CHEMICAL BIOLOGY
Nucleosides in RNA, Modified
N. Dinuka Abeydeera and Christine S. Chow, Wayne State University, Detroit, Michigan
doi: 10.1002/9780470048672.wecb688
Natural and unnatural modified nucleosides play important roles in chemistry and biology. Over 100 different functional group modifications occur on natural nucleosides, which range from sugar methylations to base isomerization reactions. Unnatural modifications are also broad in their range of possible structures and applications. Both types of modifications have been incorporated site specifically into RNAs to understand their biological roles or to be used as biophysical, structure, and mechanistic probes. More recently, modified RNAs have been applied as therapeutic agents. This article summarizes the approaches toward generation of modified nucleosides and their incorporation into RNA using chemical, biochemical, or combined (semisynthesis) approaches. Some examples of applications with modified RNAs will also be discussed.
Background to Modified Nucleosides in RNA
The expansive array of RNA functions discovered to date is highly dependent on the ability of RNA to fold into unique structures, undergo large conformational changes, or participate in specific interactions with macromolecules (e.g., RNA and proteins), metal ions, and small organic ligands. Although the core nucleotide structures are sufficient for many biological roles of RNAs, over 100 natural modifications in RNA have been discovered over the past six decades, which seem to modulate RNA function (1, 2). In addition, a wide variety of unnatural nucleosides has been synthesized and incorporated into RNA. These modifications often provide RNA with unique properties, such as expanded or altered hydrogen-bonding; van der Waals, base-stacking, or electrostatic interactions; as well as unique metal-binding sites, chemical reactivity, or fluorescent characteristics. The synthetic routes to generate modified nucleosides vary considerably, as do the routes for incorporation of the nucleosides into small oligonucleotide fragments or large, full-length RNAs. The syntheses of modified RNAs serve many purposes, such as developing a better understanding of the biological roles of natural modifications, generating tools for studies of RNA structure and function, and producing novel therapeutic agents.
The types of nucleoside or nucleotide modifications range from alterations of the base component to changes in the sugar or phosphate moieties. The natural modifications include pseudouridylation (generation of a C-glycoside), methylation, deamination, thiolation, and alkylation (1, 2). Hypermodifications also exist in which multiple components of the nucleotide are altered. Some unnatural modifications include fluorescent bases, such as 2-aminopurine, or novel nucleobases to expand the genetic code. This review summarizes some of the most common approaches taken by chemical biologists toward generation of modified nucleosides and their incorporation into RNA using chemical, biochemical, or combined (semisynthesis) approaches. It is possible to use these techniques to construct RNA molecules with a wide variety of functional groups at highly specific positions. Although the applications of modified RNAs are numerous and far reaching, it is not possible to give a comprehensive view in this short article; therefore, representative or recent examples of applications have been selected for discussion.
Synthesis of Modified Nucleosides
The chemical synthesis of novel nucleosides or those that mimic natural modifications will provide biologists with new probes for RNA function. Thousands of modified bases have been described in the literature, which range from synthesis of both natural and unnatural nucleosides to their incorporation into short oligonucleotides and full-length nucleic acids. Numerous different approaches have been taken to generate modified nucleosides (3-6), which clearly cannot all be described in this brief review. Therefore, several examples have been selected to highlight some general strategies for preparing nucleoside analogs. Traditional methods for modified nucleoside synthesis involve either a glycosylation reaction between a purine or pyrimidine base and an activated sugar, or derivatization of a standard nucleoside.
Generation of modified nucleosides—coupling strategies
Most nucleosides contain N-glycosidic linkages (i.e., linkages through the base nitrogen atom). The N -glycosides can be generated by reactions with a suitably protected and activated sugar component and a silylated base (purine or pyrimidine) fragment. The most commonly used reaction in N-glycoside synthesis is the Hilbert-Johnson-type sugar-base condensation in the presence of a Friedel-Crafts catalyst, such as SnCl4, which can be used to produce pyrimidine, purine, as well as sugar- and/or base-modified nucleosides (Fig. 1a) (7, 8). Several improvements to these methods have been made since the original reports, which include the use of microwave-assisted reactions to generate nucleoside libraries (9). Maydanovych et al. (10) have used similar coupling strategies to generate adenosine analogs for the study of RNA editing, and they reported the detailed procedures in a recent review article.
The less-common C-glycosides contain a C-C linkage between the carbohydrate moiety and the heterocyclic base. A wide range of aryl C-glycosides can be generated by using the Heck coupling reaction, as recently reviewed by Wellington and Benner (11). Pseudouridine (Fig. 1b), which is the most abundant natural modification found in RNA, is a C-glycoside generated by posttranscriptional isomerization of uridine residues at specific sites along the oligonucleotide chain. Although numerous groups have reported on the synthesis of pseudouridine, Hanessian, and Machaalani’s stereocontrolled synthesis is the most convenient and efficient route to date (12). Their synthesis involves a reaction between a suitably protected D-ribonolactone and lithiated pyrimidine, followed by sequential ring-opening and ring-closing steps to produce either α or β pseudouridine in high yields. The ratio of α/β anomers is controlled by the presence of L-selectride and zinc chloride.

Figure 1. A general scheme for the synthesis of N- and C-glycosides is shown. a) The preparation of N-glycosidic linkages for pyrimidines and purines is outlined: i) SnCl4, ClCh2CH2Cl, 22-70 °C, 4-5 hours; ii) saturated NaHCO3; iii) NH3 in CH3OH, rt, 16 hours; iv) excess NH3 (7, 8). b) The preparation of a C-glycoside, β pseudouridine, is shown: i) t-BuLi, THF, -78 °C; ii) ZnCl2, L-Selectride, DCM/THF, -78 °C to rt; iii) PPh3, DIAD, THF, 0 °C to rt, 24 hours; iv) 70% AcOH, 50 °C, 5 hours (12).
Nucleoside modifications
An alternative route to modified nucleosides involves derivatization of the standard nucleosides, guanosine, adenosine, cytidine, and uridine. Among the large repertoire of modifications identified to date, the methylated nucleotides are highly abundant and occur frequently in the functionally important regions of RNA (1, 2). Methylation typically occurs on the sugar or base portion of the nucleoside. Although many approaches have been taken to methylate nucleosides, site-selective introduction of a methyl group can be challenging. A recent report by Hobartner et al. (13) showed the synthesis of a wide variety of methylated ribonucleosides, which included 1-methylguanosine (m1G), N2-methylguanosine (m2G), N2N2-dimethylguanosine (m22G), 1-methylinosine (m1I), 3-methyluridine (m3U), N4-methylcytidine (m4C), N6-methyladenosine (m6A), and N6,N6-dimethyladenosine (m62A).
Guanosine methylation at the amido nitrogen to yield m1G can be achieved by treatment of guanosine with NaH in DMSO followed by addition of methyl iodide (Fig. 2a, steps i, ii) (13). Methylation at the N2 position on guanosine to yield m2G and m22G can be achieved following sugar and O6 protection and generation of a 2-fluoro intermediate. The 2-fluoro intermediate is treated with CH3NH2 or (CH3)2NH to yield m2G or m22G, respectively (Fig. 2a, steps iii-vii) (13). Alternatively, m2G can be generated through reduction of a suitably protected p-thiocresol intermediate (Fig. 2a, steps iii, viii-x) (14).
Methylated adenosine analogs can be obtained using inosine as a starting material. Generation of the 6-chloro intermediate allows for conversion into m6A or m62A with CH3NH2 or (CH3)2NH, respectively (Fig. 2b, steps xi, xii, vi, vii) (13).
Methylated cytidine analogs can be obtained from 5-methyluridine (m5U) as the starting material (15). Suitably protected m5U is reacted with P2S5 to yield a 4-thio intermediate that is then converted to 5-methylcytidine (m5C) during treatment with NH3 (Fig. 2c, steps xiii-xv). Uridine can be used as a starting material for the synthesis of N4-methylcytidine (m4C), N4-2'-O-dimethylcytidine (m4Cm) and 2'-O-methylcytidine (Cm) (Fig. 2d, xvi-xx). A tetrazole intermediate is generated (16,17) that can be converted to m4C or m4Cm with the appropriate methylating agent. The 2'-O -methyl group is installed through an anhydronucleoside intermediate and opening with Mg(OCH3)2 in methanol (18).
Methylated uridine analogs can be generated using related methods, such as treatment of uridine with NaH and MeI (Fig. 2d, steps i, ii) (13) to give m3U or conversion to 2’-O-methyluridine (Um) through an anhydronucleoside intermediate and opening by the appropriate nucleophile (18).
Several unnatural nucleoside phosphate modifications (Fig. 2e) have been generated, which include phosphorothioate, phosphoroamidate, phosphorothiolate, and methylphosphonate derivatives. The syntheses of these modifications have been reviewed by Verma and Eckstein (19). A modification of those procedures can be used for the generation of phosphoroselenoate RNAs (20).


Figure 2. Nucleoside base, sugar, and phosphate modifications are shown. The syntheses of methylated guanosine (a), adenosine (b), cytidine and uridine (c and d) nucleosides are highlighted. Reaction conditions: i) NaH, DMSO, rt, 2 h; ii) MeI, rt, 5 h (13); iii) Ac2O, DMAP, Et3N, CH3CN, 0.5 hours; iv) NPE, PPh3, DEAD, dioxane, rt, 2 hours; v) HBF4, NaNO2, acetone/water, -20 °C to rt, 3 hours; vi) 8 M CH3NH2, ethanol, 7-12 hours; vii) (CH3)2NH in ethanol/water, rt, 3-9 hours (13); viii) p-thiocresol, CH3CO2H, HCHO, ethanol, reflux, 4 hours; ix) NaBH4, DMSO, 100 °C, 1 hour; x) NH3 in CH3OH, rt, 16 hours (14); xi) Ac2O, DMAP, pyridine, rt, 16 hours; xii) chloromethylenedimethyliminiumchloride, CHG3, reflux, 4 hours (15); xiii) benzoyl chloride, pyridine, 55 °C, 3 days; xiv) P2S5, pyridine/water, reflux, 4 hours; xv) NH3 in CH3OH (sealed), 100 °C, 24 hours; xvi) tetrazole, TsCl, diphenyl phosphate, pyridine, rt, 36 hours (16, 17); xvii) CH3NH3 + Cl-, KOH, (C2H5)3N, CH3CN/water, rt, 24 hours; xviii) (PhO)2CO, NaHCO3, DMF, 80 °C, 3 hours; xix) Mg(OCH3)2, CH3OH, reflux, 5 hours (18); xx) NH4G, KOH, (C2H5)3N, CH3CN/water, rt, 24 hours. e) Variations of nucleoside phosphate modifications are shown.
Convertible nucleotides
Convertible nucleotides are modified nucleotides that contain reactive functionalities. They are incorporated into RNA at specific locations and allow for convenient postsynthetic introduction of chemical probes, isotope labels, or cross-linking agents, such as disulfide linkages. In this approach, the nucleotide derivative contains a leaving group on its base moiety and is incorporated into the oligonucleotide of interest at a defined location (21). Once the preparation of the desired precursor RNA is completed, the oligonucleotide is treated with an appropriate nucleophile (e.g., amine), which can displace the leaving group and become attached to the specific base moiety (Fig. 3). As will be discussed in more detail later, the convertible nucleosides have many applications, and they can be used to control the RNA structure and function, as well as to regulate RNA conformational switching and dynamics.

Figure 3. The convertible nucleoside approach is shown (21). On the left, a single convertible nucleoside that has been incorporated site-specifically into the RNA is treated with a nucleophile to displace the leaving group, X. On the right, an RNA with multiple convertible nucleosides can be used to generate a cross-linked analog.
Enzymatic approaches to nucleoside triphosphate synthesis
Nucleotides can also be prepared by using enzymatic reactions. Modified nucleoside triphosphates are useful as precursors for the synthesis of RNA through a modified DNA template and T7 RNA polymerase. The chemoenzymatic synthesis of nucleoside triphosphates was recently reviewed by Wu et al. (22). A recent example is the synthesis of 8-azaguanosine (8azaGuo), which is a guanosine analog with N8 in place of C8 that displays fluorescent properties, and the conversion to the corresponding triphosphate (8azaGTP) (23). The strategy for 8azaGTP synthesis involves the use of enzymes from the pentose phosphate and nucleotide salvaging pathways, many of which are commercially available. The presence of the triphosphate allows for the incorporation of 8azaGuo into RNA using a DNA template and T7 RNA polymerase.
Incorporation of Modified Nucleosides into RNA
Through a combination of chemical and biochemical approaches, modified nucleosides can be incorporated into RNA either globally (i.e., at many sites) or in a site-specific manner. Depending on the particular application of interest, either a global or site-specific incorporation strategy may be desirable.
Enzymatic incorporation
Modified nucleotides in RNA can be generated by using site-specific enzymes. Several naturally occurring modifying enzymes have been isolated and used to modify either small oligonucleotides or full-length RNA templates at specific sites (Fig. 4a) (24). The enzyme reactions are useful for understanding the biological roles of the modified nucleosides; however, they are not as useful for generating modified RNAs for biophysical studies or as therapeutic agents because the reactions do not go to completion, and the modified RNAs are often difficult to separate from the unmodified RNAs. RNAs in bacteria and eukaryotes are modified by different pathways. Individual modifications in bacterial RNAs are mediated by one of many unique protein enzymes that are typically site specific and target particular structures or sequences of RNA (24). The most common enzymes are methyltransferases and pseudouridine synthases. Because of the greater number of modifications in RNAs from higher organisms, an alternative pathway of insertion is used that employs fewer site-specific enzymes. Noncoding RNAs known as small nucleolar RNAs (snoRNAs) combine with a set of proteins to generate a small nucleolar ribonucleoprotein (snoRNP) complex that guides the site of enzyme modification on the target RNA (25). The discovery of snoRNAs has been extremely important for understanding the biological roles of RNA modifications, but the use of snoRNPs is not as convenient for generation of site-specifically modified RNAs for biophysical studies.
RNA segments can be modified at their 3' ends by using a bis-phosphate analog of the modified nucleoside and T4 RNA ligase (Fig. 4b) (26). This reaction is general and essentially can be applied to any RNA sequence and modification type that can be converted into a bisphosphate. The 3'-modified RNA segment can then be dephosphorylated with calf intestinal phosphatase and ligated with another 5'-phosphorylated RNA fragment to generate larger RNAs with site-specific modifications (Fig. 4b) (26). Methods for ligation include the use of a DNA “splint” with either T4 RNA ligase or T4 DNA ligase (27, 28). The presence of the complementary DNA fragment promotes formation of the desired RNA product and minimizes dimer or circular RNA formation.
A second enzymatic approach employs T7 RNA polymerase and a DNA template that directs the addition of specific ribonucleotides according to standard Watson-Crick base-pairing rules. This transcription method can be used to generate RNAs of any length and sequence. The T7 RNA polymerase will often accept modified nucleoside triphosphates. The method is useful for global modification, but typically it does not allow for site-specific incorporation of modified nucleotides. This limitation has been overcome in several cases, in which nonstandard nucleotides are used to direct the insertion of modified nucleotides through alternative base-pairing schemes (Fig. 4c) (29).


Figure 4. The site-specific incorporation of modified nucleotides into RNA is depicted. a) Modifying enzymes (E) convert standard nucleotides (N) into modified versions (N-X) (e.g., methylation, pseudouridylation) (24). Eukaryotes require a snoRNP complex along with the modifying enzyme to direct the site of modification, whereas bacteria use unique enzymes to modify each site (25). b) T4 RNA ligase is used to add a single modified nucleotide (X) in the form of a bisphosphate (pXp), or to ligate a modified fragment to another piece of RNA (26, 27). c) Enzymatic incorporation employs T7 RNA polymerase and a modified DNA template, in which deoxynucleotide Y makes a unique base pair with ribonucleotide X (in the form of XTP) to incorporate the modified nucleotide at a single, specific site on the RNA chain (29). d) The steps for incorporation of modified nucleotides by solid-phase oligonucleotide synthesis are shown (32-34). e) The most common protective groups for the phosphoramidite building blocks are highlighted.
Chemical approaches
The chemical synthesis of RNA has the distinct advantage of allowing for site-selective incorporation of modified nucleosides into the oligonucleotide chain. The early phosphodiester method developed by Khorana (30) was followed by the H-phosphonate and phosphoramidite approaches (31). Although the H-phosphonate approach has distinct advantages over phosphoramidite synthesis, it is not as widely used. Therefore, the focus of this section will be on the more commonly employed phosphoramidite method. The chemical synthesis of RNA occurs on a solid support, such as a controlled-pore glass or polystyrene bead (Fig. 4d). The synthesis typically goes in the 3' to 5' direction, building from the 5'-hydroxyl group at each cycle. The 5' OH is protected with a suitable organic moiety that can be removed quickly and efficiently. The 2' hydroxyl is also protected until the end of the synthesis. The monomer unit that reacts with the 5’ OH is a nucleoside with a phosphoramidite moiety at the 3’ hydroxyl. The phosphoramidite method involves four major steps after deprotection of the 5’-hydroxyl group from the 3’ nucleoside attached to the solid support: 1) coupling at the 5' OH with a suitably protected phosphoramidite, 2) capping the unreacted 5'-OH groups, 3) oxidation of the phosphite linkage to generate a phosphate, and 4) removal of the 5’-protecting group of the newly added nucleotide unit (Fig. 4d). The final steps involve removal of the oligonucleotide product from the solid support and deprotection of the 2’-OH groups and phosphate moieties. The final RNA product is then purified using standard methods, such as polyacrylamide gel electrophoresis or high-performance liquid chromatography.
The choice of 5’ and 2’ protecting groups is critical for the successful generation of an oligonucleotide product. The chosen phosphoramidites must have high coupling efficiencies and be stable during the entire oligonucleotide synthesis, yet easily removed at the end of the synthesis. The synthetic route to generate the phosphoramidites should also be efficient in order to reduce the cost of the oligonucleotide synthesis. A wide range of protecting groups has been introduced for RNA synthesis over the past three decades; however, only the most commonly used methods will be discussed. One of the first 2'-OH protecting groups to be employed was tertbutyldimethylsilyl (TBDMS or TBS) (32). More recently, [(triisopropylsilyl)oxy]methyl (TOM) (33) and benzhydryloxy-bis(trimethylsilyloxy)silyl (BzH) (34) have been developed for 2'-OH and 5'-OH protection (Fig. 4e). Whereas each method has distinct advantages and disadvantages, they complement one another because unique chemical modifications on the nucleoside may be more compatible with one particular method. Modifications that are chemically incompatible with these protecting groups would require alternative methods. Several key factors in choosing the type of phosphoramidite to use include: 1) fast coupling times (<2 minutes), 2) high coupling efficiencies (>99.8%), 3) high stability in solution, and 4) the ability to generate long RNA fragments (>20 nucleotides in length). Many recent reviews on RNA synthesis protocols are available that provide more details regarding phosphoramidite preparation (6, 34).
The rationale for choosing chemical synthesis to generate modified RNAs is that a wide range of both natural and unnatural modifications can be incorporated at single or multiple sites, and highly pure RNAs can be produced on a large scale (>mg). As mentioned earlier, convertible nucleotides can be generated in which a group of modified nucleosides that contain reactive functionalities are incorporated into RNA and allow for subsequent attachment of chemical probes, such as isotopic labels, tools for disulfide cross-linking, or fluorescent moieties (21). This postsynthetic modification allows for generation of site-specifically modified RNAs in fewer steps compared with complete enzymatic or chemical synthesis (Fig. 3).
Semisynthesis approaches
The enzymatic and chemical approaches can be easily combined to generate >100 nucleotide-length RNAs that contain either natural or unnatural modifications at specific sites. Either chemically or enzymatically produced fragments can be joined or ligated with T4 DNA ligase or T4 RNA ligase along with a DNA splint, as mentioned above (Fig. 4b) (27, 28). Together, these methods can be applied to generate large, full-length RNA molecules, such as 23S ribosomal RNA (approximately 3000 nucleotides in length), with site-specific modifications to study RNA function (35).
Applications of Modified RNAs
Numerous motivations can be cited for generating site-specifically modified RNAs. The first reason is to use modified RNAs for structure-function studies in either natural or model systems. For this purpose, many different biophysical approaches are employed, such as fluorescence spectroscopy, NMR spectroscopy, and X-ray crystallography. For mechanistic studies, site-specific changes to the oligonucleotide allow for single-atom or functional-group changes and subsequent biochemical studies to be conducted and compared with wild-type RNA. A second major reason for constructing modified RNAs is to use them as therapeutic agents with enhanced stability or improved biological activity. Antisense oligonucleotides were recognized long ago as having potential therapeutic properties for downregulating gene expression. More recently, RNA interference (RNAi) has been discovered as another tool for regulation of mRNA activity. Other therapeutic agents include RNA aptamers and ribozymes. Also of recent interest is the use of modified nucleosides to expand the genetic code, as well as to understand the basic principles of Watson-Crick base pairing and recognition of certain RNA structures by enzymes, proteins, and other biological ligands.
Structure studies
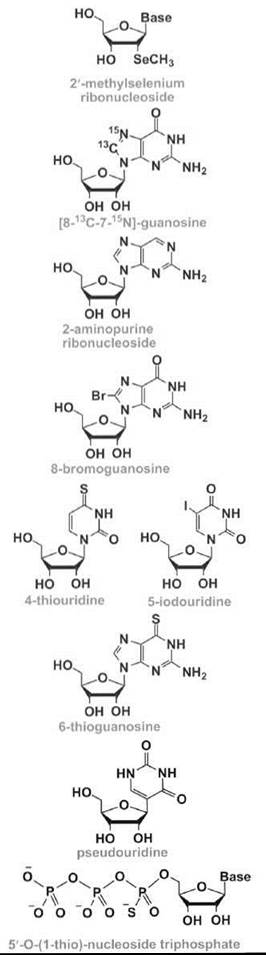
X-ray crystallography is a widely used technique for the three-dimensional structure analysis of RNA molecules. The extraction of information from X-ray diffraction patterns from RNA crystals requires proper phase determinations using methods such as multiwavelength anomalous dispersion in which placement of specific heavy atoms is required. Selenium-modified ribonucleotides have been used for these purposes by Du et al. (36), because the selenium atom does not seem to affect the stability or structure of the RNA. An enzymatic synthesis of large RNAs (up to 100 nucleotides) that contain 2'-methylselenium labels (Table 1) (36-50) was reported by Hobartner et al. (37), which was combined with chemical synthesis using the 2'-O-TOM methodology and a 2'-Semethyladenosine phosphoramidite. More recently, they have used the ACE methodology and modifications of all four standard nucleosides to generate RNAs suitable for X-ray analysis (38). In addition to the sugar modifications, Carrasco et al. (20) have also generated selenophosphate modifications.
Nuclear magnetic resonance (NMR) methods have also been developed for three-dimensional structure analysis of RNA. The major challenge in RNA structure determination by NMR is the size limitation because of spectral overlap with large RNAs. Consequently, heteronuclear NMR has become increasingly important for solving the structures of large RNAs and/or RNA complexes with proteins and other ligands. Although RNAs modified at all or multiple sites with 13C or 15N labels can be generated using enzymatic methods (51, 52), site-specific placement of these labels is often desirable. Numerous approaches have been used in which modified nucleosides with specific 13C and/or 15N labels (see for example, Reference 17) are converted into the corresponding phosphoramidites and used for site-selective incorporation into the desired RNA fragment, which includes convertible nucleotides (21). In one recent example, an RNA hairpin that represents a region of the self-splicing Tetrahymena group I intron was generated with [8-13C-7-15N]-guanosine (Table 1) at one position and [7-15N]-guanosine at another position, such that the two 15N7 NMR signals from the RNA could be unambiguously assigned (39). The specifically labeled sample was used to identify specific metal-ion binding sites that are important for RNA folding and catalysis.
Biophysical probes
A variety of nucleoside modifications can be used to monitor RNA folding, stability, and structure, as well as ligand interactions. Fluorescent analogs, such as 2-aminopurine (2-AP) (Table 1), have proven to be highly useful to monitor RNA conformational changes and dynamics in solution (40-42). The 2-AP modification causes very little perturbation to the RNA structure, and it has been used to monitor drug (e.g., aminoglycoside antibiotic) interactions with target RNA (53). In contrast, attachment of a bulky substituent, such as 8-bromoguanosine (8-BrG) (Table 1), to the RNA base may limit rotation about the glycosidic bond, favoring one conformation (e.g., syn) over another (e.g., anti). Such an approach has been used to stabilize YNMG RNA hairpin motifs and enhance the RNA folding rate, because of preorganization of the denatured state of the RNA (43).
Generation of site-specifically modified RNAs that can perform disulfide cross-linking allows for RNA folding pathways to be examined (21, 54). Photoactivated cross-linking is another strategy that uses naturally occurring or synthetic modifications, such as 4-thiouridine, 6-thioguanosine, 5-bromouridine, and 5-iodouridine (Table 1), to study RNA-RNA interactions in folded RNAs (19).
Modified nucleosides incorporated into small RNA model systems can also be used to investigate the global versus individual effects of modified nucleotides on natural RNAs, such as rRNA or tRNA. For example, in some early studies, Yarian et al. (44) demonstrated that pseudouridine (Table 1) leads to increased thermal stability of the tRNA anticodon stem-loop region. Later, Meroueh et al. (45) demonstrated that pseudouridines have opposing effects on rRNA helix 69 stability, which depends on their specific locations and sequence contexts. These effects on stability may be important for conformational switching mechanisms in functional RNAs (46, 47).
Table 1. Some representative modified nucleosides and nucleotides are shown
|
Modification |
Application |
Reference(s) |
|
|
phasing tool in X-ray crystallography |
36-38 |
|
unambiguous NMR peak assignments during 3D structure analysis |
39 |
|
|
fluorescent probe of RNA conformational changes |
40-42 |
|
|
stabilize nucleoside conformations about the glycosidic bond |
43 |
|
|
photoactivated cross-linking probe of RNA folding to study RNA-RNA interactions |
19 |
|
|
photoactivated cross-linking probe of RNA folding to study RNA-RNA interactions |
19 |
|
|
natural modified nucleoside |
44-47 |
|
|
mechanistic probe of RNA structure and function |
48 |
|
|
|
probe for RNA-mediated catalysis in functional RNAs
|
49 |
|
photolabile moiety for the temporal and spacial control of siRNA targeting
|
50 |
Site-specifically modified RNAs have been used in many applications to examine RNA structure-function relationships, RNA-protein interactions, RNA-ligand interactions, and RNA- catalysis mechanisms. Some earlier studies demonstrated the use of synthetic oligonucleotides to probe the roles of specific functional groups and detailed mechanisms in ribozyme catalysis (55). The synthesis of nucleoside analogs allows for a full-range of chemical diversity (e.g., inductive effects, space-filling capacity, etc.) to be explored, such that quantitative structure activity relationships can be determined for RNA enzymes and other biologically important RNAs (56).
Nucleotide analog interference mapping (NAIM) is an alternative method to conventional single-atom substitution experiments, which provides biochemical information related to the RNA structure and function (48). NAIM has the ability to screen rapidly the effects of chemical group substitution rapidly on RNA function. This method uses an array of modified 5'-O-(1-thio)-nucleoside triphosphates (Table 1) to assess concurrently the contributions of functional groups at every nucleotide position along the RNA backbone. The applications of this method range from mapping of RNA tertiary contacts and substrate contacts in ribozymes to quantification of the energetic contributions of RNA-ligand interactions.
Functional RNAs
RNAs have an enormous range of functions. Those functions have been the subject of numerous publications over the past several decades. One example is RNA aptamers, which are oligonucleotides that have been selected for and tailored to bind with high affinity and selectivity to a variety of ligands, which include nucleotides, peptides, proteins, vitamins, and drugs (57). Aptamers are engineered from random nucleic acid libraries through repeated rounds of in vitro selection. Modified nucleotides play important roles in aptamer function, because they provide enhanced stability to the RNA-based aptamers and can also endow them with desirable chemical reactivities or control of their folded structures, which would be beneficial for the development of novel targeting agents. Several modified aptamers have already been developed as potential therapeutics that are currently in clinical trials (57).
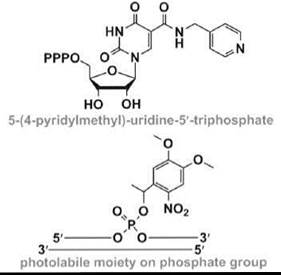
Nucleic acid selection methods have also been exploited for the development of novel RNA enzymes or ribozymes (58). An in-vitro -selected RNA that contains the modified nucleotide 5-(4-pyridylmethyl)-uridine (Table 1) can catalyze carbon-carbon bond formation in a Diels-Alder cycloaddition, with an 800-fold rate acceleration compared with a random RNA (49). Modified RNAs that contain the same uridine modification have also been selected to mediate metal-metal bond formation in the synthesis of palladium nanoparticles (59). Modified RNAs are likely to have many other applications as novel ribozymes that catalyze important biological reactions or can be used to create novel materials.
RNAi/siRNA
Short interfering RNA (siRNA) is a class of molecules composed of double-stranded RNA molecules (20-25 nucleotides) that are involved with the interference of specific genes (i.e., RNA interference or RNAi). During the RNAi process, the siRNA is incorporated into a protein complex, which unwinds the siRNA by an ATP-dependent mechanism to generate an active RNA-induced silencing complex (RISC) (60). The unwound antisense strand of RNA will then guide the RISC to the target mRNA and bind according to standard Watson-Crick base-pairing schemes. Strand scission of the target mRNA is induced by RISC, which leads to a decreased level of protein expression by the corresponding mRNA. The overall success of gene downregulation depends on the stability and structure of the duplex formed between the siRNA and target mRNA. This idea has led to the design and synthesis of numerous modified siRNAs with the anticipation that appropriate changes in the nucleotide composition will lead to enhanced stability, both in terms of chemical stability (i.e., to avoid nuclease degradation) and thermal stability (i.e., to generate a duplex with more favorable free energy, enthalpy, and/or entropy contributions). The modified oligonucleotides are also designed to have increased cellular uptake and properties that will enhance their biodistribution or entry into specific cell types (61, 62). Another type of nucleotide modification involves the addition of a photolabile moiety (Table 1), such that temporal and spatial control of siRNA targeting is possible (50).
Other applications
RNAs that employ the four standard bases have more limited opportunities for hydrogen-bonding interactions and functionality, even though the range of tertiary interactions is broad (63). The use of additional base pairs would not only increase the structural complexity of RNA but would also allow for enzymatic replication. This concept has been exploited by several research groups to expand the genetic code, as well as to generate novel, site-specifically modified RNAs (e.g., biotinylated, fluorescent, cross-linked RNAs) (6, 29, 64).
Summary and Outlook
This article highlights methods for synthesis of modified nucleosides and their site-specific incorporation into RNA. RNAs of practically any size can now be engineered with a broad range of natural or unnatural modifications to the base, sugar, or phosphate portions of the nucleotides. Using chemical, biochemical, or combined approaches, a change of a single atom at a desired location can be accomplished, as well as global modification. Because of the diversity of modifications accessible through chemical or enzymatic synthesis, the modified RNAs also have a tremendous range of applications, some of which have been summarized in the last portion of this review. Future efforts will continue to give researchers a better understanding of the importance of natural modified RNAs, provide new tools for biophysical and structure analysis for structure-function studies of RNA, and lead to novel RNA therapeutics or catalysts. On a more practical note, novel methodologies for cost-effective, large-scale preparation of highly pure modified RNAs will also be of importance (65), and improved catalysts for RNA ligation (66) will allow for larger, biologically relevant RNAs to be generated with greater ease.
We are grateful for support from the National Institutes of Health (Grants GM054632 and AI061192).
References
1. Limbach PA, Crain PF, McCloskey JA. Summary: the modified nucleosides of RNA. Nucleic Acids Res. 1994; 22:2183-2196.
2. Rozenski J, Crain PF, McCloskey JA. The RNA modification database: 1999 update. Nucleic Acids Res. 1999; 27:196-197.
3. Beaucage SL, Iyer RP. The synthesis of modified oligonucleotides by the phosphoramidite approach and their applications. Tetrahedron 1993; 49:6123-6194.
4. Eaton BE, Pieken WA. Ribonucleosides and RNA. Annu. Rev. Biochem. 1995; 64:837-863.
5. Gallo M, Montserrat JM, Iribarren AM. Design and applications of modified oligonucleotides. Braz. J. Med. Biol. Res. 2003; 36:143-151.
6. Cobb AJA. Recent highlights in modified oligonucleotide chemistry. Org. Biomol. Chem. 2007; 5:3260-3275.
7. Vorbruggen H, Bennua B. A new simplified nucleoside synthesis. Chem. Ber. 1981; 114:1279-1286.
8. Lichtenthaler FW, Voss P, Heerd A. Stannic chloride catalysed glycosidations of silylated purines with fully acylated sugars. Tetrahedron Lett. 1974; 15:2141-2144.
9. Bookser BC, Raffaele NB. High-throughput five minute microwave accelerated glycosylation approach to the synthesis of nucleoside libraries. J. Org. Chem. 2007; 72:173-179.
10. Maydanovych O, Easterwood LM, Cui T, Veliz EA, Pokharel S, Beal PA. Probing adenosine-to-inosine editing reactions using RNA-containing nucleoside analogs. Methods Enzymol. 2007; 424: 369-386.
11. Wellington KW, Benner SA. A review: synthesis of aryl C-glycosides via the Heck coupling reaction. Nucleosides Nucleotides Nucleic Acids 2006; 25:1309-1333.
12. Hanessian S, Machaalani R. A highly stereocontrolled and efficient synthesis of α- and β-pseudouridines. Tetrahedron Lett. 2003; 44:8321-8323.
13. Hobartner C, Kreutz C, Flecker E, Ottenschlager E, Pils W, Grubmayr K, Micura R. The synthesis of 2'-O-[(triisopropylsilyl)oxy] methyl (TOM) phosphoramidites of methylated ribonucleosides (m1G, m2G, m22G, m1I, m3U, m4C, m6A, m62A) for use in automated RNA solid-phase synthesis. Monatshefte fur Chemie 2003; 134:851-873.
14. Bridson PK, Reese CB. A novel method for the methylation of heterocyclic amino groups. Conversion of guanosine into its 2-N-methyl and 2-N,2-N-dimethyl derivatives. Bioorg. Chem. 1979; 8:339-349.
15. Fox JJ, Van Praag D, Wempen I, Doerr IL, Cheong L, Knoll JE, Eidinoff ML, Bendich A, Brown GB. Thiation of nucleosides. II. Synthesis of 5-methyl-2'-deoxycytidine and related pyrimidine nucleosides. J. Am. Chem. Soc. 1959; 81:178-187.
16. Reese CB, Ubasawa A. Nature of side-reactions in oligonucleotide synthesis involving arenesulphonyl derivatives of 3-nitro-1,2,4- triazole and related condensing agents. Nucleic Acids Symp. Ser. 1980; (7): 5-21.
17. Ariza X, Vilarrasa J. High-yielding preparation of [3-15N]cytidine, [4-15NH2]cytidine, and [3-15N,4-15NH2]cytidine. J. Org. Chem. 2000; 65:2827-2829.
18. Roy SK, Tang J-y. Efficient large scale synthesis of 2'-O-alkyl pyrimidine ribonucleosides. Org. Process Res. Dev. 2000; 4:170-171.
19. Verma S, Eckstein F. Modified oligonucleotides: synthesis and strategy for users. Ann. Rev. Biochem. 1998; 67:99-134.
20. Carrasco N, Caton-Williams J, Brandt G, Wang S, Huang Z. Efficient enzymatic synthesis of phosphoroselenoate RNA by using adenosine 5'-(α-β-seleno)triphosphate. Angew. Chem. Int. Ed. Engl. 2006; 45:94-97.
21. Allerson CR, Verdine GL. Synthesis and biochemical evaluation of RNA containing an intrahelical disulfide crosslink. Chem. Biol. 1995; 2:667-675.
22. Wu W, Bergstrom DE, Jo Davisson V. Chemoenzymatic preparation of nucleoside triphosphates. Curr. Protoc. Nucleic Acid Chem. 2004; Chapter 13: Unit 13.2.
23. Da Costa CP, Fedor MJ, Scott LG. 8-Azaguanine reporter of purine ionization states in structured RNAs. J. Am. Chem. Soc. 2007; 129:3426-3432.
24. Ferre-D’Amare AR. RNA-modifying enzymes. Curr. Opin. Struct. Biol. 2003; 13:49-55.
25. Ye K. H/ACA guide RNAs, proteins and complexes. Curr. Op. Struct. Biol. 2007; 17:287-292.
26. England TE, Uhlenbeck OC. Enzymatic oligoribonucleotide synthesis with T4 RNA ligase. Biochemistry 1978; 17:2069-2076.
27. Moore MJ, Sharp PA. Site-specific modification of pre-mRNA: the 2'-hydroxyl groups at the splice sites. Science 1992; 256:992-997.
28. Stark MR, Pleiss JA, Deras M, Scaringe SA, Rader SD. An RNA ligase-mediated method for the efficient creation of large, synthetic RNAs. RNA 2006; 12:2014-2019.
29. Hirao I, Kimoto M, Mitsui T, Fujiwara T, Kawai R, Sato A, Harada Y, Yokoyama S. An unnatural hydrophobic base pair system: site-specific incorporation of nucleotide analogs into DNA and RNA. Nat. Methods 2006; 3:729-735.
30. Khorana HG. Nucleic acid synthesis. Pure Appl. Chem. 1968; 17:349-381.
31. Gait MJ. An introduction to modern methods of DNA synthesis. In: Oligonucleotide Synthesis: A Practical Approach. Gait MJ, ed. 1984. Washington, D.C.: IRL Press Ltd.
32. Ogilvie KK, Theriault N, Sadana KL. Synthesis of oligoribonucleotides. J. Am. Chem. Soc. 1977; 99:7741-7743.
33. Pitsch S, Weiss PA, Jenny L, Stutz A, Wu X. Reliable chemical synthesis of oligoribonucleotides (RNA) with 2'-O-[(triisopropylsilyl)oxy]methyl(2'-O-tom)-protected phosphoramidites. Helv. Chim. Acta 2001; 84:3773-3795.
34. Scaringe SA. Advanced 5'-silyl-2'-orthoester approach to RNA oligonucleotide synthesis. Methods Enzymol. 2000; 317:3-18.
35. Erlacher MD, Lang K, Shankaran N, Wotzel B, Huttenhofer A, Micura R, Mankin AS, Polacek N. Chemical engineering of the peptidyl transferase center reveals an important role of the 2'-hydroxyl group of A2451. Nucleic Acids Res. 2005; 33:1618-1627.
36. Du Q, Carrasco N, Teplova M, Wilds CJ, Egli M, Huang Z. Internal derivatization of oligonucleotides with selenium for X-ray crystallography using MAD. J. Am. Chem. Soc. 2002; 124:24-25.
37. Hobartner C, Rieder R, Kreutz C, Puffer B, Lang K, Polonskaia A, Serganov A, Micura R. Syntheses of RNAs with up to 100 nucleotides containing site-specific 2’-methylseleno labels for use in x-ray crystallography. J. Am. Chem. Soc. 2005; 127:12035-12045.
38. Puffer B, Moroder H, Aigner M, Micura R. 2’-Methylselenomodified oligoribonucleotides for x-ray crystallography synthesized by the ACE RNA solid-phase approach. Nucleic Acids Res. 2008; 36:970-983.
39. Fan Y, Gaffney BL, Jones RA. RNA GG●UU motif binds K+ but not Mg2+. J. Am. Chem. Soc. 2005; 127:17588-17589.
40. Ward DC, Reich E, Stryer L. Fluorescence studies of nucleotides and polynucleotides. J. Biol. Chem. 1969; 244:1228-1237.
41. Millar DP. Fluorescence studies of DNA and RNA structure and dynamics. Curr. Op. Struct. Biol. 1996; 6:322-326.
42. Ballin JD, Bharill S, Fialcowitz-White EJ, Gryczynski I, Gryczynski Z, Wilson GM. Site-specific variation in RNA folding thermodynamics visualized by 2-aminopurine fluorescence. Biochemistry 2007; 46:13948-13960.
43. Proctor DJ, Ma H, Kierzek E, Kierzek R, Gruebele M, Bevilacqua PC. Folding thermodynamics and kinetics of YNMG RNA hairpins: specific incorporation of 8-bromoguanosine leads to stabilization by enhancement of the folding rate. Biochemistry 2004; 43:14004-14014.
44. Yarian CS, Basti MM, Cain RJ, Ansari G, Guenther RH, Sochacka E, Czerwinska G, Malkiewicz A, Agris PF. Structural and functional roles of the N1- and N3-protons of ψ at tRNA’s position 39. Nucleic Acids Res. 1999; 27:3543-3549.
45. Meroueh M, Grohar PJ, Qiu J, SantaLucia J Jr, Scaringe SA, Chow CS. Unique structural and stabilizing roles for the individual pseudouridine residues in the 1920 region of Escherichia coli 23S rRNA. Nucleic Acids Res. 2000; 28:2075-2083.
46. Newby MI, Greenbaum NL. A conserved pseudouridine modification in eukaryotic U2 snRNA induces a change in branch-site architecture. RNA 2001; 7:833-845.
47. Abeysirigunawardena SC, Chow CS. pH-dependent structural changes of helix 69 from Escherichia coli 23S ribosomal RNA. RNA; 2008; 14:782-792.
48. Ryder SP, Strobel SA. Nucleotide analog interference mapping. Methods 1999; 18:38-50.
49. Tarasow TM, Tarasow SL, Eaton BE. RNA-catalysed carbon-carbon bond formation. Nature 1997; 389:54-57.
50. Shah S, Rangarajan S, Friedman SH. Light-activated RNA interference. Angew. Chem. Int. Ed. Engl. 2005; 44:1328-1332.
51. Batey RT, Inada M, Kujawinski E, Puglisi JD, Williamson JR. Preparation of isotopically labeled ribonucleotides for multidimensional NMR spectroscopy of RNA. Nucleic Acids Res. 1992; 20:4515-4523.
52. Nikonowicz EP, Sirr A, Legault P, Jucker FM, Baer LM, Pardi A. Preparation of 13C and 15N labelled RNAs for heteronuclear multi-dimensional NMR studies. Nucleic Acids Res. 1992; 20:4507-4513.
53. Shandrick S, Zhao Q, Han Q, Ayida BK, Takahashi M, Winters GC, Simonsen KB, Vourloumis D, Hermann T. Monitoring molecular recognition of the ribosomal decoding site. Angew. Chem. Int. Ed. Engl. 2004; 43:3177-3182.
54. Sigurdsson ST, Tuschl T, Eckstein F. Probing RNA tertiary structure: interhelical crosslinking of the hammerhead ribozyme. RNA 1995; 1:575-583.
55. Earnshaw DJ, Gait MJ. Modified oligoribonucleotides as site-specific probes of RNA structure and function. Biopolymers 1998; 48:39-55.
56. Gordon PM, Fong R, Deb SK, Li N-S, Schwans JP, Ye J-D, Piccirilli JA. New strategies for exploring RNA’s 2'-OH expose the importance of solvent during group II intron catalysis. Chem. Biol. 2004; 11:237-246.
57. Lee JF, Stovall GM, Ellington AD. Aptamer therapeutics advance. Curr. Opin. Chem. Biol. 2006; 10:282-289.
58. Joyce GF. Forty years of in vitro evolution. Angew. Chem. Int. Ed. Engl. 2007; 46:6420-6436.
59. Gugliotti LA, Feldheim DL, Eaton BE. RNA-mediated metal-metal bond formation in the synthesis of hexagonal palladium nanoparticles. Science 2004; 304:850-852.
60. Fire A, Xu SQ, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998; 391:806-811.
61. Manoharan M. RNA interference and chemically modified small interfering RNAs. Curr. Opin. Chem. Biol. 2004; 8:570-579.
62. Beaucage SL. Solid-phase synthesis of siRNA oligonucleotides. Curr. Opin. Drug Discov. Devel. 2008; 11:203-216.
63. Hendrix DK, Brenner SE, Holbrook SR. RNA structural motifs: building blocks of a modular biomolecule. Q. Rev. Biophys. 2005; 38:221-243.
64. Piccirilli JA, Krauch T, Moroney SE, Benner SA. Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature 1990; 343:33-37.
65. Donga RA, Hassler M, Chan T-K, Damha MJ. Oligonucleotide synthesis using ionic liquids as soluble supports. Nucleosides Nucleotides Nucleic Acids 2007; 26:1287-1293.
66. Purtha WE, Coppins RL, Smalley MK, Silverman SK. General deoxyribozyme-catalyzed synthesis of native 3'-5' RNA linkages. J. Am. Chem. Soc. 2005; 127:13124-13125.
See Also
Fluorescence to Study Nucleic Acids
NMR Tools for Nucleic Acids
RNA Interference, Mechanisms and Proteins Involved in