CHEMICAL BIOLOGY
Electron Paramagnetic Resonance in Enzymology
Aimin Liu, Department of Chemistry, Georgia State University, Atlanta, Georgia
doi: 10.1002/9780470048672.wecb668
Electron paramagnetic resonance (EPR) is a versatile tool for chemical biology research. A remarkable wealth of information about the mechanistic enzymology has been obtained by EPR experiments. As an analytical method, EPR can obtain these two useful pieces of information in a study of enzymatic reaction mechanisms: 1) detection and complete description of free radicals and 2) characterization of the electronic structures of paramagnetic metal ions and their response to changes of the protein environment or substrate binding. The first feature has spurred the development of the new field known as radical enzymology; the second has enhanced the understanding of the mechanisms of metalloenzyme action. The potential of EPR spectroscopy to serve as an important biophysical tool for the future development of enzymology is highlighted by several selected examples.
This article does not intend to be a survey of electron paramagnetic resonance (EPR) spectroscopy in chemical biology, but rather the focus is to provide an entry-level introduction to chemists who are interested in the research opportunities that EPR may provide for enzymology studies. The principles of this technique will be briefly described, as well as the breadth of the technique’s applicability to free radicals and metallocenters in enzymology. Potential applications in the future will also be outlined.
History
EPR was first applied to biological materials in 1954 (1), 10 years after the discovery of an experimental approach to detecting electron spin resonance by the Russian physicist Zavoisky in 1944 (2, 3). This technique became useful for studying enzymes when a spectrometer that was capable of detecting about 10-10 moles of unpaired (nonbonding or “free”) electron spins in samples that contained about 0.15 milliliter of liquid water was developed by Townsend and colleagues in 1957 (4). The first successful application of this technique to elucidating the structure of substrate radicals in enzymatic reactions was in 1958 (5-7). In the ensuing 50 years, numerous seminal and classical contributions have been made to the development of enzymology by scientists employing EPR spectroscopy analytically.
Application
It is an advantage to analyze EPR samples of aqueous solution directly because the buffered solution is the most physiologically relevant condition. However, EPR samples are not restricted to samples in the solution state, gaseous and solid samples and single crystals can also be analyzed directly by EPR spectroscopy. The only absolute requirement for preparing an EPR sample is that it must contain unpaired electron spins or that such status can be achieved before EPR measurements by oxidation/reduction reactions. EPR spectroscopy can determine unambiguously free radicals that are present in the sample or radicals produced by oxidation or irradiation with light, X-rays, and y-rays. Because paramagnetic transition metals contain unpaired electrons, EPR spectroscopy can determine the detailed geometric and electronic environment of the metal centers. This technique can also indicate the degree of molecular motion in a sample with unpaired electrons and analyze the populations in a heterogeneous sample that has two or more paramagnetic species. Its ability to focus on the paramagnetic active sites without interference from the rest of the diamagnetic species makes this technique an ideal method for mechanistic studies. Therefore, it is commonly employed in studies of the structure of enzyme active site, interactions between enzyme and substrate, conformational dynamics, electron transfer, and reaction kinetics.
EPR Basics
As an analytical spectroscopic technique, EPR is similar in concept to the more widely used nuclear magnetic resonance (NMR) spectroscopy [see NMR: Overview of Applications in Chemical Biology]. In fact, EPR and NMR are complementary to each other. Both techniques detect magnetic moments, but NMR determines the chemical structures in solution, whereas EPR describes more precisely the electronic and chemical structures of a particular region of the biological system, such as electron transfer centers, metal ions, and an intermediate state of the enzyme or substrate. It is not possible to present a full description of the theory of EPR in an article with this scope. Therefore, only sufficient information is provided here to enable the readers to understand the practical aspects of this analytical tool in enzymology.
Resonance and the Zeeman effect
Electrons have “spin,” and when they are unpaired (or “free”), this “spin” gives them a measurable magnetic moment. In the absence of an external magnetic held, the electron’s magnetic moment will orient randomly. The electron with a spin quantum number S = ½ can have two orientations (ms = ± ½) in a magnetic held (Bo). Therefore, the electrons will align either parallel or antiparallel to the external magnetic held (Fig. 1). The orientation of unpaired electrons causes discrete energy levels (Fig. 2). The orientation antiparallel to Bo is energetically higher than the parallel. The displacement of discrete energy levels is known as the Zeeman effect.
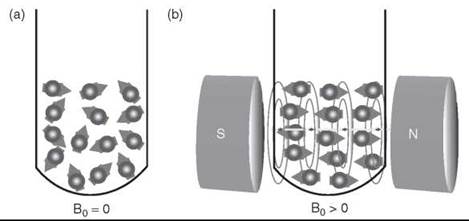
Figure 1. The orientation of electron's magnetic moment in the absence a) and presence b) of an external magnetic field.

Figure 2. Acquisition of an EPR spectrum for an unpaired S = ½ electron, a) Energy-level diagram as a function of magnetic field, b) absorption/emission of microwave energy because of the field of resonance, and c) first derivative of the energy absorption profile, for example, an EPR spectrum.
The two spin states (Ms = ± ½) of an S = ½ electron have the same energy when Bo is 0. In the presence of an external magnetic held, two discrete energy levels occur, and the electronic-Zeeman energy diverges linearly as the strength of the magnetic held increases. In the event that a constant electronmagnetic radiation frequency is present, peak absorption of the energy will occur when the magnetic held strength is varied and tuned to the two spin states so that the electronic Zeeman energy matches the energy of the allowed spin transitions. At this point, resonances are observed. The detection of the first derivative of energy absorption of the radiation for transitions results in an EPR spectrum (Fig. 2).
For a spin S > ½ system, such as a system with 5 unpaired electrons each occupying an orbital, and all 5 pointing upward (i.e., in high symmetry), application of an external magnetic field yields uniformly spaced multiplet splitting of energy levels at Ms = ± 1/2, ± 3/2, and ± 5/2. These energy differences are predominately because of the interaction of the sample’s unpaired electrons with the external magnetic field, and each Ms converges to a common origin at zero field. If the electrons are in an asymmetry environment, for example, some point upward and some downward, then the energy levels may not be uniformly spaced. A measurable parameter, zero-field splitting, has been introduced to describe the energy-level splittings of an electron spin (S > 1/2) in the absence of an applied magnetic field (see the materials in the Further Reading section).
g-factor
The difference between the energies of the unpaired electron in the initial and final spin states of an allowed transition is AE, which can be described by the fundamental equation of EPR spectroscopy:
![]()
where h is Planck’s constant (6.62606896 x 10 34 J∙s), V is the electromagnetic wave of frequency, μB is Bohr magneton (9.27400949 x 10-24 J∙T-1), and g is the electron Zeeman factor. Equation 1 shows that ∆E is proportional to B0. Gauss, tesla (T), and millitesla (mT) are commonly used units for magnetic field B0 (1 mT = 10 Gauss) in presenting an EPR spectrum. For a free electron, the electron Zeeman factor g is 2.0023193. However, g-factor can vary considerably, depending on the molecule in which the unpaired electron resides. In most EPR experiments, the frequency value V is known, and the B0 values for various EPR resonance components can be obtained from EPR spectra. Therefore, the g-factor can be calculated directly from Eq. 2, which is derived from Eq. 1.

In principle, EPR spectra can be generated by varying the frequency of the electromagnetic radiation incident on a sample while holding the magnetic field constant. In practice, it is the frequency that is kept constant and the magnetic field that is swept or varied. The so-called conventional EPR spectrometer operates at 9-10 GHz region (X-band). The majority of EPR measurements are conducted in this frequency in a continuous-wave mode. Other commonly used frequencies include L-band (1-2 GHz), S-band (~ 3 GHz), K-band (~ 25 GHz), Q-band (~ 35 GHz), W-band (~ 94 GHz), and very high-field EPR spectrometers operating at 180-600 GHz (8, 9).
At 9.5 GHz, which is in the X-band frequency range, the magnetic field where the EPR signal should occur for a free electron can be calculated from Eq. 3, for example, B0 = 714.4775 ![]() 2.002 3193 x 9.5 ≈ 3390 Gauss.
2.002 3193 x 9.5 ≈ 3390 Gauss.

The value of g-factor is a measure of the coupling between the spin of an unpaired electron and an external magnetic field. It is not only dependent on the spin species but also on its environment. A single numerical value of g is applicable only to systems that behave isotropically. With anisotropic systems, a modified term that accommodates the variability of g with orientations relative to the external field is introduced as g-tensor. Three values, gx, gy, and gz, which represent principal gxx, gyy, and gzz values of the g-matrix, are important EPR parameters.
Hyperfine coupling
The actual field at each spin species is not necessarily only the externally imposed magnetic field. In addition to this field, local fields from nearby nuclei may add to the external field. Nuclei can also have spins, and they possess an intrinsic spin angular momentum. It is common practice to represent the total angular momentum of a nucleus by the symbol I and to call it “nuclear spin.” Nonzero nucleus spins affect the local magnetic field where the unpaired electrons are located in a sample during an EPR measurement. The interaction between the electron and the nearby nuclei is called hyperfine interaction or hyperfine coupling. Such an interaction would cause additional splitting of the energy levels of unpaired electrons and, in turn, would produce multiplicity in EPR spectrum that requires spin Hamiltonian terms to describe the coupling of the spin system (see the Further Reading section). However, the number of the spectral lines in the EPR spectrum can be predicted by the following simple formula:
![]()
where n is the number of the equivalent nuclei, that are being coupled to and I is the nuclear spin number. Table 1 summarizes the nuclear spin of common elements that may be used in analyzing EPR data.
Table 1. Nuclear spin data of common elements and isotopes
|
Isotope |
Spin (I) |
Natural abundance (%) |
Isotope |
Spin (I) |
Natural abundance (%) |
|
1H |
1/2 |
99.99 |
63Cu |
3/2 |
69.20 |
|
2H |
1 |
0.01 |
65Cu |
3/2 |
30.80 |
|
12C |
0 |
98.89 |
59Co |
7/2 |
100 |
|
13C |
1/2 |
1.11 |
60Co* |
5 |
0 |
|
35Cl |
3/2 |
75.77 |
53Cr |
3/2 |
9.50 |
|
37Cl |
3/2 |
24.23 |
|
|
|
|
14N |
1 |
99.63 |
54Fe |
0 |
5.85 |
|
15N |
1/2 |
0.37 |
55Fe** |
3/2 |
0 |
|
|
|
|
56Fe |
0 |
91.72 |
|
|
|
|
57Fe |
1/2 |
2.15 |
|
|
|
|
58Fe |
0 |
0.28 |
|
16O |
0 |
99.95 |
55Mn |
5/2 |
100 |
|
17O |
5/2 |
0.04 |
|
|
|
|
18O |
0 |
0.01 |
|
|
|
|
33S |
3/2 |
0.75 |
61Ni |
3/2 |
1.13 |
|
31P |
1/2 |
100 |
95Mo |
5/2 |
15.90 |
|
|
|
|
97Mo |
5/2 |
9.60 |
|
19F |
1/2 |
100 |
50V |
6 |
0.25 |
|
|
|
|
51V |
7/2 |
99.75 |
*Radioactive.
**Half-life: 2.7 years.
For an organic free radical with a single unpaired electron, the EPR spectrum would consist of a single line as shown in Fig. 2. However, this situation is often not the case in natural systems. If the unpaired electron of the radical interacts with an I = 1/2 nucleus, such as a proton, then the single line could split into two identical lines (2I + 1 = 2 x 1/2 + 1 = 2) as illustrated in Fig. 3a. This possibility is because the nucleus (a proton in this case) has two allowed values: MI = ± 1/2. If the unpaired electron interacts with an I = 1 nucleus, such as 14N, then three lines (2I + 1 = 2 x 1 + 1 = 3) are expected in the EPR spectrum of the radical (Fig. 3b). In some cases, the hyperfine interactions originate from two distinct nuclei, one with a nuclear spin I 1, and the other with 12. Then, the number of spectral lines in the EPR spectrum is expected to be (2I1 + 1)(2I2 + 1). Fig. 3c depicts a nitrogen-based free radical with a single unpaired electron interacting with a proton in its vicinity. In this case, the hyperfine splitting lines = (2 x 1 + 1)(2 x 1/2 + 1) = 2 x 3 = 6. These spectral lines are not always fully resolved. They could partially overlap, which would result in an EPR signal with a hyperfine structure.
An important EPR parameter, hyperfine coupling constant, A, is required to describe the hyperfine interaction. The A values can be obtained directly from EPR spectra when equally spaced spectral lines are exhibited because of the hyperfine interactions (Fig. 3). In many cases, a matrix, known as A-tensor, is required to describe the A values for a more complex anisotropic system. Because of the hyperfine interaction, EPR is very sensitive to the local surroundings. From a chemical standpoint, this property provides a wealth of information such as the identity and number of atoms that makes up a molecule or complex and their distances from the unpaired electron. This information can often be translated, in favorable cases, into the molecular structure of the sample.
Much of the information EPR provides about the composition, structure, and bonding of a paramagnetic metal center is obtained through the analysis of the hyperfine coupling constants that represent interactions between the spin of the unpaired electron(s) and the spins of nuclei associated with the metal center. These coupling constants are calculated from splittings seen in the EPR spectrum. In many occasions, the splittings are not resolvable in conventional EPR spectroscopy. Advanced EPR spectroscopy, such as electron nuclear double resonance (ENDOR) and electron spin echo envelope modulation (ESEEM) spectroscopy, are necessary to retrieve the missing information (10). ENDOR and ESSEM provide an NMR spectrum of those nuclei that interact with the electron spin of the paramagnetic center, and such spectra display orders-of-magnitude-better resolution than the EPR spectrum of the center. Resolved features in these NMR spectra are characterized by frequencies, and these frequencies directly give the electron-nuclear coupling constants (see the materials in the Further Reading section).

Figure 3. Energy-level diagram and corresponding EPR signals at 9.6 GHz (X-band) with maximized resolution for an unpaired electron in an imposed magnetic field that interacts with a) a proton (nuclear spin I = 1/2), b) a 14N nucleus (I = 1), and c) a 14N nucleus in which the energy levels are additionally split by a nearby proton. The spectra were produced by the EPR simulation program SimFonia for illustration of the hyperfine splitting purpose.
Radical Enzymology
Life is in a sea of free radicals. In this aerobic world, dioxygen, the essential molecule for sustaining life, is a radical because it has two unpaired electrons with identical spins in separate orbitals in its outer shell. Nitric oxide [see Nitric Oxide, Biological Targets of], a signaling molecule in the cardiovascular system, is another biologically important free radical. The production of deoxyribonucleotides for DNA replication also depends on radicals—the protein-based radicals in enzymes. These examples are but a few examples of the tremendous number of free radicals that exist in the chemical biological world. In fact, the net effect of many biochemical oxidation-reduction reactions is the transfer of one or more electrons. If the electron transfer takes place one at a time, then a great chance of observing free radical intermediates occurs. To study these stable and transient radicals, EPR spectroscopy is the most powerful tool.
Detection and characterization of enzyme-based free radicals
In the past three decades, evidence for enzyme-based protein radicals as the catalytic driving force or the key transient intermediate in enzymatic reactions has strengthened (11). Spectroscopic data from EPR suggest that these highly reactive species can be stored with remarkable stability inside an enzyme either as a transient intermediate or as a paramagnetic center and catalyze a wide range of biological reactions in electron transfer and oxidation/reduction.
It was not known that a protein can harbor a stable free radical derived from an amino acid residue until the discovery of the first tyrosyl radical from the smaller R2 subunit of the Escherichia coli ribonucleotide reductase (RNR) in the 1970s. RNR catalyzes the first committed step in DNA synthesis by reducing the hydroxyl group of all four types of ribonucleotide to produce the corresponding deoxyribonucleotides (12). RNR, thus, is an essential enzyme for all living organisms. The observation of an EPR signal with an asymmetric doublet centered at g = 2.0047 in the EPR spectrum and a sharp absorption peak at 410 nm (ε = 3250 M-1cm-1) of the optical spectrum led to the identification of a protein-based radical (13). This free radical has a half-life time of a matter of days at room temperature and is an integral part of the R2 subunit. It is essential for the enzyme activity. RNR, thus, has been introduced as “a radical enzyme” (14). Later, it was discovered that a transient cysteine-based thiyl radical is formed at the expense of the tyrosyl radical during catalysis and that the transient thiyl radical is responsible for the catalytic reduction of the ribonucleotide substrates (15, 16).
It would be expected that all tyrosyl radicals have similar spectroscopic properties; however, the characterized tyrosyl radicals exhibit distinct spectral features. From a spectroscopic point of view, at least three different EPR signals for tyrosyl radicals are found (Fig. 4). The EPR signal shown in Fig. 4a (trace a) is observed in the active form of E. coli RNR R2 protein, which exhibits a 1.8 mT hyperfine splitting (13). EPR signals similar to trace b have been found in Mycobacterium tuberculosis and Salmonalla typhimurium RNRs, photosystem II (PSII), and many other enzyme-based tyrosyl radical intermediates (16, 19, 20). The most complex spectrum, trace c, exhibits the largest (ca. 2.1 mT) hyperfine splitting and is observed from mouse, yeast, and higher plant RNR R2 proteins. These radicals are the representative neutral tyrosyl radicals. The tyrosyl radicals in R2 proteins are stable because the free electron is delocalized to the phenyl ring and the radical center is shielded from reaction by a hydrophobic pocket.

Figure 4. a) X-band EPR spectra of tyrosyl free radical in (i) E. coli, (ii) Mycobacterium tuberculosis, and (iii) mouse ribonucleotide reductase R2 proteins (17). All spectra were obtained under nonsaturation conditions at 20 K. b) Spin density distribution of the unpaired electron obtained from isotope-labeling EPR studies. c) The distances between the phenolic oxygen of tyrosyl radical and the nearest Fe ion deduced from the relaxation properties of the tyrosyl radicals.
Most known protein-based tyrosyl radicals, either stable or transient, exhibit a line shape that resembles one of the above line shapes unless they are strongly spin-coupled with another paramagnetic center, such as that found in galactose oxidase (17, 18). Although the observed EPR spectra are distinct, the differences in EPR spectral characteristics are mainly because of the dihedral angles θH, defined by the locations of the β-methylene protons, β-methylene carbon, ring carbon C1, and its 2pz axis relative to the phenyl ring of the side chain (19). These dihedral angles (Table 2) can be determined from the hyperfine coupling constants, for example, A values of the β-methylene protons and spin density (ρ) according to the following equations (20, 21):
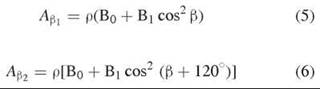
It should be noted that no structure analysis is available for the radical-containing form in the above enzymes with the exception of one radical-containing form obtained by oxidizing the existing crystals of E. coli R2 by H2O2 (22). It is rare to obtain the crystal structure of a biological radical within its catalytic site, although, indeed, some successes have been accomplished. One such case is the intermediate radical form of hydroxyethylidene-thiamine pyrophosphate (HE-TPP), which is visualized in a structure of the enzyme pyruvate ferredoxin oxidoreductase (PFOR) crystallized with HE-TPP (23). The radical is present in the structure because the thiazole ring is apparently puckered. Free radical intermediates are generally not sufficiently stable to survive crystallization and X-ray diffraction, thus EPR spectroscopy is often the most pertinent tool for comprehending the chemical properties of the radicals. Thus far, the electronic structure and orientation of the tyrosyl radicals in enzymes have all been determined by EPR-based techniques in conjunction with isotope-labeling of the radicals.
Table 2. Dihedral angles of P-methylene protons of tyrosyl radicals in PSII and R2 and the hyperfine splitting in the EPR spectra
|
Tyr• |
H+ hyperfine (mT) |
θ1 (deg)1 |
θ2 (deg) |
|
M. tb R2 |
1.0 |
52(± 6) |
68(± 6) |
|
E. coli R2 |
1.8 |
30(± 3) |
90(± 3) |
|
PSII (YD●)2 |
1.0 |
52(± 4) |
68(± 4) |
1θ1 and θ2 represent the dihedral angles between the 2pz orbital at C1 and the planes containing the C1-Cβ-Hβ1 bonds and the C1-Cβ-Hβ2 bonds, respectively.
2Yd● is one of the two tyrosyl radicals in PSII.
The interaction between radical and other paramagnetic species
EPR can often detect the interaction between radical and other paramagnetic species. The tyrosyl radical in RNR is generated by a nonheme di-iron cluster. It is visualized from the crystal structures of the enzyme that the phenolic oxygen of the tyrosine is 5.3 A away from the nearest iron ion in E. coli R2 and 6.8-6.9 A in Salmonalla typhimurium and Mycobacterium tuberculosis R2 proteins (24). This distance is unknown in the mouse R2 because the structure contains a partially occupied metal center (25). It is also uncertain whether the differences of the distance still exist in the tyrosyl radical-containing forms because the structures were determined in the absence of the radicals. EPR spectroscopy can be used to study these unresolved issues. Although the di-ferric center in R2 proteins is diamagnetic because of the antiferromagnetic coupling nature of the two high-spin ferric ions, the interaction of the di-iron center with the adjacent tyrosyl radical can be detected indirectly by EPR spectroscopy. When the tyrosyl radical was found in E. coli RNR R2 protein (a-type), it exhibited some unusual relaxation properties relative to an organic free radical that is not part of a protein. Later biochemical and biophysical studies attributed these unusual relaxation properties to the magnetic dipolar and to exchange interactions between the R2 free radical and the iron center. The interaction between the iron-center and tyrosyl radical in mouse R2 (c-type) is strongest, whereas in Salmonalla typhimurium and Mycobacterium tuberculosis R2 proteins (b-type), it is almost negligible. Therefore, the iron-radical distance must be shorter in mouse and yeast R2 proteins.
The EPR relaxation behavior can be analyzed quantitatively by measuring microwave power saturation profiles of the EPR signal of the radical at various temperatures. The EPR signal intensity increases in proportion to the square root of the microwave power until the onset of saturation of the spin system (26). The power saturation occurs when the rate of absorption of microwave exceeds the rate at which the system returns to equilibrium. A spectral parameter, P1/2, is used to describe quantitatively the microwave power saturation profile. In the RNR tyrosyl radical case, the P1/2 values at four representative temperatures are given in Table 3. The most straightforward interpretation for the easily saturated radical spectra with very small P1/2 values, as seen in M. tuberculosis R2, is that the tyrosyl radical is minimally influenced in its relaxation by the di-ferric cluster. This finding is reverse in mouse and yeast R2 proteins. To obtain the precise distance information in a biological system, advanced techniques such as ESSEM would be more pertinent than the continuous-wave EPR spectroscopy.
Table 3. The relaxation properties of the glycyl radical of anaerobic L. lactis RNR and the tyrosyl radical of aerobic M. Tuberculosis RNR
|
|
|
Temperature (K) |
|
|
|
|
10 |
77 |
100 |
273 |
|
P1/2 (mW) of Tyr● of M. tb R2 |
0.09 |
0.72 |
1.28 |
144* |
|
P1/2 (mW) of Gly● of L. lactis RNR |
< 0.01 |
0.10 |
0.14 |
7 |
*The estimated value for the half saturation because the full saturation did not occur with the applied microwave power.
Enzyme-mediated radical chemistry
The past few decades have witnessed an explosive growth in the number of enzymatic reactions found to proceed by mechanisms that involve free radicals as intermediates. For example, another rapidly growing superfamily of enzymes catalyzes free radical-based reactions in an S-adenosyl methionine-(SAM-)- dependent process (27, 28). These enzymes are named radical SAM (29). In the catalytic mechanisms proposed, a protein-bound [4Fe-4S]+ cluster reduces SAM and produces 5’-deoxyadenosyl radical and methionine in a reversible homolytic cleavage process. The 5’-deoxyadenosyl radical intermediate acts as a potent oxidant and initiates turnover by abstracting a hydrogen atom from an appropriate substrate and generating a substrate-related free radical (30). Lysine 2,3-aminomutase (LAM), which catalyzes the interconversion of L-α-lysine and L-β-lysine, is the best characterized radical enzyme in this superfamily. This enzyme catalyzes the interconversion of L-α-lysine and L-β-lysine. The catalytic 5’-deoxyadenosyl radical generates a lysyl radical at the active site of the enzyme (28, 29). Isomerization of the lysyl radical leads to the rearrangement of the substrate molecule and regeneration of the 5'-deoxyadenosyl radical. This entire process is analogous to the mechanisms of adenosylcobalamin-dependent enzymes (31, 32).
A special concern of the SAM-depended radical mechanism is to detect the key catalytic intermediate 5'-deoxyadenosyl radical, which is too high in energy to exist in the active site of an enzyme at concentrations detectable by EPR spectroscopy (29). By creatively incorporating a sulfur atom or an adjacent allylic group near the radical, Frey and Reed and colleagues have successfully trapped and characterized the closely related analogues of the 5'-deoxyadenosyl radical by EPR spectroscopy (reviewed in Refs. 27-30). The enzyme mechanism of LAM thus is established by combined bioorganic and EPR spectroscopic studies. More importantly, these studies have produced an innovative strategy for stabilizing a very short-lived radical intermediate in the catalytic cycle for EPR characterizations, which is entirely distinct from common approaches such as rapid freeze-quench and nitrone-based spin trapping (i.e., the radical will be forming an adduct with a spin trap reagent) techniques.
EPR Characterization of Metallocofactors
For the metalloenzymes [see Metallo-Enzymes and Metallo-Proteins, Chemistry of] with a paramagnetic metal center, NMR spectroscopy may have limitations because the lines of the nuclei near the metal are often broad [see NMR for Proteins]. X-ray crystallography reveals the location of atoms when the diffraction quality of crystals is obtainable; however, it does not illustrate the electronic structure, the oxidation state of the metal ion, and the charge distribution, which are the real determinants of the activity of active site in metalloenzymes. Therefore, even with the crystal structure, information about transient or intermediary steps still is needed to understand electron or atom transfer mechanisms and enzymatic reactions. For this purpose, spectroscopy is unassailable, and for obtaining information about paramagnetic centers, EPR-centered methods, including pulse techniques and the technique of electron-nuclear double resonance at high- and low-field, are uniquely suited. The application of EPR spectroscopy for the characterization of metalloenzymes has recently been reviewed in several elegant articles (see the Further Reading section). In this section, three examples will be described to highlight the potential of EPR spectroscopy for characterization of metalloenzymes.
Electronic structure of paramagnetic centers in enzymes and the response to chemical, redox, and environmental changes
In a recent combined biochemical and spectroscopic study, the RNR R2 protein from Chlamydia trachomatis (Ct), a human pathogen, is shown to employ a novel enzyme-bound MnIVFeIII cofactor to initiate catalysis in the R1 protein (33). This heterodinuclear cofactor exhibits an S = 1 ground spin state, which resulted from antiferromagnetically coupling of the MnIV (d3, S = 3/2) and FeIII (d5, S = 5/2) ions (34). It is reduced to an EPR-active MnIIIFeIII form when the Ct R2 protein is treated with the substrate analog 2'-azido 2-deoxyadenosine 5’-diphosphate (N3-ADP) in the presence of R1 and adenosine triphosphate (ATP) (33). This observation suggests that the MnIVFeIII cofactor can generate protein and substrate radicals in the R1 site during the catalysis. The MnIVFeIII cluster is proposed, therefore, to be a functional equivalent of the FeIIIFeIII/Tyr* cofactor found in other aerobic RNR enzymes (33-36). The EPR spectrum of the MnIIIFeIII form is centered at g ≈ 2 region (S = 1/2), which is originated from antiferromagnetically coupling of the MnIII (d4) and FeIII (d5) ions. Subsequent EPR and Mossbauer spectroscopic studies have suggested that the catalytic MnIVFeIII form can be generated from the oxidation of the MnIIFeII form of the protein by either O2 or H2O2 (35, 36). The mechanism of formation of the MnIVFeIII form is found to involve in an EPR-active form of a highly oxidized MnIVFeIV cluster (Fig. 5). The EPR spectrum of this MnIVFeIV cluster is also centered at the g ≈ 2 region. The MnIV (d3) ion has a spin state of 3/2, and the FeIV (d4) ion is believed to be at an unusually high spin state of 2 instead of 1 (35). The two metal ions remains to be antiferromagnetically coupled, which results in a total ground spin state of 1/2. The detection of the EPR signals of the MnInFeIII and MnIVFeIV has contributed significantly to the awareness of the existence and formation mechanism of the MnIVFeIII catalytic cofactor (33-36). The fully reduced MnIIFeII form, with d5 and d6 electrons in the two metal ions, respectively, may also be EPR-active regardless of ferromagnetic, antiferromagnetic, or noncouplings between the two metal ions. The full characterization of this reduced form has not yet been reported.

Figure 5. The heterodinuclear MnIVFeIII cluster of Chlamydia trachomatis RNR R2 protein and its proposed formation mechanism from its MnIIFeII form described in a recent combined biochemical, EPR, and Mossbauer spectroscopic study (33-36).
The catalytic 5’-deoxyadenosyl radical is also proposed to be a key intermediate in pyruvate formate lyase (PFL) and anaerobic RNR enzymes in which it produces a stable glycyl radical for initiating catalysis. In PFL and RNR, the iron-sulfur cluster that cleaves SAM is located in an activase protein. The resting state of the activase contains a [4Fe-4S]2+, which is diamagnetic and EPR-inactive. When the activase is reduced by dithionite or a photochemical reductant, a typical low spin (S = 1/2) EPR signal of a [4Fe-4S]+ cluster is observed at temperatures below 20 K (Fig. 6). The line shape of the EPR signals of the [4Fe-4S]+ cluster is sensitive to the reductants added, which suggests that the reductants either modify the structure of the protein or directly interact with the iron-sulfur center. A prolonged chemical reduction by dithionite at high concentrations, 10 mM, for example, results in a fully reduced EPR-silent activase (37). The fully reduced activase is proposed to contain an all-ferrous [4Fe-4S]0 cluster because it is a additional reduction of the [4Fe-4S]+ cluster. The putative [4Fe-4S]0 can transform into a [3Fe-4S]+ cluster by direct air exposure (Fig. 6). The fully reduced, EPR-inactive form of the L. lactis activase is not obtainable by a mild reductant such as ascorbic acid.
In the presence of SAM, the photochemically reduced activase contains a protein-bound [4Fe-4S]+ cluster capable of activating the corresponding PFL or RNR enzymes. The SAM-[4Fe-4S]+ complex in the anaerobic L. lactis RNR displays a more rhombic EPR signal at low temperatures (Fig. 6). This EPR signal has smaller principal g values (1.86, 1.92, and 2.00). A similar observation of substrate/cofactor-induced spectrum changes, for example, increased rhombicities and decreased g-values, has been found in many other enzymes such as aconitase (38). Figure 6 suggests that SAM may coordinate directly to the iron-sulfur cluster. Using advanced EPR spectroscopy, the direct coordination of the SAM molecule to the iron-sulfur cluster has been established in LAM and verified by crystallographic results. This example illustrates a general feature that the electronic structure of the metal center changes on direct coordination to the substrate or inhibitor.

Figure 6. EPR spectra of the iron-sulfur cluster in the activase protein of anaerobic RNR from L. lactis. a) (3Fe-4S)+ cluster in oxidized activase, b) (4Fe-4S)+ cluster in photochemically reduced activase, and sample b in the presence of SAM. Spectra were taken at 10 K (35).
Electron transfer
EPR spectroscopy can serve as an efficient tool for studying the delivery of electrons between biological systems. Ni- trogenase, for example, consists of an iron protein and an iron-molybdenum protein. N2-binding and reduction take place at the iron-molybdenum protein, which contains a 7Fe-9S-Mo- X-homocitrate cofactor (FeMo-co). During catalysis, FeMo-co acquires electrons one at a time from the Fe-protein. Using rapid freeze-quench EPR, Hoffman and Seefeldt et al. have trapped and characterized several reactive intermediates of the enzyme reaction (39). The relaxation properties of the intermediates have been measured by an innovative step-annealing approach, and the information obtained has helped to assign the structure of the observed intermediates with proposed structure. The direct outcome of this work is a significant step forward in understanding the nitrogenase catalytic mechanism. The step-annealing approach employed in the nitrogenase study includes rapidly warming a sample held at 77 K to 253 K by placement for a fixed time in a methanol bath held at that temperature, quench-cooling it back to 77 K, and then collecting a 2 K EPR spectrum. These procedures are based on Davydov and colleagues’ studies of the iron-oxygen intermediates that are directly generated in an EPR sample at 77 K by radiolytic cryoreduction (39).
Concluding Remarks
EPR data on enzymes and mechanisms have been accumulating rapidly, and the knowledge learned from EPR-centered techniques has significantly contributed to the development of modern enzymology. In this article, the background and technique of EPR spectroscopy is introduced, and a few selected topics are used to illustrate the potential of its application in two areas of enzymology.
EPR spectroscopy has been a pivotal tool in the identification, quantification, and characterization of both cofactor-derived and protein-based radicals. With the assistance of EPR spectroscopy, several important enzymes are now known to be radical enzymes, including the well-studied cytochrome c peroxidase, galactose oxidase, photosystem II, prostaglandin H synthase, pyruvate formate lyase, ribonucleotide reductase, quino-containing enzymes, and coenzyme B12- or SAM-dependent superfamily of enzymes. Although as a terminology “radical enzymology” was only formally introduced in 2003 (11), it was recognized long before as a major area of enzymology. EPR-centered techniques will continue to be the primary tools in the future development of radical enzymology.
The oxidation state and electronic structure of protein-based paramagnetic metal ions can also be described in great detail by EPR spectroscopy, and the structural transitions in response to substrate-binding and the redox and environmental changes can be followed by EPR spectroscopy. Therefore, this technique provides an experimental handle in the studies of metalloenzymes.
To solve more complex chemical and biological problems, EPR spectroscopy has grown into a family of techniques. In addition to the conventional X-band continuous-wave EPR spectroscopy, ENDOR and ESEEM have become popular analytic tools (see the Further Reading section). PELDOR is an emerging new technique, and its value in enzymology has received considerable attention (40, 41). EPR at a very high field (HF-EPR) is expected to characterize integer spin systems with large zero-field interactions, some of which are EPR silent at lower fields. Examples of where such an approach would be useful are Mn(III) in manganese enzymes; Fe(II) in hemoglobin; Fe(IV), Co(I) in vitamin B12-binding enzymes; Ni(II), Mo(IV) in oxidases; and W(IV) in dehydrogenases (42). The combination of pulse EPR and multifrequency EPR techniques seems to be the most promising use for enzymologists who are studying the dynamics and long-distance interactions in radical enzymes and paramagnetic metalloproteins. The development and successful application of these advanced EPR techniques will continue to play an important role in the future development of enzymology.
Acknowledgment
The NIH subaward of GM069618, an institutional American Cancer Society award, and an ORAU Faculty Enhancement Award in Life Sciences are acknowledged.
References
1. Commoner B, Townsend J, Pake GE. Free radicals in biological materials. Nature 1954; 174:689-691.
2. Kochelaev BI. Discovery of electron spin resonance. Encycl. Mag. Reson. 2007.
3. Eaton GR, Eaton SSE, Salikhov KM. Foundations of Modern EPR. 1998. World Scientific Publishing Company, Hackensack, NJ.
4. Commoner B, Heise JJ, Lippincott BB, Norberg RE, Passonneau JV, Townsend J. Biological activity of free radicals. Science 1957; 126:57-63.
5. Ehrenberg A, Ludwig GD. Free radical formation in reaction between old yellow enzyme and reduced triphosphopyridine nucleotide. Science 1958; 127:1177-1178.
6. Commoner B, Lippincott BB, Passonneau JV. Electron-spin resonance studies of free-radical intermediates in oxidation-reduction enzyme systems. Proc. Natl. Acad. Sci. USA 1958; 44:1099-1110.
7. Malmstrom BG, Vanngard T, Larsson M. An electron-spin-resonance study of the interaction of manganous ions with enolase and its substrate. Biochim. Biophys. Acta 1958; 30:1-5.
8. Grinberg O, Berliner LJ. Very high frequency (VHF) ESR/EPR; Biological Magnetic Resonance 2004; 22.
9. Robitaille PM, Berliner L. Ultra high field magnetic resonance imaging. Biological Magnetic Resonance 2006; 26.
10. Hoffman BM. Electron-nuclear double resonance spectroscopy (and electron spin-echo envelope modulation spectroscopy) in bioinorganic chemistry. Proc. Natl. Acad. Sci. U.S.A. 2003; 100: 3575-3578.
11. Banerjee R. Radical Enzymology. Chem. Rev. (special edition) 2003; 103:2081-2456.
12. Reichard P. From RNA to DNA, why so many ribonucleotide reductases? Science 1993; 260:1773-1777.
13. Ehrenberg A, Reichard P. Electron spin resonance of the iron- containing protein B2 from ribonucleotide reductase. J. Biol. Chem. 1972; 247:3485-3488.
14. Reichard P, Ehrenberg A. Ribonucleotide reductase—a radical enzyme. Science 1983; 221:514-519.
15. van der Donk WA, Stubbe J, Gerfen GJ, Bellew BF, Griffin RG. EPR investigations of the inactivation of E. coli ribonucleotide reductase with 2'-azido-2'-deoxyuridine 5'-diphosphate: Evidence for the involvement of the thiyl radical of C225-R1. J. Am. Chem. Soc. 1995 Sep 6; 117:8908-8916.
16. Stubbe J, van Der Donk WA. Protein radicals in enzyme catalysis. Chem. Rev. 1998; 98:705-762.
17. Whittaker JW. Selective isotopic labeling of recombinant proteins using amino acid auxotroph strains. Methods Mol. Biol. 2007; 389:175-188.
18. Whittaker JW. Spectroscopic studies of galactose oxidase. Methods Enzymol. 1995; 258:262-277.
19. Barry BA, El-Deeb MK, Sandusky PO, Babcock GT. Tyrosine radicals in photosystem II and related model compounds. Characterization by isotopic labeling and EPR spectroscopy. J. Biol. Chem. 1990; 265:20139-20143.
20. Liu A, Poetsch S, Davydov A, Barra A-L, Rubin H, Graslund A. The tyrosyl free radical of recombinant ribonucleotide reductase from Mycobacterium tuberculosis is located in a rigid hydrophobic pocket. Biochemistry. 1998; 37:16369-16377.
21. Sahlin M, Graslund A, Ehrenberg A, Sjoberg BM. Structure of the tyrosyl radical in bacteriophage T4-induced ribonucleotide reductase. J. Biol. Chem. 1982; 257:366-369.
22. Hogbom M, Galander M, Andersson M, Kolberg M, Hofbauer W, Lassmann G, Nordlund P, Lendzian F. Displacement of the tyrosyl radical cofactor in ribonucleotide reductase obtained by single-crystal high-field EPR and 1.4-A X-ray data. Proc. Natl. Acad. Sci. USA 2003; 100:3209-3214.
23. Chabriere E, Vernede X, Guigliarelli B, Charon MH, Hatchikian EC, Fontecilla-Camps JC. Crystal structure of the free radical intermediate of pyruvate:ferredoxin oxidoreductase. Science 2001;294:2559-2263.
24. Nordlund P, Reichard P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006; 75:681-706.
25. Kauppi B, Nielsen BB, Ramaswamy S, et al. The three-dimensional structure of mammalian ribonucleotide reductase protein R2 reveals a more-accessible iron-radical site than Escherichia coli R2. J. Mol. Biol. 1996; 262:706-720.
26. Brudvig GW. Electron paramagnetic resonance spectroscopy. Methods Enzymol. 1995; 246:536-554.
27. Reed GH, Ballinger MD. Characterization of a radical intermediate in the lysine 2,3-aminomutase reaction. Methods Enzymol. 1995; 258:362-379.
28. Frey PA, Hegeman AD, Reed GH. Free radical mechanisms in enzymology. Chem. Rev. 2006; 106(8): 3302-3316.
29. Frey PA, Magnusson OT. S-Adenosylmethionine: a wolf in sheep’s clothing, or a rich man’s adenosylcobalamin? Chem. Rev. 2003; 103:2129-2148.
30. Booker SJ, Cicchillo RM, Grove TL. Self-sacrifice in radical S-adenosylmethionine proteins. Curr. Opin. Chem. Biol. 2007; 11:543-552.
31. Banerjee R, Ragsdale SW. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem. 2003; 72:209-247.
32. Reed GH. Radical mechanisms in adenosylcobalamin-dependent enzymes. Curr. Opin. Chem. Biol. 2004; 8:477-483.
33. Jiang W, Yun D, Saleh L, Barr EW, Xing G, Hoffart LM, Maslak M-A, Krebs C, Bollinger JM Jr. A manganese(IV)/iron(III) cofactor in chlamydia trachomatis ribonucleotide reductase. Science 2007; 316:1188-1191.
34. Jiang W, Bollinger JM Jr, Krebs C. The active form of Chlamydia trachomatis ribonucleotide reductase R2 protein contains a heterodinuclear Mn(IV)/Fe(III) cluster with S = 1 ground state. J. Am. Chem. Soc. 2007; 129:7504-7505.
35. Jiang W, Hoffart LM, Krebs C, Bollinger JM Jr. A manganese(IV)/iron(IV) intermediate in assembly of the manganese (IV)/Iron(III) cofactor of chlamydia trachomatis ribonucleotide reductase. Biochemistry 2007; 46:8709-8716.
36. Jiang W, Xie J, Nqrgaard H, Bollinger JM Jr, Krebs C. Rapid and quantitative activation of Chlamydia trachomatis ribonucleotide reductase by hydrogen peroxide. 2008; 47;4477-4483.
37. Liu A, Graslund A. Electron paramagnetic resonance evidence for a novel interconversion of [3Fe-4S]+ and [4Fe-4S]+ clusters with endogenous iron and sulfide in anaerobic ribonucleotide reductase activase in vitro. J. Biol. Chem. 2000; 275:12367-12373.
38. Beinert H, Kennedy MC, Stout CD. Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev. 1996; 96:2335-2374.
39. Lukoyanov D, Barney BM, Dean DR, Seefeldt LC, Hoffman BM. Connecting nitrogenase intermediates with the kinetic scheme for N2 reduction by a relaxation protocol and identification of the N2 binding state. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:1451-1455.
40. Denysenkov VP, Prisner TF, Stubbe J, Bennati M. High-field pulsed electron-electron double resonance spectroscopy to determine the orientation of the tyrosyl radicals in ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:13386-13390.
41. Schiemann O, Prisner TF. Long-range distance determinations in biomacromolecules by EPR spectroscopy. Quarterly Rev. Biophys. 2007; 40:1-53.
42. Ubbink M, Worrall JAR, Canters GW, Groenen EJJ, Huber M. Paramagnetic resonance of biological metal centers. Ann. Rev. Biophys. Biomol. Struc. 2002; 31:393-422.
Further Reading
Weil, JA, Bolton, JR. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. 2nd ed. 2007. Wiley, New Jersey.
Rieger, PH. Electron Spin Resonance: Analysis and Interpretation. 2007. Royal Society of Chemistry, London, UK.
Van Doorslaer, S, Vinck, E. The strength of EPR and ENDOR techniques in revealing structure-function relationships in metalloproteins. Phys. Chem. Chem. Phys. 2007; 9:4620-4638.
Hagen, WR. EPR spectroscopy as a probe of metal centres in biological systems. Dalton Trans. 2006; 37:4415-4434.
Hertel, MM, Denysenkov, VP, Bennati M, Prisner TF. Pulsed 180-GHz EPR/ENDOR/PELDOR spectroscopy. Mag. Reson. Chem. 2005; 43:S248-S255.
Eaton SS, Eaton GR, Berliner LJ. Biomedical EPR - Part A: Free Radicals, Metals, Medicine and Physiology. Biological Magnetic Resonance 23. 2004.
Eaton SS, Eaton GR, Berliner LJ. Biomedical EPR - Part B: Methodology, Instrumentation, and Dynamics. Biological Magnetic Resonance 24. 2004.
Hoffman BM. ENDOR of metalloenzymes. Acc. Chem. Res. 1991; 24:164-170 and 2003; 36:522-529.
Schweiger A, Jeschke G. Principles of Pulse Electron Paramagnetic Resonance. 2001. Oxford University Press, Cambridge, MA.
Berliner LJ, Eaton SS, Eaton GR. Distance Measurements in Biological Systems by EPR. Biological Magnetic Resonance 19. 2001.
Palmer G. Electron paramagnetic resonance of metalloproteins. In: Que Jr, L. ed. Physical Methods in Bioinorganic Chemistry. 2000. University Press, Sausalito, CA.
Pilbrow JR, Hanson GR. Electron paramagnetic resonance. Methods Enzymol. 1993; 227:330-384.
Hoff AJ, ed. Advanced EPR - Applications in Biology and Biochemistry. 1989. Elsevier, Amsterdam, The Netherlands.
Berliner LJ, Reuben J. Spin Labeling, Theory and Applications; Biological Magnetic Resonance 8. 1989. Springer, New York.
See Also
Chemistry of B12-Dependent Enzyme Reactions
Metallo-Enzymes and Metallo-Proteins, Chemistry of
Nitric Oxide, Biological Targets of
NMR: Overview of Applications in Chemical Biology
NMR for Proteins