated from ovine hypothalamic extracts and characterized by Vale et al. (54) in 1981. It has a high potency for stimulating the secretion of corticotropin-like and P-endorphin-like immunoactive substances.
The sequence of ovine CRF is as follows: Ser-Gln-Glu-Pro-Pro-Ile-Ser-Leu-Asp-Leu-Thr-Phe-His-Leu-Leu-Arg-Glu-Val-Leu-Glu-Met-Thr-Lys-Ala-Asp-Gln-Leu-Ala-Gln-Gln-Ala-His-Ser-Asn-Arg-Lys-Leu-Leu-Asp-Ile-Ala-NH2.
Vale et al. (54) also synthesized the peptide, but they did not provide details of the synthesis. In 1982, Sueiras-Diaz and Coy (55) published the first detailed synthesis of ovine CRF using Na-Boc solid-phase synthesis techniques. The amino acids were assembled on a 1% cross-linked BHA resin (2.1 g; 0.49-mmol amino groups/g) using a Beckman 990 automatic synthesizer (Beckman Instruments, Fullerton, CA). The symmetric anhydride procedure in DMF was used for the coupling of amino acids, and DIC was the coupling reagent. In the case of Asn and Gln, they were coupled with an equimolar amount of HOBt. Na-Boc protection was removed at each stage by two treatments with 33% TFA in DCM for 1 minute and 25 minutes. Reactive side-chains protections were as follows: Ser and Thr were protected by Bn groups; Lys by a 2-Cl-Cbz group; His and Arg by Tos groups; Asp and Glu by 4-Cl-Bn groups. Coupling reactions were monitored at each step by the ninhydrin test and repeated if incomplete after 60 minutes. If free amino groups persisted, they were acetylated with acetylimidazole (5% in DCM, 50 minutes). (Refer to the section on “Motilin synthesis” and “Secretin synthesis” for details.)
The completed, protected peptide resin, with its N-terminal Boc group removed to avoid alkylation of Met during HF cleavage steps, was deprotected and liberated from the resin support (1 mM) by a treatment with 60 mL anhydrous HF that contained 10% anisole for 1 hour at 0 °C. After removing the HF under nitrogen, the peptide was precipitated with ether, then filtered and extracted with 50% acetic acid. After reduction in volume, the solution was applied directly onto a column (2.5 x 95 cm) of Sephadex G-50 and eluted with 2 M AcOH. Fractions were collected and aliquots were examined by TLC in the solvent system of 1-butanol:pyridine:acetic acid:water (15:10:3:12). The peptide was visualized with ninhydrin, and then pooled and lyophilized to a constant weight of 3.92 g. This material was examined by HPLC on a column (0.4 x 25 cm) of Synchropack RP-18 (10 μm, 300 A pore size) using a gradient of 25-35% isopropanol in 0.1% TFA over 30 minutes. Absorption at 215 nm revealed a major peak (Fig. 10) that contained contaminants at its leading and trailing edges.
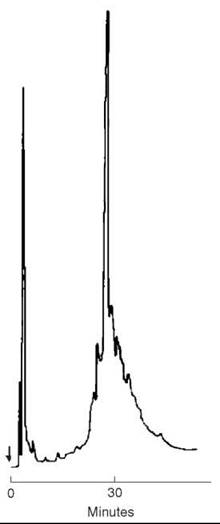
Figure 10. HPLC of 100 μg of crude CRF after Sephadex chromatography on C-18 Synchropack (10 pm, 300 A°)(0.4 x 25 cm) using a gradient of 25-35% isopropanol in 0.1% TFA over 30 minutes. Flow rate 1 mL/min., absorption at 215 nm. Taken from Sueiras-Diaz et al. (55).
Part of this material (340 mg) was purified even more by reversed-phase medium-pressure liquid chromatography procedure using a column (2.5 x 45 cm) of Whatman LRP-I (C-l8-bonded silica gel, 13-24 pm) eluted with a gradient from 15-60% acetonitrile in 0.1% TFA. Aliquots from the main peak were pooled and lyophilized to give 75 mg of almost pure peptide, which counted a 18.5% yield based on resin incorporation. The final purification was achieved by a preparative RP-HPLC procedure using a C-18, 10 μm, 300A Synchropack column (1 x 25 cm), loaded with 11.7 mg and eluted with a gradient of 23-36% isopropanol in 0.1% TFA developed over 1 h. Fractions were collected manually and aliquots were checked by HPLC. Those fractions judged pure, favoring purity rather than quantity, were then pooled, concentrated in vacuo and lyophilized to a constant weight of 4.55 mg, which gave a 7% overall yield.
Gonadotropin-releasing factor (GnRF or GnRH)
GnRH, which was originally known as luteinizing hormone (LH) and follicle-stimulating hormone (FSH) releasing hormone, is a 10-residue polypeptide isolated from porcine hypothalami by Schally et al. (56) in 1971. It acts on the gonadotrope to control the release of LH and FSH.
The sequence of porcine GnRH is as follows: (pyro)Glu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2.
Matsuo et al. (57) also have prepared this peptide via solid-phase peptide synthesis. The protected decapeptide resin ester (Na-Boc-Gln-His-Trp-Ser(OBzl)-Tyr(OBzl)-Gly-Leu-Arg (NO2)-Pro-Gly-resin ester) that corresponds to the amino acid sequence of LH-RH/FSH-RH was synthesized using Na-Boc chemistry by the method described by Stewart and Young (38), which starts with Na-Boc-Gly-resin ester (1.0 g: 0.35 mmole). The side-chain protections were as follows: Ser and Tyr were protected by Bn groups, Gln by a Np group, Arg by a NO2 group. Coupling was achieved with DCC with the single exception that Gln was coupled by means of its p-nitrophenyl ester. Stepwise synthesis was carried out in the DCM and/or DMF using a glass shaker at room temperature. The removal of the Na-Boc groups in the first seven steps was performed by treatment with 50% TFA in DCM for 20 minutes. After the incorporation of the Trp residue, 1 N HCI in acetic acid that contained 1% 2-mercaptoethanol was used for the removal of the Na-Boc group as described by Marshall (58). The neutralization was carried out by shaking with 10% triethylamine in CHCl3. In every DCC coupling step, 4 equivalents of Na-Boc-amino acid (4 x 0.35 mmole) for every equivalent of the starting glycine resin ester was used in the presence of 4 equivalents of DCC for 5 hours. An additional reaction with the same reagents was performed for another 5 hours. For the incorporation of His residue, 5 equivalents of Na-Boc-His were used, and the same reaction was carried out twice as above. Nitrophenyl ester coupling of Na-Boc-Gln was performed with 10 equivalents of the active ester for 5 hours, followed by an additional treatment for 5 hours in the presence of 5 equivalents of imidazole. The yield of the protected decapeptide resin ester was 1.38 g (about 78% based on dry weight and amino acid analysis).
To achieve cyclization of N-terminal glutaminyl group on the resin to the pyroglutamyl-ring after removal of Na-Boc group, the Na-Boc-glutaminyl-peptide resin ester was treated with 1 N HCI in acetic acid that contained 1% of 2-mercaptoethanol for 1 hour at room temperature. The peptide was cleaved by stirring in 20 mL of absolute methanol saturated with ammonia for 3 days at room temperature. After filtration, evaporation of the methanolic filtrate yielded 368 mg of the corresponding amide as a yellow semi-solid material. All protecting groups were removed by treatment with liquid HF in the presence of 2 mL of anisole for 1 hour at 0 °C.
After evaporation, the residual material was dissolved in 5 mL of acetic acid that contained 1% of 2-mercaptoethanol and heated in an evacuated sealed tube at 100 °C for 10 minutes for the completion of cyclization to the pyroglutamyl-group (59, 60). After addition of 50 mL of water, followed by lyophilization, the residual solid was dissolved in 0.2 N acetic acid and lyophilized. The resulting slightly yellow solid was purified by the counter-current distribution (CCD) in the solvent system of 0.1% acetic acid:butanol:pyridine (11:5:3) for 400 transfers. Lyophilization of fractions No. 282-343 (K = 2.0) after CCD afforded 153 mg of purified LH-RH as a white fluffy solid, which showed about 25% of the biological activity of natural porcine GnRH. On paper-chromatography (Whatman No. 1) in 1-butanol:pyridine:acetic acid:water (15:10:3:12), this material showed two spots, positive to Pauly’s reagent and negative to ninhydrin, with Rf = 0.67 and 0.79, respectively. Paper-electrophoresis (pH 6.4 pyridine acetate, 2,500 volts, 1 hour) also exhibited two spots with RLVP2 0.95 and 0.76, respectively. The main spot with Rf = 0.67 and RLVP2 = 0.95 was found to be identical with natural porcine LH-RH. Additional purification of 9-mg portions of the CCD-purified material was carried out on a column of carboxymethylcellulose (1 x 60 cm) using 0.1 M ammonium acetate pH 7.0 for elution. This process yielded 2 mg of essentially pure synthetic GnRH, which was identical chromatographically and electrophoretically with natural porcine GnRH.
Growth hormone releasing factor (GRF or GRH)
GRF has been isolated and characterized from a human tumor of the pancreas (61) as well as from rat (62), porcine (63), bovine (64), caprine (65), ovine (65), and human (65) hypothalamus stalk-median eminence. It is a 40-44-amino-acid peptide hormone produced in the arcuate nucleus of the hypothalamus and released from neurosecretory nerve terminals of these arcuate neurons, and it is carried by the hypothalamo-hypophysial portal circulation to the anterior pituitary gland where it stimulates growth hormone (GH) secretion (66).
The sequence of human GRH is as follows: Tyr-Ala-Asp-Ala-Ile-Phe-Thr-Asn-Ser-Tyr-Arg-Lys-Val-Leu-Gly-Gln-Leu-Ser-Ala-Arg-Lys-Leu-Leu-Gln-Asp-Ile-Met-Ser-Arg-Gln-Gln-Gly-Glu-Ser-Asn-Gln-Glu-Arg-Gly-Ala-Arg-Ala-Arg-Leu-NH2.
Ling et al. (65) have described the synthesis of hGRH in 1984 after they isolated and characterized the peptide from human hypothalamic tissues. The peptide was prepared by solid-phase methodology on a Beckman model 990 peptide synthesizer (Beckman Instruments, Fullerton, CA) using Na-Boc chemistry. The MBHA resin (6 g, 0.6 mmol/g) was used, and the side-chain protections were as follows: Ser, Glu, Asp, and Thr were protected by Bn groups; Arg by a Tos group; Lys by a 2-Cl-Cbz group; Tyr by a 2,6-Cl-Bn group. The C-terminal amino acid was loaded to the MBHA resin using the symmetric anhydride method, and DCC was the coupling reagent. The subsequent amino acids were coupled according to the schedule in Table 6.
After the last amino acid had been incorporated, the Na-Boc protecting groups were removed before 1 g of the peptide-resin conjugate was treated with a mixture of 14 mL of HF, 1.5 mL of anisole, and 0.25 mL of methylethyl sulfide at -20 °C for 0.5 hour and at 0 °C for 0.5 hour. The HF was removed in vacuo at 0 °C, and the resulting peptide and resin mixture was washed twice with ether and twice with CHCl3 and ether alternately. The peptide was extracted five times with 2 M CH3COOH, and the extract was lyophilized.
The first purification was done by loading the lyophilized product on a column of Sephadex G-50 developed in 30% CH3COOH to remove the truncated fragments and salt. The next step of purification was by CM-32 carboxymethylcellulose cation-exchange chromatography developed with a gradient generated by adding 2.5 volumes of 0.4 M NH4OAc at pH 6.5 to 1 volume of 0.01 M NH4OAc at pH 4.5. Final purification was achieved by partition chromatography on Sephadex G-50 using the solvent system 1-butanol:ethanol:pyridine:0.2 M CH3COOH (4:1:1:7). The chromatographic fractions were monitored by UV at 280 nm and TLC on 0.25-mm-thick precoated silica gel 60 plates with the solvent system 1-butanol:pyridine:CH3COOH: water (6:6:12:4.8), and the spots were detected with ninhydrin spray. Four-hundred twenty-six milligrams of hGRF was obtained after final purification with an overall yield of 2.3%.
Table 6. Schedule of events for assembling the peptide p-MBHA resin
|
Step |
Reagents or solvents and operations |
Time (min) |
|
1 |
DCM wash (x 2) |
1 |
|
2 |
55% TFA/5% 1,2-ethanedithiol in DCM wash, (x 1) |
0.5 |
|
3 |
Deprotect with 55% TFA/5% 1,2-ethanedithiol in DCM |
20 |
|
4 |
DCM wash, (x 3) |
1.5 |
|
5 |
CH3OH wash, (x 2) |
1 |
|
6 |
10% triethylamine in DCM wash, (x 2) |
1 |
|
7 |
CH3OH wash, (x 2) |
1 |
|
8 |
Repeat step 6, 7 |
|
|
9 |
DCM wash (x 2) |
1 |
|
10 |
Couple with symmetric anhydride* of Boc-amino acid (1 mmole/g of resin) in DCM, Leu, Arg Asn and Gln were coupled in 30% DMF in DCM** |
120 |
|
11 |
DCM wash, (x 1) |
0.5 |
|
12 |
50% DMF-DCM wash, (x 2) |
1 |
|
13 |
10% triethylamine in DCM wash |
0.5 |
|
14 |
CH3OH wash, (x 2) |
1 |
|
15 |
DCM wash, (x 2) |
1 |
|
16 |
Acetylate unreacted N “-amino group with 25% acetic anhydride in DCM |
20 |
|
17 |
DCM wash, (x 2) |
1 |
|
18 |
CH3OH wash, (x 2) |
1 |
* The symmetric anhydride was formed with one equivalent amount of DCC.
**In the cases of Asn and Gln, a 1.2 equivalents of HOBt were included.
Thyrotropin-releasing factor
TRF was isolated from porcine hypothalamus by Schally et al. (67, 68) and from ovine hypothalamus by Guillemin et al. (69) It is a tripeptide amide with the structure of pyroGlu-His-Pro-NH2. Classic synthesis of the peptide has been reported in 1970 (70); solid-phase synthesis was envisaged 1 year later, and Rivaille et al. claimed they successfully prepared the target with a better yield (71). Na-Boc chemistry was used in the synthesis, and Na-Boc was introduced into the amino acid by reacting amino acids with Boc-azide at a constant basic pH. The His side chain was protected with an o,p -dinitrophenyl group. Protected His was coupled to the proline linked on the chloromethylresin by the symmetric anhydride method using DCC (refer to the sections on “Somatostatin synthesis” and “Angiotensin II synthesis” for details). The glutamyl residue was introduced in two ways: 1) Na-Boc-pyroglutamic acid was condensed in the presence of DCC and 2) Na-Boc-glutamine dinitrophenylester was reacted with the dipeptide linked to the resin in DMF. The tripeptide was split from the resin by aminolysis, the imidazole protecting group was removed by thiolysis at pH = 8 (72), and the glutamine converted into pyroglutamic acid by boiling acetic acid (73).
The tripeptide was purified by Sephadex G10 filtration followed by silica-gel chromatography. The pure material did not react with ninhydrin because of the presence of N -terminal py- roglutamic acid. After silica-gel thin layer chromatography in several solvent systems, the product gave a single spot when detected with Pauly’s reagent with overall yield of 73%.
Liver peptide hormones
Angiotensin II
Angiotensin II is an octapeptide derived from biologically inactive decapeptide angiotensin I, which comes from angiotensino- gen through the cleavage by the kidney enzyme renin. It is responsible for essential hypertension through stimulated synthesis and for the release of aldosterone from adrenal cells.
The sequence of horse Angiotensin II is as follows: Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-OH.
Several syntheses of the peptide have been reported by solution methods (74-76). After the introduction of solid-phase peptide synthesis, Marshall and Merrifield conducted the first study of the synthesis of the peptide by using the new technique (77). Na-Boc chemistry was used, and Merrifield resin was selected as the solid support. The side chain protections were as follows: His, Arg, and Asp were protected by Bn groups; Arg by a NO2 group. The Phe was esterified onto the resin in ethanol with the presence of 1 equivalent of triethylamine. The symmetric anhydride method was used for the coupling of the amino acids, and DCC was the coupling reagent. The following cycle of reactions was used to introduce each new residue (Table 7):
Table 7. Scheme of peptide assembling for angiotensin II
|
Step |
Reagents or solvents and operations |
Time (min) |
|
1 |
Glacial acetic acid wash (x 3) |
1 |
|
2 |
Deprotect with 1 N HCl in glacial acetic acid |
30 |
|
3 |
Glacial acetic acid wash (x 3) |
1 |
|
4 |
C2H5OH wash, (x 3) |
1 |
|
5 |
DMF wash, (x 3) |
1 |
|
6 |
Neutralize the HCl with 3 mL of triethylamine in DMF |
10 |
|
7 |
DMF wash, (x 3) |
1 |
|
8 |
DCM wash, (x 3) |
1 |
|
9 |
Add 3.83 mmol of the appropriate BOC amino acid in 20 mL of DCM and allowed to mix |
10 |
|
10 |
Add 3.83 mmol of DCC to couple* |
120 |
|
11 |
DCM wash, (x 3) |
1 |
|
12 |
C2H5OH wash, (x 3) |
1 |
* For the im-benzyl-L-His and nitro-L-arginine cycles, step 8 was deleted, and DMF was substituted for DCM in steps 9-11.
Amino acid analysis showed the average value of the eight amino acid residues to be 0.13 mmol/g of peptide resin or 0.16 mmol/g of unsubstituted copolymer. Removing the protecting groups was achieved by suspending the peptide resin in 20 mL anhydrous TFA and bubbling a slow stream of HBr through the fritted disk of the reaction vessel at 25 °C. The suspension was filtered, and the resin was washed 3 times with 10 mL portions of TFA. The filtrates were evaporated in vacuo. The product was redissolved in TFA and re-evaporated. The syrupy product was then dissolved in acetic acid and lyophilized to give 1.47 g.
Purification of the peptide was done by countercurrent distribution method. A 300-mg portion of the crude octapeptide was purified by 100 transfers in a 1-butanol:acetic acid:water (4:1:5) system. Over 80% of the Sakaguchi-positive material was located in one peak which matched closely a theoretical curve with distribution constant, k = 0.30. The material in the peak was collected and the organic phase was removed by evaporation. The residual aqueous phase was removed by lyophilization; 193 mg of product was obtained, which was equivalent to a yield of 56%.
Pancreatic peptide hormones
Amylin
Amylin is a 37-residue peptide hormone first discovered independently by two research groups in 1987 (78, 79). Amylin is secreted by pancreatic P-cells at the same time as insulin (in a roughly 100:1 ratio), and it is the major component of diabetes-associated islet amyloid deposits. It inhibits basal and insulin-stimulated glucose uptake as well as glycogen synthesis by soleus muscles (80). Thus, amylin is also known as a diabetes-associated peptide.
The sequence of human amylin is as follows: Lys-c(Cys-Asn-Thr-Ala-Thr-Cys)-Ala-Thr-Gln-Arg-Leu-Ala-Asn-Phe-Leu-Val-His-Ser-Ser-Asn-Asn-Phe-Gly-Ala-Ile-Leu-Ser-Ser-Thr-Asn-Val-Gly-Ser-Asn-Thr-Tyr-NH2.
One solid-phase synthesis of amylin has been reported in 1991 by Balasubramaniam et al. (81). Standard Na-Boc chemistry was used for the synthesis, and MBHA resin was selected as the solid support. The coupling was done by using the symmetric anhydride method except for Arg, Asn, and Gln, which were coupled as their HOBt esters (refer to the section on “Corticotropin-releasing factor synthesis” for details). After completing the chain elongation, the peptide was cleaved from the resin using HF at 0 °C. The residue was then oxidized with K3Fe(CN)6 to form the disulfide bond, followed by purification on semipreparative reversed phase column. The overall yield of the synthesis was between 10-20%.
Glucagon
Glucagon is a 29-residue polypeptide secreted by pancreas α-cells. It increases lipid mobilization and glycogenolysis to increase blood glucose levels. It was first discovered by Kimball and Murlin in 1923 (82) when studying pancreatic extracts, but the sequence was not determined until 1957 (83).
The sequence of mammalian glucagons is as follows: His-Ser-Gln-Gly-Thr-Phe-Thr-Ser-Asp-Tyr-Ser-Lys-Tyr-Leu-Asp-Ser-Arg-Arg-Ala-Gln-Asp-Phe-Val-Gln-Trp-Leu-Met-Asn-Thr-OH.
Early attempts to synthesize glucagons proved to be difficult because of the unusual structure, and successful examples were not reported until Wunsch’s classic solution-phase synthesis (84). Mojsov and Merrifield (85) reported the first stepwise solid-phase synthesis of glucagon in 1981, in which they used biphenylisopropyloxycarboxyl amino-acid derivatives and t-Bu based side-chain protections, which were unstable. They improved that to Na-Boc strategy and benzyl-based side chain protections by using their newly developed PAM resin in 1984, which gave a rapid, convenient synthesis (86). The aminomethyl-resin was prepared according to Mitchell et al. (87, 88). The first amino acid Thr was loaded to the resin through 2 steps: 1) Na-Boc-Thr(Bzl)-OH was treated with powdered KF2H2O and [4-(bromomethyl) phenyl]acetic acid phenacyl ester to produce Na-Boc-Thr(Bzl)-OCH2C6H4CH2COOH and 2) the product was then dissolved in DCM and allowed to couple with aminomethyl-resin at the presence of 1 equivalent of DCC, and unreacted amino groups were blocked by acetylation. The peptide chain was then elongated by the symmetric anhydride coupling method. In all, 8 eq. of amino acid and 4 eq. of DCC were used for each coupling, except for Leu and Gly in 1.6 eq and 1.2 eq., respectively. Asn and Gln were coupled with presence of HOBt; Arg at positions 17 and 18 were coupled twice each. The side-chain protections were as follows: Ser and Thr were protected by Bn groups; His and Arg by Tos groups; Asp by a cHx group; Tyr by a Br-Cbz group; Lys by a Cl-Cbz group; Trp by a For group. One synthetic cycle consisted of the following steps: 1) DCM wash, 1 minute, x 3; 2) deprotection with 50% TFA/ DCM wash, 1 minute prewash + 20 minutes; 3) DCM wash, 1 minute, x 6; 4) neutralization with 5% iPr2EtN/DCM wash, 1 minute, x 3; 5) DCM wash, 1 minute, x 6; 6) performed symmetric anhydride coupling in DCM (4 eq.), 60 minutes; 7) DCM wash, 1 minute, x 6; 8) neutralization with 5% iPr2EtN/DCM wash, 1 minute, x 3; 9) DCM wash, 1 minute, x 6; and 10) repeat steps 6) and 7). After the completion of the peptide assemblies, the final deprotection and cleavage of the peptide was done in a Teflon vessel using the HF procedure (80). The crude peptide was then purified by preparative HPLC using C-18 column, the eluant from the major peak was collected and desalted on Sephadex G-10, obtaining pure synthetic glucagon with 48% overall yield.
Insulin
Insulin is a polypeptide hormone produced by p-cells of the pancreas; it contains two disulfide bonded peptide chains: an A chain of 21 residues and an intra-disulfide bridge and a B chain of 30 amino acids. Insulin is well known for regulating carbohydrate metabolism as well as increasing glucose uptake and utilization.
The structure of insulin is shown in Fig. 11. Insulin became one of the most demanded drugs after finding its power to treat Type 1 diabetes in the 1920s. However, it was thought almost impossible to be chemically synthesized at that time after the structure was elucidated by Sanger in 1955 because it involved three disulfide bridges, complicated higher-level structures, and so on. But these complications did not stop it from becoming one of the hottest targets for synthetic chemists. Several independent syntheses of insulin have been reported in the 1960s (89-91) using classic peptide-synthesis methods, which usually required several months to finish, and yields were low. Merrifield et al. (92) reported a fast and efficient synthesis later on by employing solid-phase peptide synthesis technique that only took several days with good yields. Na-Boc chemistry was used for the synthesis. The assembling of the fully protected insulin B chain started with 1.9 mmol of Na-Boc-Ala esterified to 8 grams of the supporting cross-linked polystyrene resin. The following 29 cycles of coupling were completed by using the symmetric anhydride method and DCC as the coupling reagent. The side-chain protections were as follows: Glu, Cys, Ser, Tyr, and His were protected by Bn groups; Lys by a Cbz group; Arg by a Tos group. Cleavage of the peptide from the resin was done by firstly bubbling HBr through a solution of resorcinol in TFA and then into a suspension of the peptide resin in TFA that contained Met (92). The peptide was precipitated from water to remove the Met. The yield of partially protected peptide was 64%, which was based on the amount of Ala originally esterified to the resin. The partially protected peptide was thoroughly dried and then treated with sodium in liquid ammonia as described by Niu et al. (93), except that the stable light blue end point was limited to exactly 15 seconds to prevent excessive cleavage of the Thr-Pro bond. Under these conditions, this cleavage was only 20-25%, whereas 80% was lost during a 60-second treatment. The shorter time was adequate for complete debenzylation of His and Cys and complete detosylation of Arg. The deprotected triacontapeptide was converted to the S-sulfonate. On electrophoresis, a major Pauly-positive spot existed with the same mobility as that of the B-chain S-sulfonate (BSSO3) obtained by sulfitolysis of natural bovine insulin, and a minor contaminant probably caused by B1-27. The overall yield from the first Ala residue was 21%.
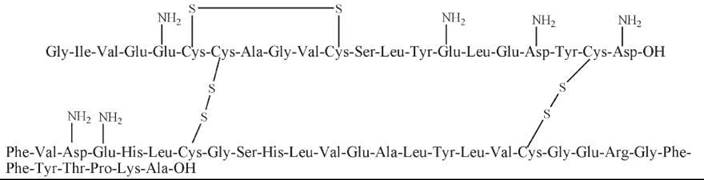
Figure 11. The structure of insulin.
The A chain was synthesized by the automated solid-phase procedure (94). Three grams of Na-Boc-Asn resin was carried through 20 reaction cycles as described for the B chain except that the reagents used for all deprotection and neutralization steps were 4M HCl in dry dioxane and triethylamine in CHCl3, respectively (95). The peptide was cleaved from the resin as described above (yield, 69%). The total time required was 8 days. The S-benzyl protecting groups were removed by treatment with sodium in liquid ammonia (96), and the four cysteine residues were converted to the S-sulfonates (ASSOB) (over all yield, 37%).
The synthetic A- and B-chain sulfonates (ASSO3 and BSSO3) were combined with each other and with the complementary natural chains. The mixtures were first reduced to the thiol derivatives with thioglycolic acid at 25 °C by a modification of the method of Du and colleagues (91, 97, 98). The reduced chains were precipitated together at pH 3.8, washed, and then oxidized in air at pH 10.0 to form insulin. The molar ratio of ASSO3:BSSO3 at the beginning of reductions was 4:1. The oxidation mixtures were extracted once by the method of Du et al. (97), and the extracts were lyophilized. On electrophoresis, these insulin preparations appeared as Pauly-positive spots with the same mobility as standard bovine insulin, and both synthetic chains could participate in the formation of biologically active insulin when they were combined with the natural chains or with each other.
Pancreatic polypeptide
Pancreatic polypeptide is a peptide hormone secreted by pancreatic polypeptide-producing cells in the islets of Langerhans in the pancreas. It consists of 36 amino acids and has a molecular weight about 4200 Da. It suppresses the pancreatic secretion and stimulates gastric secretion.
The sequence of human pancreatic polypeptide (hPP) is as follows: Ala-Pro-Leu-Glu-Pro-Val-Tyr-Pro-Gly-Asp-Asn-Ala-Thr-Pro-Glu-Gln-Met-Ala-Gln-Tyr-Ala-Ala-Asp-Leu-Arg-Arg-Tyr-Ile-Asn-Met-Leu-Thr-Arg-Pro-Arg-Tyr-NH2.
Regular solid-phase synthesis of hPP has been reported by Meyers and Coy (99). The peptide was assembled on BHA resin using Na-Boc chemistry. Amino acids were coupled as symmetric anhydrides with DIC except for Asn and Gln, which were coupled as HOBt ester. The side-chain protections were as follows: Ser and Thr were protected by Bn groups; Glu and Asp by Cl-Bn groups; Arg by a Tos group; Tyr by a 2-Br-Cbz group. Deprotection of the Na-Boc groups was done via treatment with 25% and 50% TFA in DCM. Couplings were monitored by the ninhydrin test, and they were repeated if not complete after 1 hour (refer to the sections on “Motilin synthesis” and “Secretin synthesis” for details). Free amino group found after double couplings were acetylated by acetylimidazole in DCM. After completing the synthesis, the peptide resin was treated with HF with 10% anisole; deprotected crude peptide was released from the resin and precipitated in ether. The material was applied on a column of Sephadex G-25, which was followed by a column of CM-cellulose; fractions from the major peak were collected and lyophilized. Final purification was done on a partition column of Sephadex G-50, which resulted in pure hPP that showed a single peak on HPLC. The overall yield of this synthesis was 6% based on the moles of amino groups on the resin.
Pituitary peptide hormones
Adrenocorticotropin (ACTH)
ACTH is a 39-amino acids polypeptide hormone cleaved from a precursor peptide called pro-opiomelanocortin, which is released from corticotrope cells of the anterior pituitary gland. ACTH acts through the stimulation of cell surface ACTH receptors, which are primarily located on the adrenocortical cells. It stimulates the cortex of the adrenal gland and increases the synthesis of corticosteroids. ACTH is also related to the circadian rhythm in many organisms.
The sequence of human ACTH is as follows: Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-Gly-Lys-Lys-Arg-Arg-Pro-Val-Lys-Val-Tyr-Pro-Asn-Gly-Ala-Glu-Asp-Glu-Ser-Ala-Glu-Ala-Phe-Pro-Leu-Glu-Phe-OH.
The total synthesis of ACTH attracted many researchers in the 1960s and 1970s. Schwyzer and Sieber (100) first synthesized porcine ACTH by using a conventional solution-phase peptide synthesis method. After the development of solid-phase synthesis by Merrifield (92), Yamashiro and Li (101) extended the scope of how to prepare this important biomolecule. Na-Boc chemistry was used throughout Yamashiro and Li’s synthesis. Phe was esterified to the resin by reacting the tetramethylam- monium salt of Na-Boc-Phe to Merrifield resin (0.72 mmol of Cl/g) in DMF with triethylamine at ambient temperature for 24 hours (102). The symmetric anhydride method was used for assembling the peptide chain, and DCC was the coupling reagent. The attachment of Glu was purposely limited to 0.28 mmol/g to reduce the loading on the resin and to select the most sterically favorable sites. Unreacted amines were blocked by acetylation. The remaining 37 cycles of couplings were accomplished by using a regular DCC coupling procedure, except that the deprotection of the Trp Na-Boc group was achieved by treating with 25% TFA in DCM for 30 minutes followed by treating with the 50% reagent for 6 minutes. The side-chain protections were as follows: Asp, Ser and Glu were protected by Bn groups; His by a Boc group; Arg by a Tos group, Lys by a 2-Br-Cbz group; Tyr by a 2,6-Cl-Bn group (refer to the sections on “Somatostatin synthesis” and “Angiotensin II synthesis” for details). After completing the synthesis, a portion (1.00 g) of the peptide resin was cleaved and deprotected with HF in the presence of anisole. The resulting peptide material was purified on a column of Dowex 1-X4 (acetate form), lyophilized, followed by chromatography on Sephadex G-10 in 1N acetic acid in which only one peak of 346 mg peptide material was obtained. It was then subjected to gel filtration on Sephadex G-25, which gave a major peak of 202-mg peptide material. The material was treated with dithiothreitol in 0.1 N acetic acid for 21 hours at 50 °C to convert any Met-sulfoxide to Met and then chromatographed on carboxymethylcellulose (Fig. 12) to give a major peak of 68-mg peptide material, which was very similar to that of the natural ACTH. This 32 mg of the purified material was purified even more by partition chromatography on Sephadex G-50 with a solvent system of 1-butanol:pyridine:0.1% aqueous acetic acid (5:3:11) to give 12-mg highly purified synthetic hACTH with almost the same Rf as natural hACTH (Fig. 13).
Figure 12. Carboxymethylcellulose chromatography of crude synthetic hACTH. Taken from Yamashiro et al. (101).
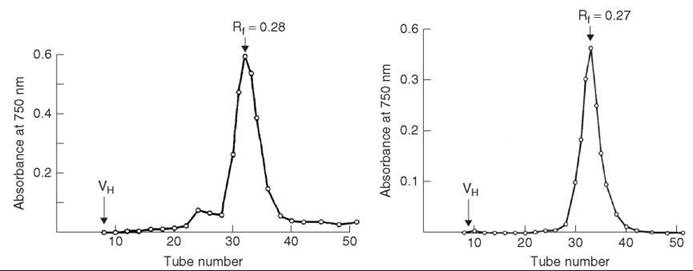
Figure 13. Partition chromatography of Sephadex G-50 of Natural hACTH (left) and highly purified synthetic hACTH (right). Taken from Yamashiro et al. (101).
Melanocyte-stimulating hormones (melanotropins, MSHs)
MSHs consist of three peptide hormones: α-MSH, β-MSH, and γ-MSH, which are secreted by intermediate lobe of the pituitary gland. They are cleaved from the same precursor peptide as ACTH. Their basic function is stimulation of melanocytes to darken skin and stimulation of melanin synthesis to darken the skin and hair. They also have been found to be released in the brain affecting appetite, sexual arousal, and many other functions.
The sequence of α-MSH is as follows: N-Acetyl-Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2.
The sequence of human β-MSH is as follows: Ala-Glu-Lys-Lys-Asp-Glu-Gly-Pro-Tyr-Arg-Met-Glu-His-Phe-Arg-Trp-Gly-Ser-Pro-Pro-Lys-Asp-OH.
The sequence of porcine β-MSH is as follows: Asp-Glu-Gly-Pro-Tyr-Lys-Met-Glu-His-Phe-Arg-Trp-Gly-Ser-Pro-Pro-Lys-Asp-OH.
The sequence of γ-MSH is as follows: Tyr-Val-Met-Gly-His-Phe-Arg-Trp-Asp-Arg-Phe-Gly-OH.
Solid-phase synthesis of α-/β-MSH were achieved in our group in 1980 (103), both using Na-Boc chemistry. In the preparation of α-MSH, amino acids were coupled successively to valine-BHA resin with threefold excess of Na-Boc amino acid and 2.4-fold excess of DCC in 1-15 hours. Removal of the Na-Boc protecting group was achieved by a treatment of 45% TFA in DCM that contained 2% anisole. The side-chain protections were as follows: Ser and Asp were protected by Bn groups; Tyr by a 2,6-Cl-Bn group; Lys by a 2,6-Cl-Cbz group; Arg and His by Tos groups; Trp by a For group. After completing all coupling cycles, the amino terminal end of the peptide was acetylated with threefold excess of N-acetylimidazole (refer to the sections on “Motilin synthesis” and “Secretin synthesis” for details). The peptide was cleaved from the resin, and all protecting groups were removed with anhydrous liquid HF with the exception of the For group on Trp. The residue was purified by gel filtration on Sephadex G-15, followed by deformylation with 4N NaOH at pH 11.5 for 3 minutes. The reaction was quenched by addition of glacial AcOH to a final pH of 4.5. Cation exchange chromatography was used for the purification of α-MSH, followed by gel filtration on Sephadex G-25. Porcine β-MSH was prepared similarly but on Asp-Merrifield resin.
The total synthesis of γ-MSH was reported by Ling et al. (104). Na-Boc chemistry was used for the synthesis. The coupling was started from loading Na-Boc-Gly to the Merrifield resin. The rest of the amino acids were coupled subsequently to the resin using DCC as coupling reagent for 1 or 2 hours, except 8 hours for Asn, which was coupled as Np ester. After each coupling was done, the unreacted amino group was blocked by acetylation with Ac2O. Peptide was cleaved and deprotected by a treatment of HF with presence of anisole and methylethyl sulfide. The residue was purified by cation-exchange chromatography on CM-32 carboxylmethyl cellulose and followed by gel filtration on Sephadex G-25. The overall yield of the synthesis was 10.3%.
Oxytocin
Oxytocin holds a special place in the development of modern hormone and neurotransmitter chemistry and biology. It was the first peptide hormone (neurotransmitter) to be isolated, its structure determined, and prepared by total synthesis. Oxytocin is a 9-amino-acid polypeptide made in magnocellular neurosecretory cells of the hypothalamus, and is released into the blood from the posterior lobe of the pituitary gland. It causes uterine contraction and milk ejection in lactating women as well as facilitates birth and breastfeeding. It is also involved in social recognition and bonding, aspects of sexual function, and might be involved in the formation of trust between people.
The sequence of oxytocin is as follows: c(Cys-Tyr-Ile-Glu-Asp-Cys)-Pro-Leu-Gly-NH2. The synthesis of oxytocin has drawn significant interest to chemists. It was first made by du Vigneaud et al. (105, 106), and many other syntheses have been reported since then in 1950s, 1960s, and beyond. One of the most high-yield and rapid syntheses was done by Manning (107). The protected nonapeptide was synthesized in a stepwise manner using Na-Boc chemistry. The symmetric anhydride method was used for the couplings, and DCC was the coupling reagent, except that Asn and Gln were coupled as their p-nitrophenyl esters. The side-chain protections were as follows: Trp and Cys were protected by Bn groups. Final Cys was coupled as N-benzyloxycarbonyl-S-benzyl derivative. After completing the synthesis, the dried resin was weighed, giving a yield of 81%. The protected nonapeptide was cleaved from the resin by ammonolysis, which was achieved by suspending the peptide resin in anhydrous methanol and bubbling with a stream of ammonia from a refluxing solution of dry ammonia at —5 °C for 2.5 hours, at 4 °C for overnight, and at 23 °C for 2 hours. The overall yield of the protected nonapeptide was 59% based on the amount of Gly originally esterified to the resin. Sodium and liquid ammonia reduction was used to remove the Bn protecting groups on Cys and Tyr and the disulfide bond was formed by K3Fe(CN)6 oxidation (108). The residue was then purified by gel filtration on Sephadex G-15, and the pure oxytocin was obtained in an overall yield of 27%.
Vasopressin
The presence of vasopressin has been known for more than 100 years when people found pressor activity in extracts of the posterior lobe of the pituitary gland (109). However, it took almost a half-century before du Vigneaud and his colleagues figured out what it is (110, 111). Arginine vasopressin is a 9-amino-acid human hormone that is responsible of reducing plasma volume and increasing plasma osmolality by inducing the kidneys to conserve water.
The sequence of arginine vasopressin (human) is as follows: c(Cys-Tyr-Phe-Gln-Asn-Cys)-Pro-Arg-Gly-NH2. The sequence of lysine vasopressin (porcine) is as follows: c(Cys-Tyr-Phe-Gln-Asn-Cys)-Pro-Lys-Gly-NH2.
The first syntheses of human and porcine vasopressin were done in du Vigneaud’s lab using solution-phase chemistry (112). Many other syntheses have been reported since then. More recent solid-phase syntheses of [8-Arg]/[8-Lys]-vasopressin through crystalline-protected nonapeptide intermediates were achieved by Meienhofer et al. (113, 114). Solid-phase Na-Boc chemistry was employed for those syntheses. In the case of [8-Lys]-vasopressin, the symmetric anhydride method was used for coupling, and DCC was the coupling reagent. Lys, Asn, and Gln were introduced into the preloaded Na-Boc-Gly-resin as their p-nitrophenyl esters. The side-chain protections were as follows: Cys was protected by a Bn group; Lys by a Tos group. Deprotection of the Na-Boc groups was done by a treatment with anhydrous TFA. After completing the peptide assembling, the protected peptide was cleaved from the resin by ammonolysis, and was crystallized from DMF as colorless needles. The crystalline-protected peptide was deprotected by sodium liquid-ammonia reduction, followed by air oxidation to form the disulfide bridge. The crude [8-Lys]-vasopressin was desalted on Amberlite IRC-50 column and lyophilized to give a powder with an overall yield of 24.7%. The synthesis of [8-Arg]-vasopressin used essentially identical methods.
Placental peptide hormones
Relaxin
Relaxin is a peptide hormone produced in the corpora lutea of pregnant mammals and is responsible for the widening of the birth canal to provide for the passage of the offspring. Human relaxin contains two peptide chains, an A chain of 24 amino acids, and a B chain of 29 amino acids, which are linked to each other by two disulfide bridges.
The structure of human relaxin is shown in Fig. 14. Tregear and colleagues (115-119) have done many studies on the synthesis of relaxin, but results were mainly published in meeting papers, which do not provide synthetic details. An interesting total synthesis of human relaxin was reported by Bullesbach and Schwabe (120). The highlight of this synthesis was their use of cysteine-protecting groups in the selective synthesis of the three disulfide links. The most stable disulfide link, the intrachain disulfide loop between A10 and A15, was synthesized first. The liberation of the cysteine side chain in position A24 and the subsequent thiolysis of the S -activated cysteine in position B23 directed the formation of the interchain disulfide bond A24-B23. The third disulfide bond was formed by oxidative removal of the corresponding protecting groups in positions A11 and B11.

Figure 14. The structure of human relaxin.
Na-Fmoc solid-phase chemistry was used for the synthesis of the A chain. HMP resin was selected as solid support, and standard DCC/HOBt coupling protocol was used to elongate the peptide chain. The side-chain protections were as follows: Glu, Asp, Tyr, Ser, and Thr were protected by t-Bu groups; His by a Trt group; Lys by a Boc group; Arg by a Pmc group. The Cys side chains were protected by S-Trt groups in positions A10 and A15, by an Acm group in A11, and by a methyl-Bn group in A24. Deprotection of the Na-Fmoc groups was done in 20% piperidine in DMF. After finishing the peptide chains synthesis, the peptide was cleaved from resin and deprotected with TFA using thiophenol as a scavenger to yield an A chain with two free thiol groups (A10, A15) and two differently protected cysteines. The formation of the intrachain disulfide loop was achieved by titration with iodine in 50% acetic acid. After purification with HPLC, the chain was stored in this form and the methyl-Bn group was removed prior to reaction with the corresponding B chain by HF, and the resulting A chain that contains one thiol group (A24), one disulfide link (A10/A15), and one Acm group (A11) was about 95% pure.
The B chain was synthesized using Na-Boc chemistry. PAM-resin was used as the solid support, and standard DCC/ HOBt coupling protocol was used to elongate the peptide chain. The side chain protections were as follows: Glu, Ser, Thr, and Asp were protected by Bn groups; Arg by a Tos group, Lys by a Cl-Cbz group, Trp by a For group; Met was protected as a sulfoxide (refer to the section on “Glucagon’s synthesis” details). Cys was protected by an Acm group in position B11 and by a 2-nitropyridinesulphonyl group in position B23. After completing the synthesis, the peptide was cleaved and partially deprotected with HF, followed by purification on Sephadex G-50. Trp(formyl) and Cys(Npys) remained untouched.
The formation of the interchain disulfide link A24/B23 was achieved in 8M guanidinium chloride at pH 4.5 using monothiol-A chain and thiol-activated B chain in a molar ratio of 1:1.1 for 24 hours at 37 °C. The third disulfide bond was synthesized by oxidation with iodine in aqueous AcOH. Final deprotection of the partially protected relaxin was achieved by reducing Met-sulfoxide in aqueous TFA that contained NH4I and removed the For group with NaOH. The resulting human relaxin was purified by reverse phase HPLC.
Thyroid peptide hormones
Calcitonin
Calcitonin is a 32-amino-acid polypeptide hormone that was first purified in 1962 by Copp and Cheney (121). It was originally thought as a product from parathyroid glands, but later it was discovered to be made by the C cells of the thyroid gland. Calcitonin participates in calcium and phosphorus metabolism, lowers plasma calcium and phosphate levels, and it has been used as a drug for bone and mineral disorders for a long time.
The sequence of human calcitonin is as follows: c(Cys-Gly-Asn-Leu-Ser-Thr-Cys)-Met-Leu-Gly-Thr-Tyr-Thr-Gln-Asp-Phe-Asn-Lys-Phe-His-Thr-Phe-Pro-Gln-Thr-Ala-Ile-Gly-Val-Gly-Ala-Pro-NH2.
Synthesis of human calcitonin was first carried out by Sieber et al. (122-125) in 1968 using solution-phase chemistry. More recently, Shi and Rabenstein (126) reported a convenient and high-yield synthesis by using Na-Fmoc solid-phase chemistry. Fmoc-PAL-PEG-polystyrene resin was selected as the solid support. The CID/HOAt combination was used as the coupling reagent, and 20% piperidine in DMF was used to deprotect the Na-Fmoc group. The side-chain protections were as follows: Cys, Gln, Asn, and His were protected by Trt groups; Ser, Thr, Tyr, and Asp by t-Bu groups; Lys by a Boc group. After completing the synthesis, the peptide resin was cleaved and deprotected by 81% TFA with phenol, thioanisole, ethanedithiol dimethylsulfide, and water, and Met sulfoxide was also reduced during the cleavage (as Met was oxidized during the synthesis). The deprotected crude peptide was then oxidized with trans-[Pt(en)2Cl2]2+ to form the intramolecular disulfide bond and to obtain human calcitonin.
Calcitonin gene-related peptid (CGRP)
Human CGRP (hCGRP) is a 37-residue polypeptide isolated by Morris et al. (127) from human medullary thyroid carcinoma. It acts as a vasodilator and stimulates the formation of cAMP.
The sequence of hCGRP is as follows: Ala-c(Cys-Asp-Thr-Ala-Thr-Cys)-Val-Thr-His-Arg-Leu-Ala-Gly-Leu-Leu-Ser-Arg-Ser-Gly-Gly-Val-Val-Lys-Asn-Asn-Phe-Val-Pro-Thr-Asn-Val-Gly-Ser-Lys-Ala-Phe-NH2.
The peptide has been made using a conventional solution- phase method (128). Improved synthesis on solid support has been reported later by Smith et al. (129), who have prepared hCGRP using Na-Boc chemistry and MBHA resin as solid support. All amino acids were coupled as HOBt active ester by using DCC/HOBt, the deprotection of the Na-Boc groups was done by treating with 30% TFA in DCM. The side-chain protections were as follows: Asp, Thr, and Ser were protected by Bn groups; His by a Bom group; Arg by a mesitylene-2-sulfonyl group; Lys by a 2-Cl-Cbz group. When the coupling yield was greater than 99%, the unreacted amino groups were acetylated by Ac2O; when the yield was lower than 99%, another coupling cycle was performed before acetylation (refer to the section on “Corticotropin-releasing factor synthesis” for details). After completing the synthesis, the peptide was cleaved with TFA, and the disulfide bond was formed by oxidation with K3Fe(CN)6 in NH4OAc solution. The crude hCGRP was purified by gel filtration on a column of Bio-Gel P6, followed by ion-exchange column chromatography on CM-Sephadex C25. Fractions that correspond to the hCGRP were collected and lyophilized, and final purification was achieved on HPLC using a Vydac C18 semipreparative column, obtaining pure hCGRP with 4.4% overall yield.
Peptide Neurotransmitters
Dynorphin
Dynorphins are a class of endogenous opioid peptides produced by many different populations of neurons. They function primarily as a K-opioid receptor agonist; therefore, they may act as antidote against addiction to cocaine. However, they also have p- and 8-opiod receptor agonist activities. Dynophin A, which is a 17-residue peptide, and Dynophin B, which is a 13-residue peptide, are found in nature.
The sequence of dynophin A (1-17) is as follows: Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Ile-Arg-Pro-Lys-Leu-Lys-Trp-Asp-Asn-Gln-OH.
The sequence of dynophin B (1-13) is as follows: Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Gln-Phe-Lys-Val-Val-Thr-OH.
Dynorphin A with a diiodotyrosine at position 1 was synthesized by using Na-Boc solid-phase chemistry (130), and many other syntheses of the peptide and its analogs also were reported. An optimized Na-Fmoc chemistry synthesis of [8-Ala]-dynorphin, which is a dynorphin-A analog particularly potent in an opiate receptor-binding assay (131), was reported by Sole and Barany (132). Fmoc-PAL-Nle-MBHA resin was selected as the solid support, which has a tris (alkoxy)-benzylamide linker that acts as side-chain anchor to prevent alkylation of the deprotected protecting groups to the peptide residues such as Cys, Met, Tyr, and Trp. Coupling was achieved by using DIC/HOBt as the coupling reagent, and the deprotection of the Na-Fmoc groups was done by treatment with 20% piperidine in DMF. After completing the synthesis, several cleavage cocktails were tried, and the best one turned out to be TFA with phenol, water, and triisopropylsilane (88:5:5:2), which gave more than 95% cleavage yield. The crude peptide was then purified by RP-HPLC, giving pure [8-Ala]-dynorphin A in 58% overall yield.
Synthesis of dynorphin B also has been reported by using fragment condensation in solution-phase (133).
Endorphin
Endorphins are endogenous opioid peptides produced by the pituitary gland and the hypothalamus in vertebrates. They are known as “natural pain killers” because of their analgesic effect. The best-known endorphins are α-, β-, and γ-endorphin, of which β-endorphin seems to be most implicated in pain relief.
The sequence of a-endorphin is as follows: Tyr-Gly-Gly-Phe-Met-Thr-Ser-Glu-Lys-Ser-Gln-Thr-Pro-Leu-Val-Thr-OH.
The sequence of human P-endorphin is as follows: Tyr-Gly-Gly-Phe-Met-Thr-Ser-Glu-Lys-Ser-Gln-Thr-Pro-Leu-Val-Thr-Leu-Phe-Lys-Asn-Ala-Ile-Ile-Lys-Asn-Ala-Tyr-Lys-Lys-Gly-Glu-OH.
The sequence of y-endorphin is as follows: Tyr-Gly-Gly-Phe-Met-Thr-Ser-Glu-Lys-Ser-Gln-Thr-Pro-Leu-Val-Thr-Leu-OH.
Many syntheses of P-endorphin have been achieved in the past, either in solution or on solid-phase. For example, an Na-Fmoc synthesis on solid-phase was reported by Atherton et al. (134). p-Alkoxybenzyl ester resin was selected as the solid support; coupling was done by using the symmetric anhydride and HOBt active ester method, except that Asn and Gln were coupled as p-nitrophenyl esters. The Na-Fmoc groups were deprotected by 20% piperidine in DMF. After completing the synthesis, the peptide was cleaved from the resin, and the side-chain protection groups were removed with anhydrous TFA in the presence of excess Met. The peptide was purified by CM-52 chromatography, with an overall yield of 41%.
α-/γ-Endorphin have been synthetically prepared by Ling (135). An Na-Boc strategy was used for the synthesis. In the case of a-endorphin, Na-Boc-Thr(OBn) was loaded on a chloromethyl-resin. The rest of the amino acids were coupled to the resin by using DCC as coupling reagent, and the deprotection of the Na-Boc groups was done using 50% TFA in DCM. After incorporation of the Met, the TFA deprotection step was modified with the inclusion of 5% 1,2-ethanedithiol. The side-chain protections were as follows: Thr, Ser, and Glu were protected by Bn groups; Tyr by a 2,6-Cl-Bn group, Lys by a 2-Cl-Cbz group. After the completion of the chain elongation, the peptide was cleaved by HF with 10% anisole at —5 °C. The crude peptide was purified by using a CM-32 car- boxymethyl cellulose column first, followed by gel filtration on Sephadex G-25F column and partition chromatography on a Sephadex G-25F column. The pure a-endorphin was obtained with an 18% overall yield. y-Endorphin was prepared in the same manner with a 30% overall yield.
Enkephalin
Enkephalins are endogenous opioid peptides that have analgesic effects. Met-enkephalin and Lys-enkephalin are found in nature, and they are pentapeptides that share the same first four residues.
The sequence of Met-enkephalin is as follows: Tyr-Gly-Gly-Phe-Met-OH.
The sequence of Leu-enkephalin is as follows: Tyr-Gly-Gly-Phe-Leu-OH.
Both peptides have been prepared in solution for the last 30 years (136, 137), and many solid-phase syntheses also have been reported. In addition, syntheses of these peptides have employed enzymatic methods, which could be done in low-water content systems on a preparative scale (138). Na-Cbz and Na-Boc protected Met-enkephalin and Leu-enkephalin were prepared by means of a-chymotrypsin, papain, thermolysin, and bromelain adsorbed on Celite. α-Chymotrypsin has a primary specificity for bulky and hydrophobic amino acids (Phe, Tyr, Trp, Leu, Met), and it was selected for making the Tyr-Gly, Phe-Leu, and Phe-Met peptide bonds. Papain was selected for Gly-Gly and Gly-Phe peptide bonds. Bromelain is very similar to papain as far as its specificity. All these proteases are serine or cysteine type, and the peptide bond formation can be done under kinetically controlled conditions. Thermolysin is an asparyl protease, and it was selected mainly for Phe-Leu and Gly-Phe bonds. MeCN, EtOAc, and methyl caproate were used as solvents with a controlled amount of buffer or at fixed water amount.
The synthetic scheme is shown in Fig. 15 (138), which was based on 4 + 1 enzymatic fragment condensation. Na-Cbz-Tyr-Gly-Gly-Phe-Leu-NH2 and Na-Boc-Tyr-Gly-Gly-Phe-Met-NH2 were prepared in good yield.
Figure 15. Enzymetic synthesis of Cbz-Enkephalin[Leu] and Na-Boc-Enkephalin[Met]. Cam = carboxamidomethyl acyl-donor esters. Taken from Clapes et al. (138).
Leumorphin
Leumorphin is a 29-amino-acid peptide first predicted by Yamamoto et al. (139, 140) when sequencing the cloned DNA complementary to porcine hypothalamic mRNA. They synthesized this peptide and found that it has potent opiate activity as predicted (140), and they also proved the existence of this peptide in humans (141).
The sequence of procine leumorphin is as follows: Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Gln-Phe-Lys-Val-Val-Thr-Arg-Ser-Gln-Glu-Asp-Pro-Asn-Ala-Tyr-Tyr-Glu-Glu-Leu-Phe-Asp-Val-OH.
Porcine leumorphin was synthesized by a solid-phase technique with Na-Boc strategy. The peptide chain was elongated on a chloromethylated resin with use of a peptide synthesizer (Beckman Model 990B). The side-chain protections were as follows: Asp, Glu, Thr, and Ser were protected by Bn groups; Tyr by a 2,6-Cl-Bn group; Lys by a 2-Cl-Cbz group; Arg by a Tos group. After the completion of the synthesis, the peptide was deprotected and cleaved from the resin with HF in the presence of anisole and ethane dithiol (refer to the sections on “Somatostatin synthesis” and “Angiotensin II synthesis” for details). The crude peptide was purified by gel chromatography on Sephadex G-10, followed by reverse-phase HPLC.
Tachykinin
Tachykinins are peptides characterized by the C -terminal ami- dated pentapeptide Phe-X-Gly-Leu-Met-NH2
Neurokinin A and B
Neurokinin A and B are both decapeptides discovered by Kimura et al. (142) from the porcine spinal cord. Both have very similar amino acid composition and sequence homology to the undecapeptide substance P, and they also have similar biological activities as substance P. They are potent bronchoconstrictors, constrict smooth muscle, and activate the micturition reflex.
The sequence of neurokinin A (substance K) is as follows: His-Lys-Thr-Asp-Ser-Phe-Val-Gly-Leu-Met-NH2.
The sequence of neurokinin B is as follows: Asp-Met-His-Asp-Phe-Phe-Val-Gly-Leu-Met-NH2.
These peptides were synthesized by solid-phase method using standard Na-Boc chemistry (143), and the couplings were performed with a Beckman Model 990 peptide synthesizer (Beckman Instruments, Fullerton, CA). The BHA resin was used as a solid support, and the amino acids were coupled to the resin using the symmetric anhydride method; DCC was the coupling reagent. The side-chain protections were as follows: Asp, Ser, and Thr were protected by Bn groups; His by a Tos group; Lys by a Cl-Cbz group (refer to the sections on “Motilin synthesis” and “Secretin synthesis” for details). After the completion of the synthesis, the peptide was cleaved and deprotected by HF, and the crude peptide was purified by gel filtration on Sephasex G-25. HPLC elution was applied as the second purification, and followed by desalting on a column of Sephadex G-10, obtaining more than 98% pure peptides.
Eledoisin, physalaemin, and kassinin
These peptides have only been reported and prepared by traditional solution-phase syntheses (144-146). However, because their structures are not complicated, they can be readily prepared by modern solid-phase techniques.
Others
Gastrin-releasing peptide (GRP)
Gastrin-releasing peptide is a 27-amino acid-containing peptide first isolated and sequenced by McDonald et al. (147) from porcine gastric mucosa. As the name itself implies, it stimulates the release of gastrin from gastric mucosa. It is also called mammalian bombesin as it has an identical C-terminal decapeptide with the exception of one residue as bombesin, which gives GRP similar biological activity as bombein. It lowers body temperature and increases plasma levels of epinephrine and glucose (148).
The sequence of GRP is as follows: Ala-Pro-Val-Ser-Val-Gly-Gly-Gly-Thr-Val-Leu-Ala-Lys-Met-Tyr-Pro-Arg-Gly-Asn-His-Trp-Ala-Val-Gly-His-Leu-Met-NH2.
Synthesis of this peptide has been achieved by Marki et al. (149). Na-Boc solid-phase synthesis techniques were employed, and MBHA resin was selected as the solid support. All the amino acids were coupled as symmetric anhydrides using DCC as the coupling reagent except for Asn, which was coupled as Na-Boc-Asn-ONp ester with HOBt. After completing each residue for one or two coupling cycles, unreacted amino groups were acetylated. The side-chain protections were as follows: His and Arg were protected by Tos groups; Thr and Ser by Bn groups; Tyr by a 2,6-Cl-Bn group; Lys by a Cl-Cbz group. After the completion of the synthesis, the crude peptide was cleaved from the resin and the side-chain protection groups were deprotected by treating with HF (refer to the section on “Corticotropin-releasing factor synthesis” for details). Gel filtration on Sephasex G-25 was applied as the first purification of the peptide, followed by semi-preparative reverse phase HPLC and lyophilization to give the TFA-peptide salt in a 5% overall yield.
Neurotensin
Neurotensin is a tridecapeptide discovered by Carrawy and Leeman (150) from bovine hypothalami in 1973. Neutotensin is distributed mainly in the central nervous system and in some regions of the digestive tract in various mammals. It has a broad spectrum of biological activities, including kinin activities such as inducing hypotension, contraction of ileum and uterus, and relaxation of the duodenum; it also can cause a rapid increase of glucose levels in blood.
The sequence of neurotensin is as follows: Glu-Leu-Tyr-Glu-Asn-Lys-Pro-Arg-Arg-Pro-Tyr-Ile-Leu-OH.
Carrawy and Leeman also sequenced (151) and synthesized (152) this peptide. Standard solid-phase synthesis using Na-Boc chemistry was employed for the synthesis, and the Merrified chloromethylated resin was selected as the solid support. Amino acids were coupled as symmetric anhydrides using DCC as the coupling reagent, except that Asn and Gln were coupled as p-nitrophenyl esters. The side-chain protections were as follows: Glu and Tyr were protected by Bn groups; Lys by a Cbz group; Arg by a NO2 group. The deprotection of the Na-Boc groups was achieved by 50% TFA in DCM. After completing the synthesis, the peptide was cleaved and partially deprotected by treatment with HBr-TFA, and the remaining NO2 group was removed by hydrogenation (refer to the sections on “Somatostatin synthesis” and “Angiotensin II synthesis” for details). The crude peptide was first purified on Sephadex LH-20 column, followed by cation exchange chromatography on sulfoethyl-Sephadex column. Pure neurotensin was obtained with 7% overall yield.
Bradykinin
Bradykinin is a 9-residue peptide discovered by Silva et al. (153). It is found to be a potent endothelium-dependent vasodilator, to cause contraction of nonvascular smooth muscle, and to increase vascular permeability; it also is involved in the mechanism of pain.
The sequence of bradykinin is as follows: Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH.
Bradykinin has been prepared synthetically many times in the 1960s (154-156). The first solid-phase synthesis was achieved by Merrifield (157) using Na-Boc chemistry. More recent modifications of bradykinin synthesis were reported by Khan et al. (158), who also used Na-Boc solid-phase synthesis technique. Amino acids were coupled as active esters to the preloaded Na-Boc-Arg(NO2)-OCH2-resin. Phe and Gly were coupled as O-Tcp esters, Pro was coupled as O-Pcp ester, and Ser was coupled as an O-NSu ester. The side-chain protections were as follows: Ser was protected by a Bn group, Arg by a NO2 group. After completing the synthesis and before removing the last Na-Boc protecting group, the peptide resin was submitted to hydrogenation using Pd(OAc)2 as catalyst, which resulted in only the Na-Boc protected crude peptide. Deprotection of the Na-Boc groups with TFA followed by purification on carboxymenthyl-cellulose column gave bradykinin in 20% overall yield.
Summary and Conclusions
The total synthesis of peptide natural products, especially hormones and neurotransmitters, is a highly developed area of organic synthesis. Although peptides with numerous ^-substituted amino acids, highly constrained peptides, highly hydrophobic peptides, and N-alkyl substituted amino acids can present synthetic challenges, numerous synthetic strategies have been developed to overcome these problems. Furthermore, despite all the mythology about the applications of peptides as drugs for a wide variety of diseases, peptide drugs are becoming increasingly important in medicine because of their general lack of toxicity and their high efficacy especially for hormones and neurotransmitters. These trends are likely to accelerate into the future as numerous alternative methods of drug delivery are developed and optimized even more. This trend will be especially true for hormones and neurotransmitters, in which oral delivery is often a very poor choice for numerous reasons. Furthermore, the cost of peptide synthesis has decreased dramatically in recent years, and several companies have been developing increasingly efficient syntheses of peptides for use as pharmaceutical reagents in up to metric ton quantities.
As outlined in this article, several very effective solution- and solid-phase synthetic methods have been developed for the total synthesis of peptides. We have covered most synthetic strategies and tactics that have been developed and found to be useful. In general, successful peptide synthesis depends on careful attention to the details of the synthesis and careful analytical monitoring of the progress of the synthesis. If this care is taken, then successful synthesis of most peptides can readily be accomplished by a competent synthetic chemist.
Additional readings about peptide synthesis and its practical and mechanistic considerations can be found in many books, chapters in books and reviews. A good starting point for the interested person includes the items listed within the Further Reading section.
References
1. IUPAC-IUB Joint Commission on Biochemical Nomenclature. Nomenclature and symbolism for amino acids and peptides. Recommendations 1983. Eur. J. Biochem. 1984; 138:9-37.
2. de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981; 28:89-94.
3. Ruskoaho H. Atrial natriuretic peptide: synthesis, release, and metabolism. Pharmacological Rev. 1992; 44:479-602.
4. Von Geldern TW, Rockway TW, Davidsen SK, Budzik GP, Bush EN, Chu-Moyer MY, Devine EM Jr, Holleman WH, Johnson MC, Lucas SD, Pollock DM, Smital JM, Thomas AM, Opgenorth TJ. Small atrial natriuretic peptide analogs: design, synthesis, and structural requirements for guanylate cyclase activation. J. Med. Chem. 1992; 35:808-816.
5. Ivy AC, Oldberg E. Hormone mechanism for gall-bladder contraction and evacuation. Am. J. Physiol. 1928; 86:599-613.
6. Kurano Y, Kimura T, Sakakibara S. Total synthesis of porcine cholecystokinin-33 (CCK-33). J. Chem. Soc.: Chem. Commun. 1987; 323-325.
7. Penke B, Zarandi M, Zsigo J, Toth GK, Kovacs K, Telegdy G. Synthesis of cholecystokinin peptides. Pept. Proc. Eur. Pept. Symp., 19th. 1987; 447-450.
8. Penke B, Nyerges L. Solid-phase synthesis of porcine cholecystokinin-33 in a new resin via Fmoc strategy. Peptide Res. 1991; 4:289-295.
9. Gregory RA. A review of some recent developments in the chemistry of the gastrins. Bioorg. Chem. 1979; 8:497-511.
10. Anderson JC, Barton MA, Gregory RA, Hardy PM, Kenner GW, MacLeod JK, Preston J, Sheppard RC, Morley JS. Antral hormone gastrin. Synthesis of gastrin. Nature 1964; 204:933-934.
11. Brown E, Williams BJ, Sheppard RC. An efficient general method for the synthesis of gastrins. J. Chem. Soc.: Chem. Commun. 1980; 22:1093-1095.
12. Adelhorst K, Hedegaard BB, Knudsen LB, Kirk O. Structure-activity studies of glucagon-like peptide-1. J. Biol. Chem. 1994; 269:6275-6278.
13. Gausepohl H, Kraft M, Frank R. In situ activation of Fmoc amino acids by BOP in solid-phase peptide synthesis. Pept. Proc. Eur. Pept. Symp., 20th. 1989: 241-243.
14. Brown JC, Dryburgh Jr. Gastric inhibitory polypeptide. II. Complete amino acid sequence. Can. J. Biochem. 1971; 49:867-872.
15. Yajima H, Ogawa H, Kubota M, Tobe T, Fujimura M, Henmi K, Torizuka K, Adachi H, Imura H, Taminato T. Synthesis of the tritetracontapeptide corresponding to the entire amino acid sequence of gastric inhibitory polypeptide. J. Am. Chem. Soc. 1975; 97:5593-5594.
16. Jornvall H, Carlquist M, Kwauk S, Otte SC, McIntosh CHS, Brown JB, Mutt V. Amino acid sequence and heterogeneity of gastric inhibitory polypeptide (GIP). FEBS Lett. 1981; 123:205-210.
17. Mochizuki T, Yanaihara C, Mutt V, Brown JC, Yanaihara N. 1982. Synthesis of porcine GIP (Gastric Inhibitory Polypeptide). PeptideChem. 1981; 19:109-112.
18. Brown JC, Cook MA, Dryburgh Jr. Motilin, a gastric motor activity-stimulating polypeptide: the complete amino acid sequence. Can. J. Biochem. 1973; 51:533-537.
19. Brown JC, Cook MA, Dryburgh Jr. Motilin, a gastric motor activity-stimulating polypeptide. Final purification, amino acid composition, and C-terminal residues. Gastroenterology 1972; 62:401-404.
20. Ikota N, Shioiri T, Yamada S, Tachibana S. Amino acids and peptides. XXXI. Phosphorus in organic synthesis. XVIII. Synthesis of porcine motilin by the solid-phase method using diphenyl phosphorazidate (DPPA) and diethyl phosphorocyanidate (DEPC). Chem. Pharm. Bull. 1980; 28:3347-5336.
21. Coy DH, Coy EJ, Lee KY, Chey WY. Solid-phase synthesis and biological activities of gastrointestinal hormones: secretin and motilin. Peptides 1982; 3:137-141.
22. Coy DH, Gardner J. Solid-phase synthesis of porcine vasoactive intestinal peptide. In t. J. Peptide Protein Res. 1980; 15:73-78.
23. Tatemoto K, Carlquist M, Mutt V. Neuropeptide Y. A novel brain peptide with structural similarities to peptide YY and pancreatic polypeptide. Nature 1982; 296:659-660.
24. Tatemoto K. Neuropeptide Y: complete amino acid sequence of the brain peptide. Proc. Natl. Acad. Sci. USA. 1982; 79:5485-5489.
25. Krstenansky JL, Buck SH. The synthesis, physical characterization and receptor binding affinity of neuropeptide Y (NPY). Neuropeptides 1987; 10:77-85.
26. Tatemoto K, Mutt V. Isolation of two novel candidate hormones using a chemical method for finding naturally occurring polypeptides. Nature 1980; 285:417-418.
27. Tatemoto K. Isolation and characterization of peptide YY (PYY), a candidate gut hormone that inhibits pancreatic exocrine secretion. Proc. Natl. Acad. Sci. U.S.A. 1982; 79:2514-2518.
28. Tatemoto K, Nakano I, Makk G, Angwin P, Mann M, Schilling J, GoVLW. Isolation and primary structure of human peptide YY. Biochem. Biophys. Res. Commun. 1988; 157:713-717.
29. Van Zon A, Beyerman HC. Repetitive excess mixed anhydride (REMA) synthesis of peptides. Synthesis of the gastrointestinal hormone secretin. Pept., Proc. Eur. Pept. Symp., 13th. 1975; 165-172.
30. Hemmasi B, Bayer E. The solid-phase synthesis of porcine secretin with full biological activity. Int. J. Peptide Protein Res. 1977; 9:63-70.
31. Coy DH, Gardner J. Solid-phase synthesis of porcine vasoactive intestinal peptide. Int. J. Peptide Protein Res. 1980; 15:73-78.
32. Brazeau P, Vale W, Burgus R, Ling N, Butcher M, Rivier J, Guillemin R. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 1973; 179:77-79.
33. Burgus R, Ling N, Butcher M, Guillemin R. Primary structure of somatostatin, a hypothalamic peptide that inhibits the secretion of pituitary growth hormone. Proc. Nat. Acad. Sci. U.S.A. 1973; 70:684-688.
34. Sarantakis D, McKinley WA. Total synthesis of hypothalamic somatostatin. Biochem. Biophys. Res. Commun. 1973; 54:234-238.
35. Yamashiro D, Li CH. Synthesis of a peptide with full somatostatin activity. Biochem. Biophys. Res. Commun. 1973; 54:882- 888.
36. Coy DH, Coy EJ, Arimura A, Schally AV. Solid-phase synthesis of growth hormone-release inhibiting factor. Biochem. Biophys. Res. Commun. 1973; 54:1267-1273.
37. Rivier JE. Somatostatin. Total solid-phase synthesis. J. Am. Chem. Soc. 1974; 96:2986-2992.
38. Stewart JM, Young JD. Solid-phase Peptide Synthesis. 1969. Freeman WH & Co. San Francisco, CA.
39. Nicolas P, Delfour A, Boussetta H, Morel A, Rholam M, Cohen P. Solid-phase synthesis of somatostatin-28 II. A new biologically active octacosapeptide from anglerfish pancreatic islets. Biochem. Biophys. Res. Commun. 1986; 140:565-573.
40. Hagenmaier H, Frank H. Increased coupling yields in solid-phase peptide synthesis with a modified carbodiimide coupling procedure. Hoppe-Seyler’s Zeitschrift für physiologische Chemie. 1972; 353:1973-1976.
41. Blake J, Li CH. The synthesis and biological activity of (165, 182, 189-S-carbamidomethylcysteine)-human growth hormone- (140-191). Int. J. Protein Peptide Res. 1975; 7:495-501.
42. Ellman GL. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959; 82:70-77.
43. Von Euler US, Gaddum JH. An unidentified depressor substance in certain tissue extracts. J. Physiol. 1931; 72:74-87.
44. Fisher GH, Humphries J, Folkers K, Pernow B, Bowers CY. Synthesis and some biological activities of substance P. J. Med. Chem. 1974; 17:843-846.
45. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951; 193:265-275.
46. Atherton E, Logan CJ, Sheppard RC. Peptide synthesis. Part 2. Procedures for solid-phase synthesis using Na-fluorenylmethoxy-carbonyl amino acids on polyamide supports. Synthesis of substance P and of acyl carrier protein 65-74 decapeptide. J. Chem. Soc.: Perkin transaction I. 1981; 2:538-546.
47. Said SI, Mutt V. Polypeptide with broad biological activity: isolation from small intestine. Science 1970; 169:1217-1218.
48. Bodanszky M, Klausner YS, Lin CY, Mutt V, Said SI. Synthesis of the vasoactive intestinal peptide (VIP). J. Am. Chem. Soc. 1974; 96:4973-4938.
49. Klausner YS, Bodanszky M. Synthesis of the vasoactive intestinal peptide (VIP). IV. Sequence 14-28. Bioorg. Chem. 1973; 2:354-362.
50. Bodanszky M, Lin CY, Yiotakis AE, Mutt V, Said SI. Vasoactive intestinal peptide (VIP) from chicken. Synthesis and properties of the C-terminal hendecapeptide. Bioorg. Chem. 1976; 5:339-350.
51. Schaaper WM, Beyerman HC. Synthesis of vasoactive intestinal peptide (VIP) via the mixed anhydride method. Peptides 1984; 5:167-168.
52. Fournier A, Saunders JK, St-Pierre S. Synthesis, conformational studies and biological activities of VIP and related fragments. Peptides 1984; 5:169-177.
53. Fournier A, Couture R, Magnan J, Gendreau M, Regoli D, St-Pierre S. Synthesis of peptides by the solid-phase method. V. Substance P and analogs. Can. J. Biochem. 1980; 58:272-280.
54. Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and β-endorphin. Science 1981; 213:1394-1397.
55. Sueiras-Diaz J, Coy DH, Vigh S, Redding TW, Huang WY, Torres-Aleman I, Schally AV. Synthesis and biological properties of ovine corticotropin-releasing factor (CRF). Life Sci. 1982; 31:429-435.
56. Schally AV, Arimura A, Baba Y, Nair RMG, Matsuo H, Redding TW, Debeljuk L. White WF. Isolation and properties of the FSH and LH-releasing hormone. Biochem. Biophys. Res. Commun. 1971; 43:393-399.
57. Matsuo H, Arimura A, Nair RMG, Schally AV. Synthesis of the porcine LH- and FSH-releasing hormone by the solid-phase method. Biochem. Biophys. Res. Commun. 1971; 45:822-827.
58. Marshall GR. In: Pharmacology of Hormonal Polypeptides and Proteins. Back N, Martini L, Paoletti R, eds. 1968. Plenum Press, New York.
59. Gilbert JB, Price VE, Greenstein JP. Effect of anions on the nonenzymic desamidation of glutamine. J. Biol. Chem. 1949; 180:209-218.
60. Chillemi F. Synthesis of L-glutaminyl-L-propyl-L-seryl-L-lysyl-L-aspartyl-L-alanyl-L-phenylalanyl-L-isoleucylglycyl-L-leucyl-L-methioninamide. Gazz. Chim. Ital. 1963; 93:1079-1092.
61. Guillemin R, Brazeau P, Bohlen P, Esch F, Ling N, Wehrenberg WB. Growth hormone-releasing factor from a human pancreatic tumor that caused acromegaly. Science 1982; 218:585-587.
62. Spiess J, Rivier J, Vale W. Characterization of rat hypothalamic growth hormone-releasing factor. Nature 1983; 303:532-535.
63. Bohlen P, Esch F, Brazeau P, Ling N, Guillemin R. Isolation and characterization of the porcine hypothalamic growth hormone releasing factor. Biochem. Biophys. Res. Commun. 1983; 116:726-734.
64. Esch F, Bohlen P, Ling N, Brazeau P, Guillemin R. Isolation and characterization of the bovine hypothalamic growth hormone releasing factor. Biochem. Biophys. Res. Commun. 1972; 117:772-779.
65. Ling N, Esch F, Bohlen P, Brazeau P, Wehrenberg WB, Guillemin R. Isolation, primary structure, and synthesis of human hypothalamic somatocrinin: growth hormone-releasing factor. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:4302-4306.
66. Obal F Jr, Krueger JM. The somatotropic axis and sleep. Revue Neurol. 2001; 157:S12-15.
67. Schally AV, Bowers CY, Redding TW, Barrett JF. Isolation of thyrotropin releasing factor (TRF) from porcine hypothalamus. Biochem. Biophys. Res. Commun. 1966; 25:165-169.
68. Schally AV, Redding TW, Bowers CY, Barrett JF. Isolation and properties of porcine thyrotropin-releasing hormone. J. Biol. Chem. 1969; 244:4077-4088.
69. Guillemin R, Burgus R, Sakiz E, Ward DN. Further data on the purification of the TSH-hypophysiotropic hypothalamic hormone, TRF. Comptes rendus hebdomadaires des seances de l’Academie des sciences. Serie D: Sciences Natur. 1966; 262:2278-2280.
70. Gillessen D, Felix AM, Lergier W, Studer RO. Synthesis of thyrotropin-releasing hormone (TRH) and related peptides. Helv. Chim. Acta. 1970; 53:63-72.
71. Rivaille P, Milhaud G. An efficient synthesis of thyrotropin releasing hormone (TRH). Helv. Chim. Acta. 1971; 54:355-357.
72. Chillemi F, Merrifield RB. Use of N-im-dinitrophenylhistidine in the solid-phase synthesis of the tricosapeptides 124-146 of human hemoglobin beta chain. Biochemistry 1969; 8:4344-4346.
73. Folkers K, Chang JK, Currie BL, Bowers CY, Weil A, Schally AV. Synthesis and relationship of L-glutaminyl-L-histidyl-L-prolinamide to the thyrotropin releasing hormone. Biochem. Biophys. Res. Commun. 1970; 39:110-113.
74. Schwarz H, Bumpus FM, Page IH. Synthesis of a biologically active octapeptide similar to natural isoleucine angiotonin octapeptide. J. Am. Chem. Soc. 1957; 79:5697-5703.
75. Arakawa K, Bumpus FM. An improved synthesis of isoleucine angiotensin octapeptide. J. Am. Chem. Soc. 1961; 83:728-732.
76. Bumpus FM, Schwarz H, Page IH. Synthesis and pharmacology of the octapeptide angiotonin. Science 1957; 125:886-887.
77. Marshall GR, Merrifield RB. Synthesis of angiotensins by the solid-phase method. Biochemistry. 1965; 4:2394-2401.
78. Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:8628-8632.
79. Westermark P, Wernstedt C, Wilander E, Hayden DW, O’Brien TD, Johnson KH. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:3881-3885.
80. Leighton B, Cooper GJS. Pancreatic amylin and calcitonin gene related peptide causes resistance to insulin in skeletal muscle in vitro. Nature 1988; 335:632-635.
81. Balasubramaniam A, Renugopalakrishnan V, Stein M, Fischer JE, Chance WT. Syntheses, structures and anorectic effects of human and rat amylin. Peptides 1991; 12:919-924.
82. Kimball CP, Murlin Jr. Aqueous extracts of pancreas. III. Some precipitation reactions of insulin. J Biol. Chem. 1923; 58:337-346.
83. Bromer WW, Sinn LG, Behrens OK. Amino acid sequence of glucagon. V. Location of amide groups, acid-degradation studies, and summary of sequential evidence. J. Am. Chem. Soc. 1957; 79:2807-2810.
84. Wunsch E, Wendlberger G. Zur Synthese des Glucagons, XVIII. Darstellung der Gesamtsequenz. Chem. Ber. 1968; 101:3659-3663.
85. Mojsov S, Merrifield RB. Solid-phase synthesis of crystalline glucagon. Biochemistry 1981; 20:2950-2956.
86. Mojsov S, Merrifield RB. An improved synthesis of crystalline mammalian glucagon. Eur. J. Biochem. 1984; 145:601-605.
87. Mitchell AR, Erickson BW, Ryabtsev MN, Hodges RS, Merrifield RB .Tert-butoxycarbonylaminoacyl-4-(oxymethyl)-phenyl-acetamidomethyl-resin, a more acid-resistant support for solid- phase peptide synthesis. J. Am. Chem. Soc. 1976; 98:7357-7362.
88. Mitchell AR, Kent SBH, Engelhard M, Merrifield RB. A new synthetic route to tert-butyloxycarbonylaminoacyl-4-(oxymethyl) phenylacetamidomethyl-resin, an improved support for solid- phase peptide synthesis. J. Org. Chem. 1978; 43:2845-2852.
89. Meienhofer J, Schnabel E, Bremer H, Brinkhoff O, Zabel R, Sroka W, Klostermeyer H, Brandenburg D, Okuda T, Zahn H. Synthesis of the insulin chain and the combination to insulin active preparation. Zeitschrift fuer Naturforschung. 1963; 18b:1120-1121.
90. Katsoyannis PG, Fukuda K, Tometsko A, Suzuki K, Tilak M. Insulin peptides. X. Synthesis of the B-chain of insulin and its combination with natural or synthetic A-chain to generate insulin activity. J. Am. Chem. Soc. 1964; 86:930-932.
91. Kung YT, Du YC, Huang WT, Chen CC, Ke LT. Total synthesis of crystalline bovine insulin. Scientia Sinica 1965; 14:1710-1716.
92. Merrifield RB. Solid-phase peptide synthesis. III. An improved synthesis of bradykinin. Biochemistry 1964; 3:1385-1389.
93. Niu CI, Kung YT, Huang WT, Ke LT, Chen CC. Synthesis of crystalline insulin from its natural A-chain and the synthetic B-chain. Scientia Sinica 1966; 15:231-244.
94. Merrifield RB, Stewart JM. Automated peptide synthesis. Nature 1965; 207:522-523.
95. Stewart JM, Woolley DW. All-D-bradykinin and the problem of peptide antimetabolites. Nature 1965; 206:619-620.
96. Meienhofer J, du Vigneaud V. Preparation of lysine-vasopressin through a crystalline protected nonapeptide intermediate and purification of the hormone by chromatography. J. Am. Chem. Soc. 1960; 82:2279-2282.
97. Du YC, Zhang YS, Lu ZX, Tsou CL. Resynthesis of insulin from its glycyl and phenylalanyl chains. Scientia Sinica 1961; 10:84-104.
98. Du YC, Jiang RQ, Tsou CL. Conditions for successful resynthesis of insulin from its glycyl and phenylalanyl chains. Scientia Sinica 1965; 14:229-236.
99. Meyers CA, Coy DH. Solid-phase synthesis of human pancreatic polypeptide. Inter. J. Peptide Protein Res. 1980; 16:248-253.
100. Schwyzer R, Sieber P. Total synthesis of adrenocorticotropic hormone. Nature 1963; 199:172-174.
101. Yamashiro D, Li CH. Adrenocorticotropins. 44. Total synthesis of the human hormone by the solid-phase method. J. Am. Chem. Soc. 1975; 95:1310-1315.
102. Marglin A. Solid-phase peptide synthesis. Simple esterification procedure. Tetrahedron Lett. 1971; 12:3145-3148.
103. Yang YCS, Hruby VJ, Heward CB, Hadley ME. Synthesis of α-and β-melanocyte stimulating hormones. Int. J. Peptide Protein Res. 1980; 15:130-138.
104. Ling N, Ying S, Minick S, Guillemin R. Synthesis and biological activity of four γ-melanotropin peptides derived from the cryptic region of the adrenocorticotropin/β-lipotropin precursor. Life Sci. 1979; 25:1773-1780.
105. du Vigneaud V, Ressler C, Swan JM, Roberts CW, Katsoyanis PG, Gordon S. The synthesis of an octapeptide amide with the hormonal activity of oxytocin. J. Am. Chem. Soc. 1953; 75:4879-4880.
106. du Vigneaud V, Ressler C, Swan JM, Roberts CW, Katsoyanis PG. The synthesis of oxytocin. J. Am. Chem. Soc. 1954; 76:3115-3121.
107. Manning M. Synthesis by the Merrifield method of a protected nonapeptide amide with the amino acid sequence of oxytocin. J. Am. Chem. Soc. 1968; 90:1348-1349.
108. Walti M, Hope DB. Synthesis of oxytocin: suppression of dimer formation. Experientia 1973; 29:389-389.
109. Oliver G, Schafer EA. On the physiological action of extracts of pituitary body and certain other glandular organs: preliminary communication. J. Physiol. 1895; 18:277-279.
110. Popenoe EA Jr, du Vigneaud V. Degradative studies on vasopressin and performic acid-oxidized vasopressin. J. Biol. Chem. 1953; 205:133-143.
111. du Vigneaud V, Lawler HC, Popenoe EA. Enzymic cleavage of glycinamide from vasopressin and a proposed structure for this Pressor-antidiuretic hormone of the posterior pituitary. J. Am. Chem. Soc. 1953: 75:4880-4881.
112. du Vigneaud V, Gish DT, Katsoyannis PG. Synthetic preparation possessing biological properties associated with arginine vasopressin. J. Am. Chem. Soc. 1954; 76:4751-4752.
113. For [8-Lys]-vasopressin, see: Meienhofer J, Sano Y. A solid-phase synthesis of [lysine]-vasopressin through a crystalline protected nonapeptide intermediate. J. Am. Chem. Soc. 1968; 90: 2966-2997.
114. For [8-Arg]-vasopressin, see: Meienhofer J, Trzeciak A, Havran RT, Walter R. Solid-phase synthesis of [8-arginine]-vasopressin through a crystalline protected nonapeptide intermediate and biological properties of the hormone. J. Am. Chem. Soc. 1970; 92:7199-7202.
115. Wade JD, Lin F, Talbo G, Otvos L Jr, Tan YY, Tregear GW. Synthesis and biological activity of rat relaxin. Biomed. Pept. Proteins Nucleic Acids 1996-1997; 2:89-92.
116. Tregear GW, Kemp B, Borjesson B, Thompson A, Scanlon D, Collier M, John M, Niall H, Bryant-Greenwood G. Approaches to the solid-phase synthesis of porcine relaxin. Pept., Struct. Biol. Funct., Proc. Am. Pept. Symp., 6th. 1979; 445-453.
117. Tregear GW, Fagan C, Reynolds H, Scanlon D, Jones P, Kemp B, Niall HD, Du YC. Porcine relaxin: synthesis and structure activity relationships. Pept.: Synth., Struct., Funct., Proc. Am. Pept. Symp., 7th. 1981; 249-252.
118. Tregear GW, Du Y, Wang K, Southwell C, Jones P, John M, Gorman J, Kemp B, Niall HD. The chemical synthesis of relaxin. International Congress Series. 1983; 610(Biol. Relaxin Its Role Hum.): 42-55.
119. Tregear GW, Du Y, Wang K, Wade J, Lambert P, Jones P, Southwell C, McDonald M, Niall H. The synthesis of human relaxin. Pept. Chem. 1984; 22:13-18.
120. Bullesbach EE, Schwabe C. Total synthesis of human relaxin and human relaxin derivatives by solid-phase peptide synthesis and site-directed chain combination. J. Biol. Chem. 1991; 266:10754-10761.
121. Copp DH, Cheney B. Calcitonin-a hormone from the parathyroid which lowers the calcium level of the blood. Nature 1962; 193:381-382.
122. Sieber P, Brugger M, Kamber B, Riniker B, Rittel W. Human calcitonin. IV. Synthesis of calcitonin M. Helv. Chim. Acta. 1968; 51:2057-2061.
123. Kamber B, Briicker H, Riniker B, Sieber P, Rittel W. Human calcitonin V. Synthesis and characterization of the protected intermediate calcitonin M decapeptide sequence 1-10. Helv. Chim. Acta. 1970; 53:556-564.
124. Sieber P, Riniker B, Brugger M, Kamber B, Rittel W. Human calcitonin. VI. Synthesis of calcitonin M. Helv. Chim. Acta. 1970; 53:2135-2150.
125. Kamber B, Hartman A, Eisler K, Riniker B, Rink H, Sieber P, Rittel W. The synthesis of cystine peptides by iodine oxidation of S-tritylcysteine and S-acetamidomethylcysteine peptides. Helv. Chim. Acta. 1980; 63:899-915.
126. Shi T, Rabenstein DL. Convenient synthesis of human calcitonin and its methionine sulfoxide derivative. Bioorg. Med. Chem. Lett. 2002; 12:2237-2240.
127. Morris HR, Panico M, Etienne T, Tippins J, Girgis SI, MacIntyre I. Isolation and characterization of human calcitonin gene-related peptide. Nature 1984; 308:746-748.
128. Nishimura O, Kitada C, Fujino M. New method for removing the S-p-methoxybenzyl and S-tert-butyl groups of cysteine residues with mercuric trifluoroacetate. Chem. Pharm. Bull. 1978; 26:1576-1585.
129. Smith DD, Li J, Wang Q, Murphy RF, Adrian TE, Elias Y, Bockman CS, Abel PW. Synthesis and biological activity of C-terminally truncated fragments of human-a-calcitonin gene-related peptide. J. Med. Chem. 1993; 36:2536-2541.
130. Houghten RA. Tritiated porcine Dynorphin (1-17): synthesis and characterization. Life Sci. 1982; 31:1805-1808.
131. Turcotte A, Lalonde JM, St. Pierre S, LeMaire S. Dynorphin-(1-13). I. Structure-function relationships of Ala-containing analogs. Int. J. Peptide Protein Res. 1984; 23:361-367.
132. Sole NA, Barany G. Optimization of solid-phase synthesis of [Ala8]-Dynorphin A. J. Org. Chem. 1992; 57:5399-5403.
133. Yoshiaki K, Masatoshi I, Kouki K, Tadashi A, Hideki M. Synthesis of a newly isolated opioid tridecapeptide, rimorphin, from pituitary using a trifluoroacetic acid-thioanisole deprotection system. Chem. Pharm. Bull. 1983; 31:1818-1820.
134. Atherton E, Fox H, Harkiss D, Sheppard RC. Application of polyamide resins to polypeptide synthesis: an improved synthesis of β-endorphin using fluorenylmethoxycarbonylamino-acids. J. Chem. Soc.: Chem. Commun. 1978; 13:539-540.
135. Ling N. Solid-phase synthesis of porcine α-endorphin and γ-endorphin, two hypothalamic-pituitary peptides with opiate activity. Biochem. Biophys. Res. Commun. 1977; 74:248-255.
136. Voelter W, Burvenich C, Horn H, Kalbacher H, Pietrzik E. Total synthesis of methionine-enkephalin. Angew. Chem. Int. Engl. 1976; 15:297-298.
137. Voelter W, Pietrzik E, Kalbacher H. Total synthesis of leucine-enkephaline. Tetrahedron Lett. 1976; 17:2119-2120.
138. Clapes P, Torres JL, Adlercreutz P. Enzymatic peptide synthesis in low water content systems: preparative enzymatic synthesis of [Leu]- and [Met]-enkephalin derivatives. Bioorg. Med. Chem. 1995; 3:245-255.
139. Kakidani H, Furutani Y, Takahashi H, Noda M, Morimoto Y, Hirose T, Asai M, Inayama S, Nakanishi S, Numa S. Cloning and sequence analysis of cDNA for porcine beta-neo-endorphin/ Dynorphin precursor. Nature 1982; 298:245-249.
140. Yamamoto Y, Yanaihara C, Katsumaru Y, Mochizuki T, Tobe A, Egawa M, Imura H, Numa S, Yanaihara N. Synthesis of porcine leumorphin and some of its biological activities. Regul. Pept. 1983; 6:163-168.
141. Suda M, Nakao K, Sakamoto M, Yoshimasa T, Morii N, Yanaihara N, Numa S, Imura H. Leumorphin is a novel endogenous opioid peptide in man. Biochem. Biophys. Res. Commun. 1984; 123:148-155.
142. Kimura S, Okada M, Sugita Y, Kanazawa I, Munekata E. Novel neuropeptides, neurokinin a and |3, isolated from porcine spinal cord. Proc. Japan Acad., Series B: Phys. Biol. Sci. 1983; 59:101-104.
143. Folkers K, Lu YA, Rosell S. Synthesis and biological activities of neurokinin α and β. Biochem. Biophys. Res. Commun. 1984; 118:405-408.
144. For eledoisin, see: Sandrin Ed, Biossonnas RA. Synthesis of eledoisin. Experientia 1962; 18:59-61.
145. For physalaemin see: Bernardi L, Bosisio G, Goffredo O, De Cas- tiglione R. Synthesis of physalaemin. Experientia 1964; 20:490-492.
146. For kassinin: Yajima H, Sasaki T, Ogawa H, Fujii N, Segawa T, Nakata Y. Studies on peptides. LXXVI. Synthesis of kassinin, a new frog skin peptide. Chem. Pharm. Bull. 1978; 26:1231-1235.
147. McDonald TJ, Jornvall H, Nilsson G, Vagne M, Ghatei M, Bloom SR, Mutt V. Characterization of a gastrin releasing peptide from porcine non-antral gastric tissue. Biochem. Biophys. Res. Commun. 1979; 90:227-233.
148. Brown M, Marki W, Rivier J. Is gastrin releasing peptide mammalian bombesin? Life Sci. 1980; 27:125-128.
149. Marki W, Spiess J, Tache Y, Brown M, Rivier JE. Total solid-phase synthesis of porcine gut gastrin releasing peptide (GRP), a mammalian bombesin. J. Am. Chem. Soc. 1981; 103:3178-3185.
150. Carraway R, Leeman SE. Isolation of a new hypotensive peptide, neurotensin, from bovine hypothalami. J. Biol. Chem. 1973; 248:6854-6861.
151. Carraway R, Leeman SE. Amino acid sequence of a hypothalamic peptide, neurotensin. J. Biol. Chem. 1975; 250:1907-1911.
152. Carraway R, Leeman SE. Synthesis of neurotensin. J. Biol. Chem. 1975; 250:1912-1918.
153. Silva MR, Beraldo WT, Rosenfeld G. Bradykinin, a hypotensive and smooth-muscle-stimulating factor released from plasma globulin by snake venoms and by trypsin. Am. J. Physiol. 1949; 156:261-273.
154. Boissonnas RA, Guttmann St, Jaquenoud PA. Synthesis of a nonapeptide with properties of bradykinin. Helv. Chim. Acta. 1960; 43:1349-1358.
155. St. Guttmann Pless J, Boissonnas RA. A new synthesis of bradykinin. Helv. Chim. Acta. 1962; 45:170-177.
156. Nicolaides ED, De Wald HA. Synthesis of polypeptides. Bradykinin. Studies on the synthesis of polypeptides. Bradykinin. J. Org. Chem. 1961; 26:3872-3876
157. Merrifield RB. Solid-phase peptide synthesis. II. The synthesis of bradykinin. J. Am. Chem. Soc. 1964; 86:304-305.
158. Khan SA, Sivanandaiah KM. Catalytic transfer hydrogenation in solid-phase peptide synthesis: synthesis of bradykinin. Synthesis 1978; 10:750-751.
Further Reading
Barany G, Merrifield RB. Solid-phase peptide synthesis. In: The Peptides: Analysis, Synthesis, Biology, Volume 2. Gross E, Meienhofer S, eds. 1979. Academic Press, New York. pp. 1-284.
Benoiton NL. Chemistry of Peptide Synthesis. 2006. Taylor & Francis, CRC Press, Boca Raton, FL.
Bodanszky M. Principles of Peptide Synthesis, 2nd edition. 1993. Springer-Verlag, New York.
Chan WC, White PD, eds. FMOC Solid-phase Synthesis. 2000. Oxford University Press, Oxford, UK.
Grant GA, ed. Synthetic Peptides. A User’s Guide. 1992. W.H. Freeman and Co., New York.
Hruby VJ, Meyer JP., Chemical synthesis of peptides. In: Bioorganic Chemistry: Peptide and Protein. Hecht, SM, ed. 1998. Oxford University Press, New York. pp. 27-64.
Muir TW, Dawson PE, Kent SBH. Protein synthesis by chemical ligation of unprotected peptides in aqueous solution. Methods Enzymol. 1997; 289:266-298.