CHEMICAL BIOLOGY
Schizophrenia, Biological Mechanisms of
Nora I. Perrone-Bizzozero and W. Michael Bullock, Department of Neurosciences, University of New Mexico School of Medicine.
doi: 10.1002/9780470048672.wecb669
Schizophrenia is a chronic and severely debilitating mental disorder that affects approximately 1 % of the world's population. Although schizophrenia has been recognized for over 100 years, the causes and pathophysiological mechanisms of this illness remained rather elusive until recently. Evidence obtained during the last 3 decades suggests that schizophrenia is a neurodevelopmental disorder that affects the structure and function of distributed brain regions. Multiple neurotransmitter systems have been implicated, as have both gray and white matter abnormalities. These structural alterations result in synaptic miscommunication at local neuronal circuits and long-distance functional disconnectivity, with both genetic and environmental factors contributing to these deficits. This article will discuss our current understanding of the biological and neurochemical bases of schizophrenia and will describe new pharmacological, genetic, and lesion models used for testing the mechanisms that underlie this devastating disease.
Schizophrenia was first identified in 1893 by the German psychiatrist Emil Kraepelin, who termed the disorder dementia praecox because of its early onset and irreversible mental decline. Then, 15 years later, the Swiss psychiatrist Eugen Bleuler recognized that the illness affected both the judgment and emotional state of the patient and referred to this disorder as schizophrenia, from the Greek schiz- to split and phren- mind. Bleuler identified the cardinal symptoms of the illness as loosening of associations, flat affect, social withdrawal, and ambivalence, and these criteria are still used for diagnostic purposes today. The onset of symptoms characteristically occurs during late adolescence and early adulthood. According to the Diagnostic and Statistical Manual IV (American Psychiatric Association, 1994), symptoms are categorized as positive or negative, and patients manifest these symptoms at various degrees during the course of their illness. Positive symptoms are manifestations of psychosis and include unusual behaviors such as paranoid or bizarre delusions, auditory hallucinations, and disorganized speech and thinking. In contrast, negative symptoms represent a loss of normal behaviors such as flat or blunted affect and emotion, poverty of speech (alogia), inability to experience pleasure (anhedonia), and lack of motivation (avolition). In addition, patients exhibit unremitting cognitive deficits. The clinical course and outcome of schizophrenia shows great variability; however, typically, positive symptoms fluctuate and negative symptoms remain more stable over time.
Alterations in Brain Structure and Function
Unlike neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease, the brains of patients with schizophrenia reveal neither gross anatomical changes nor the presence of pathological structures such as amyloid plaques or Lewy bodies. Thus, for much of the 1900s, psychiatry textbooks classified schizophrenia as a “functional” psychosis, for example, a condition that had no underlying physical brain disease. The technological advances in the past 25 years made it possible for investigators to re-evaluate the biological bases of schizophrenia systematically and provided evidence of subtle but consistent neuropathological and molecular alterations. Although most of these studies focused on the dorsolateral prefrontal cortex (DLPFC) and hippocampus, other brain regions including the thalamus, cerebellum, and its connecting white matter tracts have been implicated in schizophrenia (Fig. 1).
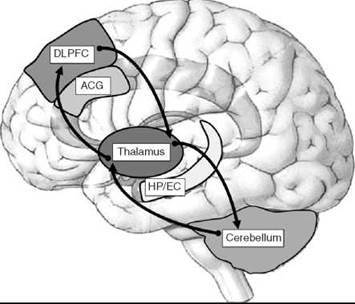
Figure 1. Schizophrenia affects the structure and function of distributed brain regions. Multiple brain regions are affected in schizophrenia including the dorsolateral prefrontal cortex, anterior cingulate gyrus, thalamus hippocampus/entorhinal cortex, and cerebellum. Arrows show the cortico-cerebellar-thalamic-cortical circuit, one of the networks implicated in schizophrenia (22).
Prefrontal cortex and anterior cingulate gyrus
Both postmortem tissue and neuroimaging analyses of the brain of patients with schizophrenia show small but reproducible gray matter reductions. In the prefrontal cortex (PFC), these changes were shown to be related to decreases in neuronal cell size and neuropil volume but not to a reduction in cell numbers (1). In the PFC, lamina-specific reductions in dendritic spine density were observed in the layer III pyramidal neurons (2) (Fig. 2). Because these cells play a critical role in cortical-cortical communications between adjacent and distant regions of the neocortex, a reduction in spine density suggests an impaired connectivity of the prefrontal cortex in the patients. Supporting this idea, the levels of the synaptic protein synaptophysin were shown to decrease in the PFC of patients along with increases in the growth-associated protein GAP-43, a marker of immature and/or “plastic” synapses (3, 4). The ratio of synaptophysin to GAP-43 is considered a putative index of synaptic maturation. Therefore, it is likely that this process is impaired in schizophrenia. Interestingly, GAP-43 mRNA levels were shown to be decreased in the PFC (5), which suggests that the observed increases in GAP-43 protein may be restricted to projections coming from other cortical and subcortical brain regions. In agreement with these structural and molecular alterations, patients with schizophrenia show signs of “hypofrontality,” which is characterized by working memory deficits and decreased cerebral blood flow in the frontal lobe (6, 7).
An altered distribution of neurons and axons was also seen in the anterior cingulate gyrus (8). Both within the prefrontal cortex and the anterior cingulate gyrus, the number of specific subtypes of inhibitory gamma-amino butyric acid (GABA) interneurons, such as the basket cells and chandelier neurons depicted in Fig. 2, was found to be decreased (9). In contrast, an increased number of interstitial white matter neurons were found in deep layers of the prefrontal cortex and other brain regions (10), which suggests the possibility of neuronal migration defects in schizophrenia.

Figure 2. Neurochemical and morphological alterations in the prefrontal cortex of patients with schizophrenia. The diagram shows some of the cell types in the layers I—VI of the dorsolateral prefrontal cortex, including glutamatergic pyramidal neurons (light gray) and GABAergic somatostatin-containing neurons, basket/wide arbor neurons and chandelier interneurons (dark gray), along with the changes in cell structure, gene expression, and neurotransmission observed in this region. These changes include decreased mRNA levels for several markers of GABAergic interneurons and reductions in the GAT-1 GABA transporter immunoreactivity in axon cartridges from chandelier cells with increased levels of postsynaptic GABAA receptors in pyramidal cells. Adapted from Ref. 9 and other references in the text.
Hippocampus and entorhinal cortex
Convergent evidence from neuroimaging and neuropathological studies indicates that the structure and function of the hippocampus is compromised in schizophrenia. Patients with schizophrenia have decreased hippocampal volume (11, 12) and several cytoarchitectural abnormalities in both the hippocampus proper and entorhinal cortex (13). In addition, neuropathological examination of postmortem hippocampal tissue revealed a decreased number of GABAergic interneurons and several synaptic protein deficits in patients with schizophrenia (14-16). In contrast to the PFC, the levels of GAP-43 protein and the ratio of GAP-43 to synaptophysin mRNAs was decreased in the hippocampus of patients with schizophrenia (16, 17). These changes were correlated with decreased levels of another marker of excitatory synapses, the synaptic protein complexin II (18). Along with these structural and molecular alterations, patients with schizophrenia exhibit reduced activation of the hippocampus during the encoding and retrieval of episodic and relational information, two well-characterized hippocampal dependent tasks (19-21).
Cerebellum and thalamus
Traditionally, most research performed in the field of schizophrenia has focused on brain regions directly implicated in the symptomatology of the disease, namely the PFC and limbic areas. Work by Andreasen et al. (22) first implicated the cerebellum as an affected structure in schizophrenia through the cortico-cerebellar-thalamic-cortical circuit (CCTCC, see arrows in Fig. 1). Dysfunction in one area of this circuit such as the thalamus (23) is thought to affect all other areas of the circuit. As a component of the CCTCC, the lateral hemispheres of the cerebellum have been implicated in cognitive and emotional functioning (24). Retroviral tracing and neuroimaging studies demonstrated cerebellar-prefrontal connections (25), which may contribute to cognitive dysfunction in schizophrenia. Intrinsic to these connections, a forward modeling system of the cerebellum has been proposed in which information from the motor cortex or the PFC is transferred to the cerebellum, and the cerebellum acts as a predictor of the outcome for both motor and cognitive functioning (26). Clinically, patients exhibit cerebellar neurological signs (27), deficits in eyeblink conditioning (28), and shortfalls in response timing (29). Neuroimaging studies have shown increases in blood flow and in glucose consumption in the cerebellum of schizophrenic patients relative to that of other brain regions (7, 30-32). In accordance with increased cerebellar activity, a recent study found that the levels of GAP-43 and brain-derived neurotrophic factor (BDNF), which are expressed in cerebellar granule cells in an activity-dependent manner, are upregulated in the patients (33). Additional molecular studies have shown decreased expression of the developmental marker reelin and the GABA synthesizing enzymes GAD65 and GAD67 and increases in the axonal chemorepellant protein semaphorin 3A (34-37). This finding is interesting considering the fact that GABA dysfunction in the cerebellum may lead to increases in granule cell firing and thus account for the increases seen in blood flow, glucose use, and the expression of activity-dependent genes. These changes ultimately could impact cerebellar contributions to cognitive (and motor) functioning in the patients.
White matter alterations
As described above and shown in Fig. 1, schizophrenia pathology is not restricted to a single brain region but affects multiple distributed areas. Brain regions considered important in the pathology of schizophrenia, such as the prefrontal cortex and temporal lobe, are linked normally to one another by tracts of dense and reciprocally afferent white matter, which thus suggests that white matter alterations could have a role in schizophrenia pathophysiology. Supporting this idea, white matter volume reductions have been reported in patients with schizophrenia (38-40). Analysis of frontal lobe white matter and corpus callosum of patients with diffusion tensor imaging revealed specific alterations in myelin structure (41). These changes were also observed in children and adolescent with schizophrenia (42), which indicates that white matter abnormalities may be present early in the disease. Postmortem tissue analysis at the light and electron microscopic levels also demonstrated the presence of myelin and oligodendrocyte abnormalities in schizophrenia (43-45). Along with these morphological abnormalities, microarray analysis of PFC tissue from subjects with schizophrenia also demonstrated downregulation of myelin-related and oligodendrocyte-related genes in the patients (46, 47). Finally, recent genetic studies identified polymorphisms in two important regulators of myelination, the transcription factor OLIG2 and the RNA-binding protein QKI, which are associated with increased risk for schizophrenia (48, 49).
Neurodevelopmental Hypothesis of Schizophrenia
As described above, the brains of patients with schizophrenia show alterations in the levels of the developmental markers reelin, GAP-43, and semaphorin 3A. In addition, evidence of disrupted neuronal distribution was found in several cortical areas and the hippocampal formation. These findings together with the absence of neuronal cell loss and concomitant reactive gliosis in these brain regions, both hallmarks of neurodegenerative disorders, led investigators to propose the neurodevelopmental hypothesis of schizophrenia (6, 50), which is still the prevailing theory in this disorder (51, 52). This hypothesis states that the illness is related to abnormal brain development and is supported by several pieces of evidence, including increased frequency of obstetric complications, viral infections, and other developmental stressors in patients with schizophrenia (see the sections below titled “Genetic and Environmental Factors” and “Developmental Models”) and the presence of soft neurological signs, cognitive impairment, and behavioral dysfunction in children long before the first psychotic episode.
Neurotransmitter Systems
As described in the previous sections, neuropathological studies demonstrated alterations in the levels of several synaptic proteins in the PFC, hippocampus, and cerebellum of patients with schizophrenia (13, 15, 53). These observations have lead to the hypothesis that the clinical symptoms of schizophrenia are manifestations of abnormal neural circuitry and dysfunctional communication between different brain regions (22, 51). These abnormalities affect multiple neurotransmitter systems. Although dopamine dysfunction in schizophrenia is widely accepted, a growing body of evidence suggests the involvement of glutamate, GABA, and other neurotransmitters in schizophrenia.
Dopamine
The discovery that the first antipsychotic drugs in the early 1950s, such as chlorpromazine, work in vitro by blocking dopamine receptors led to the hypothesis that schizophrenia was the result of excessive dopaminergic neurotransmission (54, 55). Supporting this hypothesis, drugs that enhance dopamine action (e.g., cocaine, amphetamines, and L-DOPA) worsen the symptoms of schizophrenia. However, it is clear that 1) not all patients respond to neuroleptic treatment and 2) not all symptoms are reversed by the medication.
Classical treatments for schizophrenia involve the administration of typical antipsychotics, such as haloperidol and chlorpromazine, which primarily bind to dopamine D2 receptors with high affinity, and atypical antipsychotics, such as clozapine and risperidone, which bind to a broader range of receptors including serotonergic and noradrenergic receptors among others. Although antipsychotics are effective at relieving the positive symptoms via their actions on D2 receptors, they are not effective in ameliorating the negative and cognitive symptoms. Interestingly, recent studies suggest that patients with schizophrenia may have hypofunctional D1 dopamine receptors in the prefrontal cortex (Fig. 2) and that agonists to this subtype of receptor may be effective in treating the working memory deficits associated with this illness (56, 57). In addition to drugs that work on monoamine receptors, new drugs that target glutamate, GABA, and cholinergic receptors are now being tested for ameliorating the cognitive dysfunction in schizophrenia (58, 59).
Glutamate
The glutamate hypothesis of schizophrenia was derived from the fact that drugs that block the N-methyl-D-aspartate (NMDA) subtype of glutamate receptors, such as phencyclidine (PCP) and ketamine, cause schizophrenia-like symptoms in humans and animal models. Furthermore, these drugs mimic not only the positive (psychotic) but also the negative and cognitive symptoms of the disease, which suggests that they act on the same basic pathophysiological mechanisms that are affected in schizophrenia.
NMDA receptor dysfunction has been characterized in different brain regions of patients with schizophrenia. Neuropathological studies revealed altered expression of receptor subunits in the prefrontal cortex (60), and single positron emission tomography studies have shown decreased NMDA receptor binding in the hippocampus of patients (61). Studies using NMDA receptor antagonists such as PCP, ketamine, and MK-801 additionally implicate hypofunction of these channels in schizophrenia (62). NMDA receptor antagonists have been shown to block NMDA channels located on GABAergic interneurons selectively (63, 64), which suggests that NMDA receptor dysfunction in a specific subset of these interneurons may be central to schizophrenia (62, 65).
GABA
GABA is the main inhibitory neurotransmitter in the brain, and dysfunction in certain subsets of GABAergic interneurons is one of the most consistent findings in the study of schizophrenia (9, 37, 66-68). Reductions in mRNA and protein levels of the 67 kD form of glutamic acid decarboxylase (GAD67), one of the GABA synthesizing enzymes, have been observed in the prefrontal cortex (69-71), the hippocampus (53, 72), the cerebellum (34, 35, 37), and other brain regions (73). As shown in Fig. 2, chandelier and basket interneurons in the PFC show decreased mRNA levels for the GAT-1 GABA transporter (74) and the calcium-binding protein parvalbumin (70), whereas other subtypes of interneurons have reduced levels of somatostastin (SST) mRNA (73). Postsynaptic changes such as increases in GABAA α2 receptor density and GABAA receptor radioligand binding in the PFC and anterior cingulate cortex were also observed (75-77). In addition to these findings, single nucleotide polymorphisms in the promoter region of the GAD67 gene were shown to be associated with reductions in gray matter in patients with childhood-onset schizophrenia (78). Considering the role of GABAergic interneurons in the modulation of excitatory output, it can be hypothesized that dysfunction in these cells may mediate some positive, negative, and cognitive symptoms seen in schizophrenia (79).
Other neurotransmitters: acetylcholine
Evidence of the involvement of acetylcholine in the pathophysiology of schizophrenia comes not only from the findings of decreased availability of cholinergic muscarinic receptors in patients (80, 81) but also from genetic studies that link specific polymorphisms in the gene for the alpha 7 nicotinic receptor (CHRNA7) with this illness. The CHRNA7 receptor is one of the ligand-gated ion channels that mediate fast cholinergic transmission at synapses. The CHRNA7 gene is located at chromosome 15q13-14, a locus implicated in the genetic transmission of schizophrenia (82, 83). Specific polymorphisms in CHRNA7 promoter region were shown to correlate with sensory gating alterations in patients with schizophrenia as measured by the P50 inhibition in auditory evoked response (84, 85). Furthermore, a recent study demonstrated that two additional single nucleotide polymorphisms (SNPs) in the CHRNA7 gene correlate with patterns of brain activation in schizophrenia patients during an auditory oddball task (86). The same study also linked an SNP in the gene coding for choline acetyltransferase, the acetylcholine synthesizing enzyme, with these abnormalities.
Genetic and Environmental Factors
Although the etiology of schizophrenia is not completely understood, it is becoming apparent that schizophrenia is a neurodevelopmental disorder that involves both genetic and environmental risk factors. The contribution of genetic factors was demonstrated by twin (87) and adoption studies (88) and by the higher prevalence of schizophrenia-like personality disorders in relatives of patients with schizophrenia (89). Other factors such as season of birth (90) and prenatal or perinatal complications such as ischemia (91) and viral infections (92) have also been identified as risk factors, although to a much lesser degree.
The influence of genetic factors is evidenced by the findings that about 50-75% of monozygotic twins with schizophrenia will have an affected twin and approximately 10% of first-degree relatives are also affected (93). Several genes have been associated with increased vulnerability for schizophrenia, including those encoding proteins associated with NMDA receptor function, synaptic plasticity, mitochondria energy metabolism, oxidative stress, development, and myelination (94, 95). Recent studies demonstrate that specific polymorphisms in some of these genes correlate with cognitive and neuroimaging abnormalities in patients (96). The best example of these polymorphisms is an SNP coding for the substitution of a valine for a methionine in position 108 of the short form (158 in long form) of the catechol-O-methyltransferase (COMT, Val 108/158 Met SNP) protein. The amino acid substitution results in a protein that has increased stability and, thus, increased rate of dopamine inactivation (97). This polymorphism has been associated with impaired performance in working memory tests and abnormal patterns of prefrontal cortex activation in both patients with schizophrenia and healthy volunteers (98). In addition, polymorphisms in the genes for BDNF and the metabotropic glutamate receptor 3 (GRM3), among others, have been associated with subtle but consistent alterations in PFC and hippocampal structure and function (96).
Animal Models
Schizophrenia is a purely human disease, which makes it hard to model the behavioral manifestations of this illness in animals. Nonetheless, animal models have been shown to reproduce specific aspects of the illness such as its effects on brain structure and function. Currently, several animal models are available to investigators. These animal models can be classified as developmental, pharmacological, genetic, and lesion models. Examples of these animal models are described below, and a complete listing of current animal models can be found in a recent review (99).
Developmental models
Given the evidence of perinatal stressors in some patients with schizophrenia, animal models have been generated to examine the influence of these factors in adult behavior. These studies demonstrated that animals exposed to prenatal viral infections, maternal deprivation, and other stressors exhibit several behavioral abnormalities consistent with schizophrenia, including disrupted prepulse inhibition, enhanced response to amphetamine, and impaired social interactions (100-102). Furthermore, some models show molecular and morphological deficits in the neocortex and hippocampus that mimic the alterations seen in patients with schizophrenia (103).
Pharmacological models
Pharmacological models that exploit the GABA/glutamate system have proven useful in studying the underlying pathophysiology of schizophrenia. These models include the picrotoxin-induced antagonism of GABAA receptors in rats (104) and the antagonism of NMDA receptors in both rodents and nonhuman primates (105-109). All these models affect the GABA/glutamate balance in different ways, but only the phencyclidine model has shown both the GABAergic and NMDA receptor changes seen in patients with schizophrenia (108, 110, 111).
Because PCP administration leads to many symptoms inherent to schizophrenia, studies are now being conducted that administer the compound to rodents and primates to induce a schizophrenic-like phenotype (106, 112). Acute and chronic dosing regimens show differential and often opposing effects in rodents. Immediately after administration of PCP to rats, neurons of the medial PFC show an initial excitation as seen by activation of early immediate genes (113). This effect is likely because of the preferential blockage of receptors in GABAergic interneurons by PCP and other NMDA receptor antagonists (64). This initial activation then is followed by a period of cortical depression as described by glucose use studies (114), presumably as a compensatory mechanism. Acute PCP administration also produces schizophrenia-like symptoms including social withdrawal (115), impaired sensory motor gating (116), and cognitive dysfunction (105, 107). Chronic intermittent exposure to low dose of PCP in rodents results in decreased metabolic activity in the prefrontal cortex, auditory cortex, hippocampus, and reticular nucleus of the thalamus (109), all regions affected in schizophrenia. Along with this decrease in metabolic function, decreases in parvalbumin expression were also seen (109), which mirror the chandelier and basket cell dysfunction seen in the prefrontal cortex of patients with schizophrenia (70). Taken together, the data suggest that the chronic intermittent PCP exposure model is one of the most functionally and neurochemically relevant animal models of unremitting schizophrenia.
Genetic models
Like the pharmacological model presented above, a genetic model also targets the NMDA receptor. This model was created by knocking down the NR1 subunit of the NMDA receptor, which is obligatory for receptor function, in mice so that only 5% of the protein is expressed (117). These animals, also known as NR1 hypomorphs, show NMDA receptor hypofunction and display several schizophrenia-like behaviors, such as reduced social interactions, increased locomotion, stereotypic movements, and sensorimotor gating deficits (117, 118). Interestingly, treatment of these mice with the atypical antipsychotic clozapine ameliorates some of these abnormal behaviors (117). Finally, similarly to schizophrenic patients, NR1 deficient mice show decreased metabolism in the medial prefrontal and anterior cingulate cortices (119).
Another well-characterized genetic model of schizophrenia consists of mice with target mutations in the Disrupted-inSchizophrenia 1 (DISC1) gene. This gene was discovered in a Scottish family with a high incidence of mental illness, including schizophrenia (120), which has a balanced translocation in this chromosome 1q42.1 locus. This protein is known to be critical for normal development (121), and mice that express mutant DISC1 protein exhibit brain and behavioral abnormalities suggestive of schizophrenia, such as impaired learning and memory processes and altered neuronal development (122-125).
Lesion models
Although no clear indication of a brain lesion is found in schizophrenia, developmental lesion models, such as the neonatal ventral hippocampal lesion (NVHL) and the neonatal amygdala lesion models, have been shown to reproduce several aspects of this illness (126, 127). For example, NVHL rats exhibit increased responses to dopamine agonists and NMDA receptor antagonists, which are manifested only after puberty. These animals also show impaired social interactions, altered sensorimotor gating, and cognitive deficits (128). At the molecular level, these animals show decreased numbers of GAD67 expressing interneurons in the medial PFC, which is similar to the findings observed in patients (71). Overall, this model also reproduces multiple aspects of schizophrenia behavior and pathophysiology.
Concluding Remarks
In summary, the work reviewed in the previous sections suggests that schizophrenia is a neurodevelopmental disorder that affects the structure and function of distributed brain regions and their connecting white matter, with both genetic and environmental factors contributing to these alterations. As shown in Fig. 1, affected regions include frontal lobe and limbic system structures involved in cognition and emotion and areas that participate in sensorimotor integration such as the thalamus and cerebellum. Structural and molecular abnormalities in these regions result in synaptic alterations at local neuronal circuits and long-distance functional disconnectivity. Besides dopamine, multiple neurotransmitter systems have been implicated including glutamate, GABA, and acetylcholine. Based on these findings, drugs targeting specific subtypes of these receptors are now being tested in animal models and patients. It is expected that these new developments will help researchers not only to understand the etiology and basic pathophysiological mechanisms that lead to schizophrenia but also to develop better treatment strategies for this devastating illness.
References
1. Selemon LD, Goldman-Rakic PS. The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol. Psychiatry 1999; 45:17-25.
2. Kolluri N, Sun Z, Sampson AR, Lewis DA. Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am. J. Psychiatry 2005; 162:1200-1202.
3. Perrone-Bizzozero NI, Sower AC, Bird ED, Benowitz LI, Ivins KJ, Neve RL. Levels of the growth-associated protein GAP-43 are selectively increased in association cortices in schizophrenia. Proc. Natl. Acad. Sci. USA 1996; 93:14182-14187.
4. Glantz LA, Lewis DA. Reduction of synaptophysin immunore-activity in the prefrontal cortex of subjects with schizophrenia. Regional and diagnostic specificity. Arch. Gen. Psychiatry 1997; 54:660-669.
5. Weickert CS, Webster MJ, Hyde TM, Herman MM, Bachus SE, Bali G, Weinberger DR, Kleinman JE. Reduced GAP-43 mRNA in dorsolateral prefrontal cortex of patients with schizophrenia. Cereb. Cortex 2001; 11:136-147.
6. Weinberger DR, Berman KF. Prefrontal function in schizophrenia: confounds and controversies. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1996; 351:1495-1503.
7. Andreasen NC, O’Leary DS, Flaum M, Nopoulos P, Watkins GL, Boles Ponto LL, Hichwa RD. Hypofrontality in schizophrenia: distributed dysfunctional circuits in neuroleptic-naive patients. Lancet 1997; 349:1730-1734.
8. Benes FM: Neurobiological investigations in cingulate cortex of schizophrenic brain. Schizophr. Bull. 1993; 19:537-549.
9. Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005; 6:312-324.
10. Akbarian S, Bunney WE, Jr., Potkin SG, Wigal SB, Hagman JO, Sandman CA, Jones EG. Altered distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Arch. Gen. Psychiatry 1993; 50:169-177.
11. Bogerts B, Lieberman JA, Ashtari M, Bilder RM, Degreef G, Lerner G, Johns C, Masiar S. Hippocampus-amygdala volumes and psychopathology in chronic schizophrenia. Biol. Psychiatry 1993; 33:236-246.
12. Heckers S. Neuroimaging studies of the hippocampus in schizophrenia. Hippocampus 2001; 11:520-528.
13. Arnold SE. Cellular and molecular neuropathology of the parahippocampal region in schizophrenia. Ann. N. Y. Acad. Sci. 2000; 911:275-292.
14. Benes FM, Berretta S. Amygdalo-entorhinal inputs to the hippocampal formation in relation to schizophrenia. Ann. N. Y. Acad. Sci. 2000; 911:293-304.
15. Harrison PJ, Eastwood SL. Neuropathological studies of synaptic connectivity in the hippocampal formation in schizophrenia. Hippocampus 2001; 11:508-519.
16. Chambers JS, Thomas D, Saland L, Neve RL, Perrone-Bizzozero NI. Growth-associated protein 43 (GAP-43) and synaptophysin alterations in the dentate gyrus of patients with schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005; 29:283-290.
17. Eastwood SL, Harrison PJ. Hippocampal and cortical growth-associated protein-43 messenger RNA in schizophrenia. Neuroscience 1998; 86:437-448.
18. Eastwood SL, Harrison PJ: Decreased expression of vesicular glutamate transporter 1 and complexin II mRNAs in schizophrenia: more evidence for a synaptic pathology affecting glutamate neurons. Schizophr. Res. 2005; 73:159-72.
19. Barch DM, Csernansky JG, Conturo T, Snyder AZ. Working and long-term memory deficits in schizophrenia: is there a common prefrontal mechanism? J. Abnorm. Psychol. 2002; 111:478-494.
20. Weiss AP, Schacter DL, Goff DC, Rauch SL, Alpert NM, Fischman AJ, Heckers S. Impaired hippocampal recruitment during normal modulation of memory performance in schizophrenia. Biol. Psychiatry 2003; 53:48-55.
21. Jessen F, Scheef L, Germeshausen L, Tawo Y, Kockler M, Kuhn KU, Maier W, Schild HH, Heun R. Reduced hippocampal activation during encoding and recognition of words in schizophrenia patients. Am. J. Psychiatry 2003; 160:1305-1312.
22. Andreasen NC, Nopoulos P, O’Leary DS, Miller DD, Wassink T, Flaum M: Defining the phenotype of schizophrenia: cognitive dysmetria and its neural mechanisms. Biol. Psychiatry 1999; 46:908-920.
23. Andreasen NC. The role of the thalamus in schizophrenia. Can. J. Psychiatry 1997; 42:27-33.
24. Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain 1998; 121:561-579.
25. Middleton FA, Strick PL. Cerebellar projections to the prefrontal cortex of the primate. J. Neurosci. 2001; 21:700-712.
26. Ramnani N. The primate cortico-cerebellar system: anatomy and function. Nat. Rev. Neurosci. 2006; 7:511-522.
27. Ho BC, Mola C, Andreasen NC. Cerebellar dysfunction in neuroleptic naive schizophrenia patients: clinical, cognitive, and neuroanatomic correlates of cerebellar neurologic signs. Biol. Psychiatry 2004; 55:1146-1153.
28. Sears LL, Andreasen NC, O’Leary DS. Cerebellar functional abnormalities in schizophrenia are suggested by classical eyeblink conditioning. Biol. Psychiatry 2000; 48:204-209.
29. Brown SM, Kieffaber PD, Carroll CA, Vohs JL, Tracy JA, Shekhar A, O’Donnell BF, Steinmetz JE, Hetrick WP. Eyeblink conditioning deficits indicate timing and cerebellar abnormalities in schizophrenia. Brain Cogn. 2005; 58:94-108.
30. Kim JJ, Mohamed S, Andreasen NC, O’Leary DS, Watkins GL, Boles Ponto LL, Hichwa RD. Regional neural dysfunctions in chronic schizophrenia studied with positron emission tomography. Am. J. Psychiatry 2000; 157:542-548.
31. Malaspina D, Harkavy-Friedman J, Corcoran C, Mujica-Parodi L, Printz D, Gorman JM, Van Heertum R. Resting neural activity distinguishes subgroups of schizophrenia patients. Biol. Psychiatry 2004; 56:931-937.
32. Potkin SG, Alva G, Fleming K, Anand R, Keator D, Carreon D, Doo M, Jin Y, Wu JC, Fallon JH. A PET study of the pathophysiology of negative symptoms in schizophrenia. Positron emission tomography. Am J. Psychiatry 2002; 159:227-237.
33. Paz RD, Andreasen NC, Daoud SZ, Conley R, Roberts R, Bustillo J, Perrone-Bizzozero NI. Increased expression of activity- dependent genes in cerebellar glutamatergic neurons of patients with schizophrenia. Am. J. Psychiatry 2006; 163:1829-1831.
34. Fatemi SH, Stary JM, Earle JA, Araghi-Niknam M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67kDa and Reelin proteins in cerebellum. Schizophr. Res. 2005; 72:109-122.
35. Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch. Gen. Psychiatry 2000; 57:1061-1069.
36. Eastwood SL, Law AJ, Everall IP, Harrison PJ: The axonal chemorepellant semaphorin 3A is increased in the cerebellum in schizophrenia and may contribute to its synaptic pathology. Mol. Psychiatry 2003; 8:148-155.
37. Bullock WM, Cardon K, Bustillo JR, Roberts RC, Perrone-Bizzozero NI. Altered Expression of Genes Involved in GABAergic Transmission and Neuromodulation of Granule Cell Activity in the Cerebellum of Patients with Schizophrenia. Am. J. Psychiatry. In press.
38. Wright IC, Rabe-Hesketh S, Woodruff PW, David AS, Murray RM, Bullmore ET. Meta-analysis of regional brain volumes in schizophrenia. Am. J. Psychiatry 2000; 157:16-25.
39. Bartzokis G, Beckson M, Lu PH, Nuechterlein KH, Edwards N, Mintz J. Age-related changes in frontal and temporal lobe volumes in men: a magnetic resonance imaging study. Arch. Gen. Psychiatry 2001; 58:461-465.
40. Honea R, Crow TJ, Passingham D, Mackay CE. Regional deficits in brain volume in schizophrenia: a meta-analysis of voxel-based morphometry studies. Am. J. Psychiatry 2005; 162:2233-2245.
41. Kubicki M, Westin CF, McCarley RW, Shenton ME. The application of DTI to investigate white matter abnormalities in schizophrenia. Ann. N. Y. Acad. Sci. 2005; 1064:134-148.
42. White T, Kendi AT, Lehericy S, Kendi M, Karatekin C, Guimaraes A, Davenport N, Schulz SC, Lim KO. Disruption of hippocampal connectivity in children and adolescents with schizophrenia-a voxel-based diffusion tensor imaging study. Schizophr. Res. 2007; 90:302-307.
43. Uranova N, Orlovskaya, DD. Ultrastructural pathology of neuronal connectivity in postmortem brain of schizophrenic patients. Ann. Psychiatry 1996; 6:55-72.
44. Uranova N, Orlovskaya D, Vikhreva O, Zimina I, Kolomeets N, Vostrikov V, Rachmanova V. Electron microscopy of oligodendroglia in severe mental illness. Brain Res. Bull. 2001; 55:597-610.
45. Chambers JS, Perrone-Bizzozero NI. Altered myelination of the hippocampal formation in subjects with schizophrenia and bipolar disorder. Neurochem. Res. 2004; 29:2293-2302.
46. Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V, Fienberg AA: Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. USA 2001; 98:4746-51.
47. Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB, Starkey M, Webster MJ, Yolken RH, Bahn S. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 2003; 362:798-805.
48. Aberg K, Saetre P, Lindholm E, Ekholm B, Pettersson U, Adolfsson R, Jazin E: Human QKI, a new candidate gene for schizophrenia involved in myelination. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2006b; 141:84-90.
49. Georgieva L, Moskvina V, Peirce T, Norton N, Bray NJ, Jones L, Holmans P, Macgregor S, Zammit S, Wilkinson J, Williams H, Nikolov I, Williams N, Ivanov D, Davis KL, Haroutunian V, Buxbaum JD, Craddock N, Kirov G, Owen MJ, O’Donovan MC. Convergent evidence that oligodendrocyte lineage transcription factor 2 (OLIG2) and interacting genes influence susceptibility to schizophrenia. Proc. Natl. Acad. Sci. USA 2006; 103:12469-12474.
50. Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 1987; 44:660-669.
51. Benes FM. Emerging principles of altered neural circuitry in schizophrenia. Brain Res. Brain Res. Rev. 2000; 31:251-269.
52. Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu. Rev. Neurosci. 2002; 25:409-32.
53. Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 2001; 25:1-27.
54. Meltzer HY, Stahl SM. The dopamine hypothesis of schizophrenia: a review. Schizophr. Bull 1976; 2:19-76.
55. Carlsson A. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1988; 1:179-186.
56. Abi-Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y, Hwang DR, Keilp J, Kochan L, Van Heertum R, Gorman JM, Laruelle M. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J. Neurosci. 2002; 22:3708-3719.
57. Mu Q, Johnson K, Morgan PS, Grenesko EL, Molnar CE, Anderson B, Nahas Z, Kozel FA, Kose S, Knable M, Fernandes P, Nichols DE, Mailman RB, George MS. A single 20 mg dose of the full D1 dopamine agonist dihydrexidine (DAR-0100) increases prefrontal perfusion in schizophrenia. Schizophr. Res. 2007; 94:332-341.
58. Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat. Med. 2007; 13:1102-1107.
59. Tamminga CA. The neurobiology of cognition in schizophrenia. J. Clin. Psychiatry 2006; 67:9-13.
60. Akbarian S, Sucher NJ, Bradley D, Tafazzoli A, Trinh D, Hetrick WP, Potkin SG, Sandman CA, Bunney WE, Jr., Jones EG. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 1996; 16:19-30.
61. Pilowsky LS, Bressan RA, Stone JM, Erlandsson K, Mulligan RS, Krystal JH, Ell PJ: First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol. Psychiatry 2006; 11:118-119.
62. Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell. Mol. Neurobiol. 2006.
63. Grunze HC, Rainnie DG, Hasselmo ME, Barkai E, Hearn EF, McCarley RW, Greene RW. NMDA-dependent modulation of CA1 local circuit inhibition. J. Neurosci. 1996; 16:2034-2043.
64. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J. Neurosci. 2007; 27:11496-11500.
65. Lewis DA, Moghaddam B. Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch. Neurol. 2006; 63:1372-1376.
66. Akbarian S, Huang HS. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res. Brain Res. Rev. 2006; 52:293-304.
67. Torrey EF, Barci BM, Webster MJ, Bartko JJ, Meador-Woodruff JH, Knable MB: Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol. Psychiatry 2005; 57:252-260.
68. Woo TU, Walsh JP, Benes FM. Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch. Gen. Psychiatry 2004; 61:649-657.
69. Lewis DA, Volk DW, Hashimoto T. Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: a novel target for the treatment of working memory dysfunction. Psychopharmacology (Berl) 2004; 174:143-150.
70. Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 2003; 23:6315-6326.
71. Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA. Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch. Gen. Psychiatry 2000; 57:237-245.
72. Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM. Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen. Psychiatry 2002; 59:521-529.
73. Hashimoto T, Bazmi HH, Mirnics K, Wu Q, Sampson AR, Lewis DA. Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am. J. Psychiatry 2008; 165:479-489.
74. Volk D, Austin M, Pierri J, Sampson A, Lewis D. GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am. J. Psychiatry 2001; 158:256-265.
75. Benes FM, Vincent SL, Alsterberg G, Bird ED, SanGiovanni JP. Increased GABAA receptor binding in superficial layers of cingulate cortex in schizophrenics. J. Neurosci. 1992; 12:924-929.
76. Volk DW, Pierri JN, Fritschy JM, Auh S, Sampson AR, Lewis DA. Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb. Cortex 2002; 12:1063-1070.
77. Benes FM, Vincent SL, Marie A, Khan Y. Up-regulation of GABAA receptor binding on neurons of the prefrontal cortex in schizophrenic subjects. Neuroscience 1996; 75:1021-1031.
78. Addington AM, Gornick M, Duckworth J, Sporn A, Gogtay N, Bobb A, Greenstein D, Lenane M, Gochman P, Baker N, Balkissoon R, Vakkalanka RK, Weinberger DR, Rapoport JL, Straub RE. GAD1 (2q31.1), which encodes glutamic acid decarboxylase (GAD67), is associated with childhood-onset schizophrenia and cortical gray matter volume loss. Mol. Psychiatry 2005; 10:581-588.
79. Spencer KM, Nestor PG, Perlmutter R, Niznikiewicz MA, Klump MC, Frumin M, Shenton ME, McCarley RW. Neural synchrony indexes disordered perception and cognition in schizophrenia. Proc. Natl. Acad. Sci. USA 2004; 101:17288-17293.
80. Raedler TJ, Knable MB, Jones DW, Urbina RA, Gorey JG, Lee KS, Egan MF, Coppola R, Weinberger DR. In vivo determination of muscarinic acetylcholine receptor availability in schizophrenia. Am. J. Psychiatry 2003; 160:118-127.
81. Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Towards a muscarinic hypothesis of schizophrenia. Mol. Psychiatry 2007; 12:232-246.
82. Freedman R, Leonard S, Gault JM, Hopkins J, Cloninger CR, Kaufmann CA, Tsuang MT, Farone SV, Malaspina D, Svrakic DM, Sanders A, Gejman P. Linkage disequilibrium for schizophrenia at the chromosome 15q13-14 locus of the alpha7-nicotinic acetylcholine receptor subunit gene (CHRNA7). Am. J. Med. Genet. 2001; 105:20-22.
83. De Luca V, Wang H, Squassina A, Wong GW, Yeomans J, Kennedy JL: Linkage of M5 muscarinic and alpha7-nicotinic receptor genes on 15q13 to schizophrenia. Neuropsychobiology 2004; 50:124-127.
84. Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, Rosenthal J, Waldo MC, Reimherr F, Wender P, Yaw J, Young DA, Breese CR, Adams C, Patterson D, Adler LE, Kruglyak L, Leonard S, Byerley W. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc. Natl. Acad. Sci. USA 1997; 94:587-592.
85. Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, Drebing C, Berger R, Venn D, Sirota P, Zerbe G, Olincy A, Ross RG, Adler LE, Freedman R. Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch. Gen. Psychiatry 2002; 59:1085-1096.
86. Liu J, Pearlson G, Windemuth A, Ruano G, Perrone-Bizzozero NI, Calhoun V. Combining fMRI and SNP data to investigate connections between brain function and genetics using parallel ICA. Hum. Brain Mapp. 2007.
87. Onstad S, Skre I, Torgersen S, Kringlen E. Subtypes of schizophrenia-evidence from a twin-family study. Acta. Psychiatr. Scand. 1991; 84:203-206.
88. Kety SS. Schizophrenic illness in the families of schizophrenic adoptees: findings from the Danish national sample. Schizophr. Bull. 1988; 14:217-222.
89. Kendler KS, McGuire M, Gruenberg AM, O’Hare A, Spellman M, Walsh D. The Roscommon Family Study. III. Schizophrenia-related personality disorders in relatives. Arch. Gen. Psychiatry 1993; 50:781-788.
90. Torrey EF, Miller J, Rawlings R, Yolken RH. Seasonality of births in schizophrenia and bipolar disorder: a review of the literature. Schizophr. Res. 1997; 28:1-38.
91. Zornberg GL, Buka SL, Tsuang MT. Hypoxic-ischemia-related fetal/neonatal complications and risk of schizophrenia and other nonaffective psychoses: a 19-year longitudinal study. Am. J. Psychiatry 2000; 157:196-202.
92. Mednick SA, Machon RA, Huttunen MO, Bonett D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch. Gen. Psychiatry 1988; 45:189-192.
93. Gottesman II, Erlenmeyer-Kimling L. Family and twin strategies as a head start in defining prodromes and endophenotypes for hypothetical early-interventions in schizophrenia. Schizophr. Res. 2001; 51:93-102.
94. Harrison PJ, Owen MJ. Genes for schizophrenia? Recent findings and their pathophysiological implications. Lancet 2003; 361:417-419.
95. Carter CJ. Schizophrenia susceptibility genes converge on interlinked pathways related to glutamatergic transmission and long-term potentiation, oxidative stress and oligodendrocyte viability.Schizophr. Res. 2006; 86:1-14.
96. Roffman JL, Weiss AP, Goff DC, Rauch SL, Weinberger DR. Neuroimaging-genetic paradigms: a new approach to investigate the pathophysiology and treatment of cognitive deficits in schizophrenia. Harv. Rev. Psychiatry 2006; 14:78-91.
97. Chen J, Lipska BK, Halim N, Ma QD, Matsumoto M, Melhem S, Kolachana BS, Hyde TM, Herman MM, Apud J, Egan MF, Kleinman JE, Weinberger DR. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am. J. Hum. Genet. 2004; 75:807-821.
98. Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, Goldman D, Weinberger DR: Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc. Natl. Acad. Sci. USA 2001; 98:6917-6922.
99. Carpenter WT, Koenig JI. The evolution of drug development in schizophrenia: past issues and future opportunities. Neuropsychopharmacology 2007.
100. Heidbreder CA, Weiss IC, Domeney AM, Pryce C, Homberg J, Hedou G, Feldon J, Moran MC, Nelson P. Behavioral, neurochemical and endocrinological characterization of the early social isolation syndrome. Neuroscience 2000; 100:749-768.
101. Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J. Neurosci. 2003; 23:297-302.
102. Lee PR, Brady DL, Shapiro RA, Dorsa DM, Koenig JI. Prenatal stress generates deficits in rat social behavior: reversal by oxytocin. Brain Res. 2007; 1156:152-167.
103. Fatemi SH, Emamian ES, Kist D, Sidwell RW, Nakajima K, Akhter P, Shier A, Sheikh S, Bailey K. Defective corticoge- nesis and reduction in Reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol. Psychiatry 1999; 4:145-154.
104. Berretta S, Munno DW, Benes FM. Amygdalar activation alters the hippocampal GABA system: “partial” modelling for postmortem changes in schizophrenia. J. Comp. Neurol. 2001; 431: 129-138.
105. Jentsch JD, Redmond DE, Jr., Elsworth JD, Taylor JR, Youngren KD, Roth RH. Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science 1997; 277:953-955.
106. Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1999; 20:201-225.
107. Jentsch JD, Tran A, Le D, Youngren KD, Roth RH: Subchronic phencyclidine administration reduces mesoprefrontal dopamine use and impairs prefrontal cortical-dependent cognition in the rat. Neuropsychopharmacology 1997; 17:92-99.
108. Morris BJ, Cochran SM, Pratt JA. PCP: from pharmacology to modelling schizophrenia. Curr. Opin. Pharmacol. 2005; 5:101-106.
109. Cochran SM, Kennedy M, McKerchar CE, Steward LJ, Pratt JA, Morris BJ. Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology 2003; 28:265-275.
110. Lindahl JS, Keifer J: Glutamate receptor subunits are altered in forebrain and cerebellum in rats chronically exposed to the NMDA receptor antagonist phencyclidine. Neuropsychopharmacology 2004; 29:2065-2073.
111. Rujescu D, Bender A, Keck M, Hartmann AM, Ohl F, Raeder H, Giegling I, Genius J, McCarley RW, Moller HJ, Grunze H. A pharmacological model for psychosis based on N-methyl-D- aspartate receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol. Psychiatry 2006; 59:721-729.
112. Mouri A, Noda Y, Enomoto T, Nabeshima T. Phencyclidine animal models of schizophrenia: approaches from abnormality of glutamatergic neurotransmission and neurodevelopment. Neurochem. Int. 2007; 51:173-84.
113. Gao XM, Hashimoto T, Tamminga CA. Phencyclidine (PCP) and dizocilpine (MK801) exert time-dependent effects on the expression of immediate early genes in rat brain. Synapse 1998; 29:14-28.
114. Gao XM, Shirakawa O, Du F, Tamminga CA. Delayed regional metabolic actions of phencyclidine. Eur. J. Pharmacol. 1993; 241:7-15.
115. Sams-Dodd F: Effect of novel antipsychotic drugs on phencyclidine-induced stereotyped behaviour and social isolation in the rat social interaction test. Behav. Pharmacol. 1997; 8:196-215.
116. Mansbach RS, Geyer MA: Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology 1989; 2:299-308.
117. Mohn AR, Gainetdinov RR, Caron MG, Roller BH: Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 1999; 98:427-436.
118. Duncan GE, Moy SS, Perez A, Eddy DM, Zinzow WM, Lieberman JA, Snouwaert JN, Roller BH: Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behav. Brain Res. 2004; 153:507-19.
119. Duncan G, Miyamoto S, Gu H, Lieberman J, Roller B, Snouwaert J. Alterations in regional brain metabolism in genetic and pharmacological models of reduced NMDA receptor function. Brain Res. 2002; 951:166-176.
120. Millar JR, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, Clair DM, Muir WJ, Blackwood DH, Porteous DJ. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 2000; 9:1415-1423.
121. Ishizuka R, Paek M, Ramiya A, Sawa A. A review of Disrupted-In-Schizophrenia-1 (DISC1): neurodevelopment, cognition, and mental conditions. Biol. Psychiatry 2006; 59:1189-1197.
122. Hikida T, Jaaro-Peled H, Seshadri S, Oishi R, Hookway C, Rong S, Wu D, Xue R, Andrade M, Tankou S, Mori S, Gallagher M, Ishizuka R, Pletnikov M, Rida S, Sawa A. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc. Natl. Acad. Sci. USA 2007; 104:14501-14506.
123. Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, Mori S, Moran TH, Ross CA. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol. Psychiatry 2008; 13:173-186.
124. Rvajo M, McRellar H, Arguello PA, Drew LJ, Moore H, Mac-Dermott AB, Rarayiorgou M, Gogos JA. A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc. Nat. Acad. Sci. 2008; 105:7076-7081.
125. Li W, Zhou Y, Jentsch JD, Brown RA, Tian X, Ehninger D, Hennah W, Peltonen L, Lonnqvist J, Huttunen MO, Raprio J, Trachtenberg JT, Silva AJ, Cannon TD. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc. Natl. Acad. Sci. USA 2007; 104:18280-18285.
126. Lipska BR, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology 1993; 9:67-75.
127. Hanlon FM, Sutherland RJ. Changes in adult brain and behavior caused by neonatal limbic damage: implications for the etiology of schizophrenia. Behav. Brain Res. 2000; 107:71-83.
128. Sams-Dodd F, Lipska BR, Weinberger DR. Neonatal lesions of the rat ventral hippocampus result in hyperlocomotion and deficits in social behaviour in adulthood. Psychopharmacology (Berl) 1997; 132:303-10.
Further Reading
Andreasen NC. Brave New Brain: Conquering Mental Illness in the Era of the Genome. 2003. Cambridge, MA: Oxford University Press, p 368.
Animal models of schizophrenia, Available: http://www.
schizophreniaforum.org/res/models/default.asp
Benes FM. Searching for unique endophenotypes for schizophrenia and bipolar disorder within neural circuits and their molecular regulatory mechanisms. Schizophr. Bull. 2007; 33:932-936.
Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry 2005; 10:40-68.
Lewis DA, Gonzalez-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008; 33:141-165.
Schizophrenia Forum S, Available: http://www.schizophreniaforum.org/Schizophrenia gene, Available: http://www.schizophreniaforum.org/res/sczgene/default.asp
See Also
Brain Development, Neurochemistry of
Neurotransmitter Release
Synaptic Chemistry