CHEMICAL BIOLOGY
Cofactor Biosynthesis
Ilka Haase and Markus Fischer Institute of Food Chemistry, University of Hamburg, Hamburg, Germany
Adelbert Bacher, Wolfgang Eisenreich and Felix Rohdich
Institute of Biochemistry, Department Chemie, Technische Universitat MUnchen, MUnchen, Germany
doi: 10.1002/9780470048672.wecb094
Whereas plants and certain microorganisms can generate all required coenzymes from CO2 or simple organic precursors, animals must obtain precursors (designated as vitamins) for a major fraction of their coenzymes from nutritional sources. Still, most vitamins must be converted into the actual coenzymes by reactions catalyzed by animal enzymes. The structures and biosynthetic pathways of some coenzymes are characterized by extraordinary complexity. Enzymes for coenzyme biosynthesis have frequently low catalytic rates, and some of them catalyze reactions with highly unusual mechanisms.
Many coenzymes (cofactors) involved in human and animal metabolism were discovered in the first half of the twentieth century, and their isolation and structure elucidation were hailed as milestones as shown by the impressive number of Nobel prizes awarded for research in that area. Studies on coenzyme biosynthesis were typically initiated in the second half of the twentieth century and have generated a massive body of literature that continues to grow rapidly because the area still involves many incompletely resolved problems. In parallel, numerous novel coenzymes were discovered relatively recently by studies of microorganisms. In this article, the terms “cofactor” and “coenzyme” are used as synonyms.
Cofactor biosynthesis is a very broad and multifaceted topic. This article summarizes basic concepts of major cofactors. Detailed status reports on the biosynthesis of individual coenzymes can be found in excellent recent reviews that are quoted (1).
General Aspects
Biosynthetic origin of coenzymes in autotrophic and heterotrophic organisms
Plants, algae, and certain microorganisms can generate all their organic components, including their entire coenzyme repertoire, from CO2. Certain microorganisms can also generate their entire biomass from single organic nutrients, such as carbohydrates or carboxylic acids. Animals, on the other hand, depend on plants and bacteria for the supply of many coenzymes or coenzyme precursors (designated vitamins) but synthesize others de novo from basic precursors derived from central intermediary metabolism (Table 1). The distribution of coenzyme biosynthetic pathways in eubacteria and fungi is very complex; whereas certain species are auxotrophic with regard to coenzymes, the range is from complete self-sufficiency to virtually complete dependence on exogenous coenzymes or coenzyme precursors. However, even organisms that depend on exogenous vitamins must convert those into the actual coenzymes by more or less complex enzymatic transformations.
Table 1. Origin of coenzymes in humans
|
Coenzyme |
Partial synthesis from |
De novo synthesis from |
Nutritional |
|
FMN, FAD |
Riboflavin (Vitamin B2), ATP |
|
|
|
Tetrahydrofolate Tetrahydrobiopterin |
Folate, Dihydrofolate |
GTP |
|
|
Molybdopterin Thiamine pyrophosphate |
Thiamine (Vitamin B1) |
GTP |
|
|
Pyridoxal phosphate |
Pyridoxine, Pyridoxal (Vitamin B6) |
|
|
|
Coenzyme B12 |
Vitamin B12, ATP |
|
|
|
Coenzyme A, Pantetheine Porphyrins |
Pantothenate, ATP |
Succinyl-CoA, Glycine |
|
|
Fe/S Clusters |
|
Cysteine |
|
|
Coenzyme Q10 (ubiquinone) Vitamin K (menaquinone) |
|
Acetyl-CoA, Tyrosine |
Nutritional |
|
Vitamin C NADH, NADPH |
Nicotinate, Nicotinamide, Nicotinamide riboside, ATP |
|
Nutritional |
|
|
|
|
|
|
Biotin |
|
|
Nutritional |
|
Retinal |
Carotenoids |
|
Nutritional |
Basic precursors for coenzyme biosynthesis
Precursors for cofactor biosynthesis are drawn from all major pools of central intermediary metabolism, including carbohydrates, amino acids, purine nucleotides, and carboxylic acids.
Convergent biosynthetic pathways
Numerous cofactors are generated via convergent pathways starting from two or more different precursors (e.g., the biosynthesis of vitamins B1, B2, and tetrahydrofolate). Occasionally, a single precursor is sufficient for biosynthesis, for example, in case of vitamin C and tetrahydrobiopterin.
Alternative pathways exist for several coenzymes
Whereas the biosyntheses of some coenzymes (e.g., tetrahy-drobiopterin) seem to proceed via unique pathways, several different pathways have been described for the biosynthesis of others. In certain cases, the different pathways are variations on a common theme; for example, in the biosynthesis of vitamin B2, a sequence of a deamination and reduction can proceed in different order. In other cases, entirely different reactions afford a given intermediate, for example in the case of nicotinic acid biosynthesis. Another example is the biosynthesis of isoprenoid building blocks and their downstream products, in which the existence of a second pathway besides the classic mevalonate pathway had been ignored until recently. Notably, higher plants use both isoprenoid pathways but for different final products.
Some coenzymes develop by very complex pathways and/or reactions
Whereas tetrahydrobiopterin is biosynthesized from GTP via just three enzyme-catalyzed steps (2), some coenzyme biosynthetic pathways are characterized by enormous complexity. Thus, the biosynthesis of vitamin B12 requires five enzymes for the biosynthesis of the precursor uroporhyrinogen III (16) from succinyl-CoA (10) and glycine (11) that is then converted into vitamin B12 via the sequential action of about 20 enzymes (3). Additional enzymes are involved in the synthesis of the building blocks aminopropanol and dimethylbenzimidazole (4, 5). Vitamin B12 from nutritional sources must then be converted to coenzyme B12 by mammalian enzymes. Ultimately, however, coenzyme B12 is used in humans by only two enzymes, albeit of vital importance, which are involved in fatty acid and amino acid metabolism (6). Notably, because plants do not generate corrinoids, animals depend on bacteria for their supply of vitamin B12 (which may be obtained in recycled form via nutrients such as milk and meat) (7).
However, the sheer complexity of a pathway is not an indicator for the vitamin status of a given class of compounds, as opposed to endogenous biosynthesis in mammals. Thus, animals biosynthesize molybdopterin, which is a cofactor involved in certain redox reactions, from basic building blocks using at least eight enzymes, whereas folic acid has vitamin status and must be obtained by animals from nutritional sources (8).
It should also be noted that some pathways to be discussed below depend on highly unusual chemical reactions. To give just one example, the formation of the pyridine ring system of vitamin B6 depends on a protein that catalyzes a complex series of reactions, including carbohydrate isomerization, imine formation, ammonia addition, aldol-type condensation, cycliza- tion, and aromatization (9).
Specific Biosynthetic Pathways
Biosynthesis of iron/sulfur clusters
Iron/sulfur clusters are inorganic cofactors that are used in all cells (10). They comprise S2- ions and iron ions in the +2 or +3 state (Fig. 1). Iron/sulfur clusters are essential cofactors for numerous redox and nonredox enzymes, alone or in tandem with organic cofactors such as flavocoenzymes and/or pyridine nucleotides. The simplest structural type is the rhombic [2Fe-2S] cluster (3). [3Fe-4S] (4) and [4Fe-4S] (5) clusters are characterized by distorted cubic symmetry (10, 11). Clusters can form aggregates, and other metal ions can replace iron ions or can be present additionally.
Whereas many cognate apoenzymes can be reconstituted with iron/sulfur clusters by simple and essentially alchemistic procedures using Fe2+ and sulfide ions under anaerobic conditions, a highly complex enzymatic machinery is used in vivo for the synthesis of iron/sulfur clusters and their transfer to the target enzymes. Sulfide ions required for cluster synthesis are obtained from cysteine (1) via a persulfide of a protein-bound cysteine residue (2); pyridoxal phosphate is required for the formation of the persulfide intermediate (Fig. 1) (12).
In eukaryotes, the formation of iron/sulfur clusters proceeds inside mitochondria (13). The mitochondrial enzymes are orthologs of the eubacterial isc proteins and are characterized by very slow rates of evolution. Iron/sulfur clusters are initially assembled on IscU protein (prokaryotic) or Isu protein (eukaryotic) that serves as a scaffold. They can be exported to the cytoplasm in which they can become part of cytoplasmic enzymes by the assistance of proteins that serve as iron chaperones.
The persulfide intermediate 2 can also serve as a sulfur source for the biosynthesis of thiamine (6), lipoic acid (7), molybdopterin (8), and biotin (9) (Fig. 1).
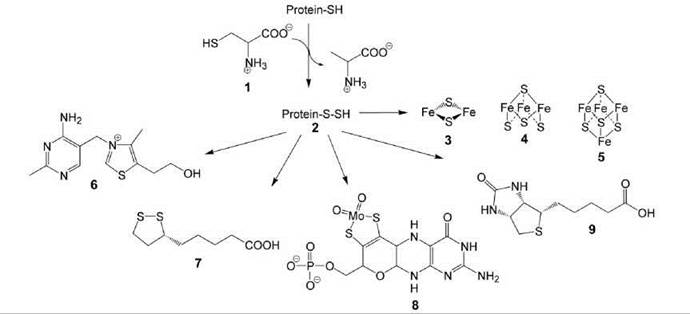
Figure 1. Formation of protein-bound persulfide and its delivery to sulphur-containing natural compounds.1, cysteine; 2, persulfide of a protein bound cysteine; 3, rhombic [2Fe-2S] cluster; 4, [3Fe-4S] cluster; 5, [4Fe-4S] cluster; 6, thiamine; 7, lipoic acid; 8, molybtopterin; 9, biotin.
Tetrapyrroles
A large and structurally complex family of coenzymes, including various hemes, chlorophylls, corrinoids (including coenzyme B12) and the archaeal coenzyme F430 is characterized by their macrocyclic tetrapyrrole structure (14, 15). These coenzymes contain a metal ion (Fe, Mg, Co, or Ni) at the center of the tetrapyrrole macrocycle, which is specifically introduced by enzyme catalysis. These compounds are all derived from 8-aminolevulinic acid (12) that can be biosynthesized by two independent pathways, that is, from glycine (11) and succinyl-CoA (10) in animals and some bacteria (e.g., Rhodobacter) or from glutamyl-tRNA (13) in plants, many eubacteria, and archaea (Fig. 2) (16, 17). Two molecules of 8-aminolevulinic acid (12) are condensed under formation of porphobilinogen (14). Oligomerization of porphobilinogen affords hydroxymethylbilane (15), in which all pyrrole rings share the same orientation of their substituents. Ring D is then inverted by a rearrangement that affords uroporphyrinogen III (16) (3). Side-chain modification and the incorporation of iron by ferrochelatase (18) afford the various heme cofactors, including heme a (18), that carries an isoprenoid side chain. Starting from 16, a sequence of partial reduction, side-chain modification and incorporation of Mg2+ affords chlorophyll (17), which also carries an isoprenoid side chain. Partial reduction, ring contraction, and incorporation of Co2+ afford vitamin B12 (20) as well as several analogs that are found in archaea. Moreover, archaea incorporate nickel into the corrinoid coenzyme F430 (19) that plays a central role in the biosynthesis of methane (19).
Because plants are devoid of vitamin B12, the supply of humans and animals is ultimately of bacterial origin (although humans can obtain vitamin B12 via animal products such as meat, milk, and milk products). A glycoprotein designated intrinsic factor that is secreted by the human gastric mucosa enables the take-up of the vitamin, which is then converted into coenzyme B12 by a series of enzyme reactions that occur in the human host (7).
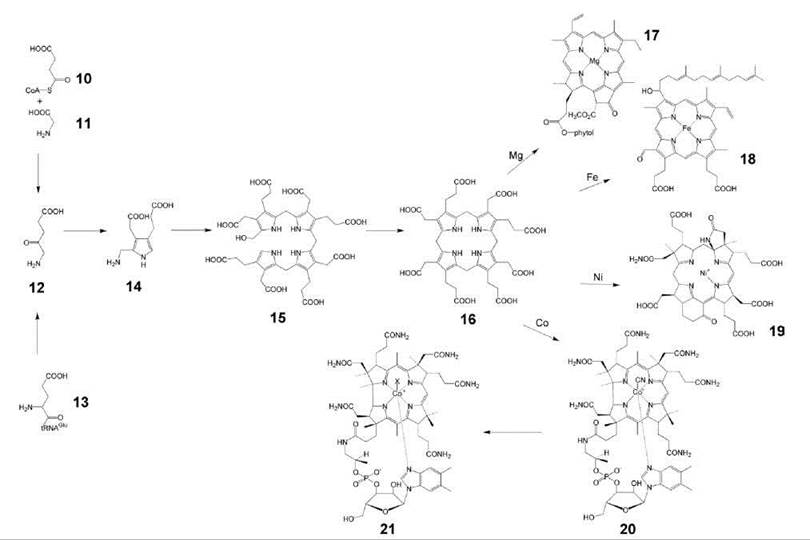
Figure 2. Biosynthesis of tetrapyrroles. 10, succinyl-CoA; 11, glycine; 12, 8-aminolevulinic acid; 13, glutamyl-tRNA; 14, porphobilinogen; 15, hydroxymethylbilane; 16, uroporphyrinogen III; 17, chlorophyll a; 18, heme a; 19, coenzyme F430; 20, vitamin B12; 21, coenzyme B12.
A family of coenzymes biosynthesized from GTP: tetrahydrofolate, tetrahydrobiopterin, flavocoenzymes, molybdopterin
Several coenzymes comprising a pyrimidine ring motif are derived from GTP (22) (Fig. 3). Specifically, this group comprises two members of the B vitamin group, riboflavin (vitamin B2) (24) and folic acid/tetrahydrofolate (33). Two other members of the group, tetrahydrobiopterin (31) and molybdopterin (8), are biosynthesized de novo in animals and do not have vitamin status (20, 21).
The first committed step in the biosyntheses of these compounds is the hydrolytic opening of the imidazole ring of GTP, which affords a diaminopyrimidine-type intermediate. In the biosynthetic pathways of folate, tetrahydrobiopterin, and methanopterin (34), the respective diaminopyrimidine intermediate undergoes ring closure by means of an intramolecular condensation that involves parts of the ribose side chain of GTP, which affords a 2-amino-4-pteridinone compound (29).
The 4-aminobenzoate moiety of tetrahydrofolic acid is obtained from the shikimate pathway of aromatic amino acid biosynthesis via chorismate. Interestingly, apicomplexan protozoa may have conserved the complex shikimate pathway for the single purpose to generate 4-aminobenzoate as a tetrahydrofolate precursor, whereas aromatic amino acids are obtained from external sources.
The formation of intermediate 27 (compound Z) in the biosynthesis of molybtopterin (8) proceeds a rearrangement that involves the ribose side chain as well as C-8 of GTP for the formation of the tetracyclic ring system.
The carbocyclic moiety of vitamin B2 is assembled from two molecules of a deoxytetrulose phosphate (22). The carbocyclic moiety of the deazaflavin-type coenzymes (36) is provided by the tyrosine precursor, 4-hydroxyphenylpyruvate.
Notably, the GTP cyclohydrolases that catalyze the first committed step in the pathways of tetrahydrofolate biosynthesis in plants and microorganisms and of tetrahydrobiopterin in animals are orthologs.
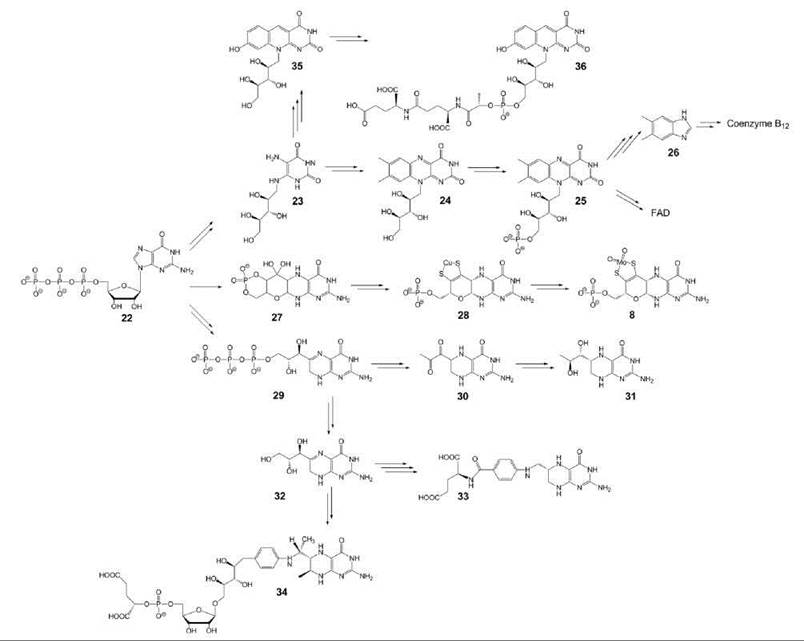
Figure 3. Coenzymes biosynthesized from GTP. 8, molybdopterin; 22, GTP; 23, 5-amino-6-ribitylamino-2,4(1 H,3H)-pyrimidinedione; 24, riboflavin; 25, FMN; 26, 5,6-dimethylbenzimidazole; 27, precursor Z; 28, metal containing pterin; 29, dihydroneopterin triphosphate; 30, 6-pyruvoyl-tetrahydropterin; 31, 6(R)-5,6,7,8-tetrahydrobiopterin; 32, dihydroneopterin; 33, 6(S)-5,6,7,8-tetrahydrofolate; 34, 5,6,7,8-tetrahydromethanopterin; 35, 5-deaza-7,8-didemethyl-8-hydroxyribo-flavin; 36, coenzyme F420.
Thiamine pyrophosphate
The biosynthesis of thiamine pyrophosphate (46) in microorganisms and plants is characterized by extraordinary complexity (21, 23). Animals are dependent on nutritional sources but can convert unphosphorylated thiamine (that is not an intermediate of the bacterial biosynthetic pathway) into thiamine pyrophosphate in two steps.
In bacteria, the pyrimidine precursor 38 is derived from 5-aminoimidazole ribotide (37), an intermediate of the basic branch of purine biosynthesis, which supplies all carbon atoms for 38 by a complex rearrangement reaction (the fate of the individual carbon atoms is indicated by Greek letters in Fig. 4). In yeasts, a totally unrelated reaction sequence uses carbon atoms from vitamin B6 (39) that are indicated by roman letters in Fig. 4 for the assembly of the thiamine precursor 38, whereas the nitrogen atoms and one additional carbon atom are introduced from histidine (40).
In bacteria, the thiazole moiety (42) of thiamine is derived from 1-deoxy-D-xylulose 5-phosphate (43) that can also serve as a precursor for pyridoxal in many eubacteria (Fig. 5) and for isoprenoids via the nonmevalonate pathway (cf. isoprenoid cofactors). The sulfur atom is derived from the persulfide that also serves as precursor for iron/sulfur clusters and for biotin (6) and thiooctanoate (7) (Fig. 1). C2 and N3 of the thiazole moiety of thiamine have been reported to stem from tyrosine in Escherichia coli and from glycine in Bacillus subtilis, respectively. Yeasts use ADP-ribulose (44) derived from NAD as precursor (24).
In plants, little is known about the basic building blocks and the reactions involved in thiamine biosynthesis. An early study with chloroplasts of spinach indicated that 1-deoxy-D-xylulose 5-phosphate, tyrosine, and cysteine act as precursors of the thiazole moiety in analogy to the pathway in E. coli. More recently, it has been shown that a homolog of the THIC protein that converts 5-aminoimidazole ribotide into 38 is essential (25). These results suggest that the plant pathway is similar to the pathway in prokaryotes but not to that in yeast.
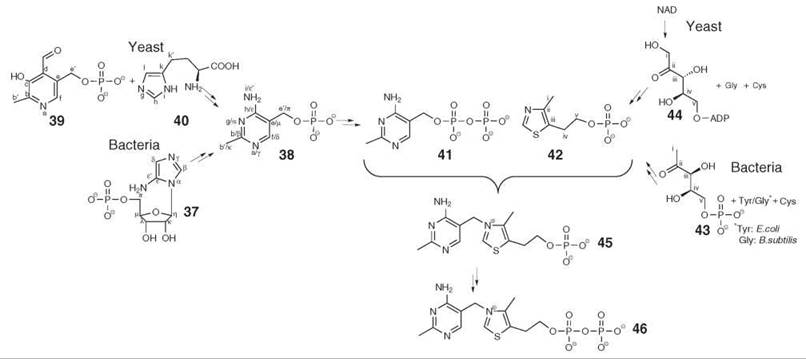
Figure 4. Biosynthesis of thiamine (vitamin B1). 37, aminoimidazole ribotide; 38, 2-methyl-4-amino-5-hydroxymethyl-pyrimidine phosphate; 39, pyridoxal 5’-phosphate; 40, histidine; 41, 2-methyl-4-amino-5-hydroxymethyl-pyrimidine pyrophosphate; 42, 4-methyl-5-β-hydroxyethylthiazole phosphate; 43, 1-deoxy-D-xylulose 5-phosphate; 44, 5-ADP-D-ribulose; 45, thiamine phosphate; 46, thiamine pyrophosphate.
Pyridoxal phosphate
In many eubacteria, 1-deoxy-D-xylulose phosphate (43) serves as a common precursor for the biosynthesis of vitamins B1 and B6 and for the biosynthesis of isoprenoids via the nonmeval-onate pathway. Condensation of 43 with 3-amino-1-hydroxy-acetone phosphate (47) (biosynthesized from D-erythrose 4-phosphate) affords pyridoxine 5'-phosphate (48, Fig. 5A). A sequence of elimination, tautomerization, and water addition precedes cyclization via an aldol condensation (26, 27). Then, pyridoxine 5'-phosphate can be converted into pyridoxal 5'-phosphate (39) by oxidation with molecular oxygen.
A more recently discovered second pathway starts from ribulose 5-phosphate (49) that is condensed with ammonia and glyceraldehyde phosphate or its isomerization product dihydroxy-acetone phosphate (50), which affords pyridoxal 5'-phosphate in a single enzyme-catalyzed reaction step (Fig. 5B) (28, 29). This pathway seems to be widely distributed; it is used in plants (30) and has also been shown to proceed in fungi, archaea, and most eubacteria.
In mammals, dietary vitamin forms, including pyridoxal, pyridoxol, and pyridoxamine, can all be converted to the respective coenzyme forms by phosphorylation.
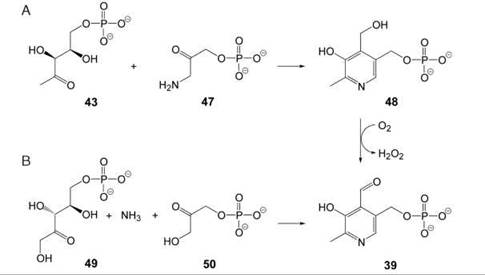
Figure 5. Formation of the pyridoxine ring in vitamin B6. (A) deoxyxylulose phosphate-dependent pathway; (B) deoxyxylulose phosphate-independent pathway. 43, 1-deoxy-D-xylulose 5-phosphate; 47, 3-amino-1-hydroxyacetone 1-phosphate; 48, pyridoxine 5’-phosphate; 49, ribulose 5-phosphate; 50, dihydroxyacetone phosphate; 39, pyridoxal 5'-phosphate.
Pyridine nucleotides
Animals and yeasts can synthesize nicotinamide from tryptophan via hydroxyanthranilic acid (52) and quinolinic acid (53, Fig. 6A) (31), but the biosynthetic capacity of humans is limited. On a diet that is low in tryptophan, the combined contributions of endogenous synthesis and nutritional supply of precursors, such as nicotinic acid, nicotinamide, and nicotinamide riboside, may be insufficient, which results in cutaneous manifestation of niacin deficiency under the clinical picture of pellagra. Exogenous supply of nicotinamide riboside was shown to promote NAD+-dependent Sir2-function and to extend life-span in yeast without calorie restriction (32).
Bacteria and plants use aspartate (54) and dihydroxyacetone phosphate (50) as precursors for the biosynthesis of nicotinamide via quinolinic acid (53, Fig. 6B) (33).
The transformation of precursors into NAD (56) and NADP (57) follow the same pathway in all organisms. A ribosyl phosphate residue can be transferred to biosynthetic quinolinic acid or to preformed nicotinamide affording 55 (or its amide) that is converted to NAD by adenylation. Subsequent phosphorylation yields NADP.
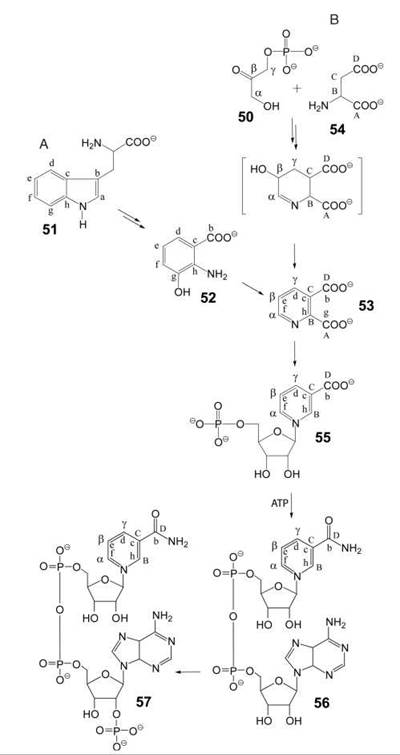
Figure 6. Biosynthesis of pyridine nucleotides. A) in animals and yeasts; B) in plants and bacteria. 50, dihydroxyacetone phosphate; 51 tryptophan; 52, hydroxyanthranilic acid; 53, quinolinic acid; 54, aspartate; 55, nicotinic acid mononucleotide; 56, NAD, 57, NADP.
Pantothenate
Pantothenate (also designated vitamin B5, 64, Fig. 7) is biosynthesized de novo in plants and many microorganisms but must be obtained from nutritional sources by animals (20, 34). The branched carboxylic acid 63 is obtained from α-ketoisovalerate (61), which is an intermediate of valine biosynthesis, via 62. β-Alanine (60) is obtained by decarboxylation of aspartate (54) in microorganisms. Plants and yeasts can biosynthesize β-alanine from spermine (58) (35). An additional pathway to 60 starting from uracil (59) has been reported in plants, where the downstream steps that lead to pantothenate are understood incompletely.
Organisms of all biological kingdoms convert 64 into the cysteamine derivative phosphopantetheine (65) using L-cysteine as substrate. 65 is converted to coenzyme A (66) by attachment of an adenosine moiety via a pyrophosphate linker and phosphorylation of the ribose moiety. Phosphopantetheine can be attached covalently to serine residues of acyl carrier proteins that are parts of fatty acid synthases and polyketide synthases.
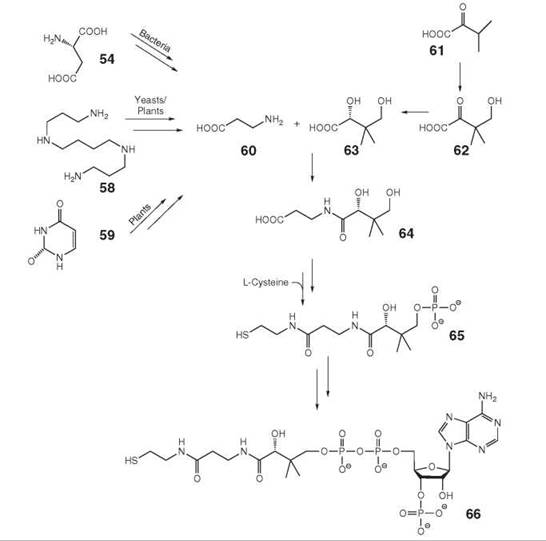
Figure 7. Biosynthesis of pantothenate. 54, aspartate; 58, spermine; 59, uracil; 60, β-alanine; 61, α-ketoisovalerate; 62, ketopantoate; 63, pantoate; 64, pantothenate; 65, pantetheine 4-phosphate; 66, coenzyme A.
Vitamin C
Whereas most mammals can synthesize ascorbic acid (vitamin C, 75) from D-glucose 1-phosphate (67) via the pathway shown in Fig. 8B, humans and guinea pigs lack the last enzyme of that pathway and are therefore dependent on nutritional sources (36, 37). Plants use the pathway shown in Fig. 8A that has been elucidated relatively recently (38, 39). Yeasts produce and use a five-carbon analog, which is called erythroascorbic acid, instead of ascorbate. The biosynthetic pathway of erythroascorbic acid involves the oxidation of D-arabinose to D-arabino-1,4-lactone, which is then oxidized to erythroascorbic acid.
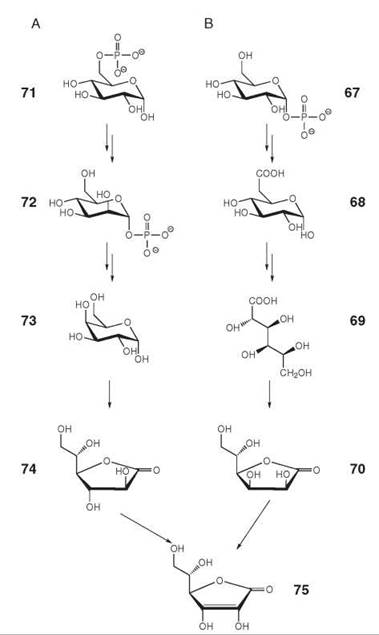
Figure 8. Biosynthesis of ascorbic acid (vitamin C). (A) in plants; (B) in mammals. 67, d-glucose 1-phosphate; 68, d-glucuronate; 69, l-gulonate; 70, l-gulono-1,4-lactone; 71, d-glucose 6-phosphate; 72, d-mannose 1-phosphate; 73, l-galactose; 74, l-galactono-1,4-lactone; 75, ascorbic acid. The enzyme converting 70 into 75 is missing in primates and guinea pigs.
Pyrroloquinoline quinone
Pyrroloquinoline quinone (77, Fig. 9; PQQ) serves as cofactor of bacterial oxidoreductases (40). The heated debate whether PQQ has vitamin character for animals is still inconclusive (41-43).
PQQ is derived from a peptide precursor that contains conserved glutamate (13) and tyrosine (76) residues. All carbon and nitrogen atoms of the precursor amino acids are incorporated into the product (44). Gene clusters involved in this pathway have been studied in considerable detail. The X-ray structure of the enzyme that catalyzes the final reaction step has been determined, and reaction mechanisms have been proposed on that basis (45). However, details of the biosynthetic pathway are still incompletely understood.
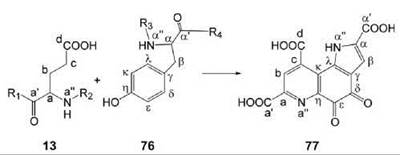
Figure 9. Biosynthesis of PQQ. 13, glutamate (protein bound); 76, tyrosine (protein bound); 77, PQQ.
Biotin
Biotin (vitamin H, 6, Figs. 1 and 10) acts a cofactor of carboxylases. It can be produced in bacteria, plants, and some fungi (46). The biosynthetic pathway involves four steps that start from alanine (78) and pimeoyl-CoA (79). Carboxylation and cyclization of 81 affords dethiobiotin (82), which is then converted into biotin (6) by the iron/sulfur protein, biotin synthase, in an unusual radical mechanism (47).
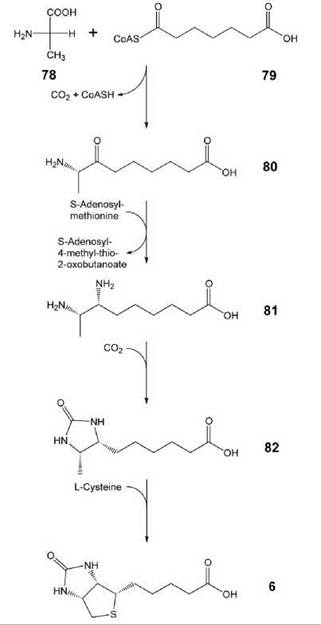
Figure 10. Biosynthesis of biotin. 78, alanine; 79, pimeloyl-CoA; 80, 7-keto-8-amino-pelargonic acid; 81, 7,8-diamino-pelargonic acid; 82, dethiobiotin; 6, biotin.
Isoprenoid cofactors
Isoprenoids are one of the largest classes of natural products that comprise at least 35,000 reported members (48). Many of these compounds play crucial roles in human metabolism as hormones, vitamins (vitamins A, D, E and K), quinine-type cofactors of respiratory chain enzymes (ubiquinone), membrane constituents, and functionally important side chains of signal cascade proteins (Fig. 11). Chlorophyll (17, Fig. 2) and heme a (18) have isoprenoid side chains.
Whereas vitamin E (99) and vitamin A or its biosynthetic precursor, |3-carotene, must be obtained by animals from dietary sources, many other isoprenoids, including the quinone type coenzyme Q family (where individual representatives differ by the length of their side chains), can be synthesized de novo by vertebrates. 3-Hydroxy-3-methylglutaryl-CoA reductase, the enzyme that catalyzes the conversion of (S )-3-hydroxy-3-methylglutaryl-CoA (84) to mevalonate (85), is one of the most important drug targets for the prevention of cardiovascular disease (see below) (49, 50).
All isoprenoids are biosynthesized from two isomeric 5- carbon compounds, isopentenyl diphosphate (IPP, 86) and dimethylallyl diphosphate (DMAPP, 87) (Fig. 11). The mammalian pathway for the biosynthesis of these key biosynthetic precursors from three acetyl-CoA units (83) via mevalonate (85) had been elucidated in the 1950s (51). In the wake of that pioneering work, it became established dogma that all terpenoids are invariably of mevalonate origin, even in the face of significant aberrant findings.
The existence of a second pathway that affords IPP and DMAPP was discovered in the 1990s. The details of that non- mevalonate pathway were then established in rapid sequence by the combination of isotope studies, comparative genomics, and enzymology (52). The delayed discovery of the nonmevalonate pathway can serve as paradigm for pitfalls in the elucidation of biosynthetic pathways.
The mevalonate pathway starts with a sequence of two Claisen condensations that afford (S)-3-hydroxy-3-methylglutaryl-CoA (84) from three acetyl-CoA moieties. The pathway affords IPP that can be converted into DMAPP by isomerization. The first committed intermediate of the nonmevalonate pathway is 2C-methyl-D-erythritol 4-phosphate (90) obtained from 1-deoxy-D-xylulose 5-phosphate (43), which is a compound also involved in the biosynthesis of vitamins B1 (46, cf. Fig. 4) and B6 (39, cf. Fig. 5), by rearrangement and subsequent reduction. Three enzyme-catalyzed steps are required to convert the compound into the cognate cyclic diphosphate 91 that is then converted reductively into a mixture of IPP and DMAPP by the consecutive action of two iron/sulfur proteins.
It is now firmly established that green plants use both isoprenoid biosynthesis pathways (52). More specifically, sterols and triterpenes are generated in the cytoplasm via the mevalonate pathway, whereas monoterpenes and diterpenes are generated in plastids via the nonmevalonate pathway. These assignments are not absolute because there is a level of crosstalk between the compartments.
Oligomerization of isoprenoids under elimination of pyrophosphate affords the precursors for the biosynthesis of monoterpenes, sesquiterpenes, diterpenes, triterpenes, and tetraterpenes (93). Long-chain oligomer pyrophosphates also supply the side chains of vitamin E (99, α-tocopherol, Fig. 11), heme a (18), chlorophyll (17, Fig. 2), and the quinone type cofactors, including vitamin K (menaquinone, 98) and coenzyme Q10 (ubiquinone, 97). The quinone moieties are derived from hydroxybenzoate that is synthesized from tyrosine in animals or from chorismate in microorganisms (53, 54).
The tetraterpene, β-carotene (95), is biosynthesized in microorganisms and in the chloroplasts of higher plants where it serves as an important component of the light-harvesting apparatus. In plants, the isoprenoid precursor units of carotenoids are predominantly of nonmevalonate origin. In vertebrates, β-carotene serves as provitamin. A that can be converted into the vitamin retinol (96) by oxidative cleavage. Whereas vitamin A functions as a component of retinal photoreceptors and in signal transmission in vertebrates, it is also involved in bacterial photosynthesis, in which it serves as prosthetic group in a light-driven proton pump. The halobacterial proton pump and the human photoreceptor proteins are structurally related 7-helix membrane proteins.
Vitamin D exerts its numerous effects via the binding to a receptor protein that serves as a transcription factor. It is included in this review in light of its essential status for human health.
Humans can generate vitamin D3 (cholecalciferol, 94, Fig. 11) by endogenous biosynthesis but require dietary sources under certain environmental conditions. More specifically, 94 that can be obtained via the endogenous mevalonate pathway can be photochemically converted into provitamin D in light-exposed skin areas. The transformation requires ultraviolet light. At higher geographical latitudes, light exposure of the skin can be a limiting factor. The fact that skin pigmentation retards the photochemical formation of vitamin D may have acted as a selective factor that favored pale skin when modern humans migrated to areas with higher geographic latitude.
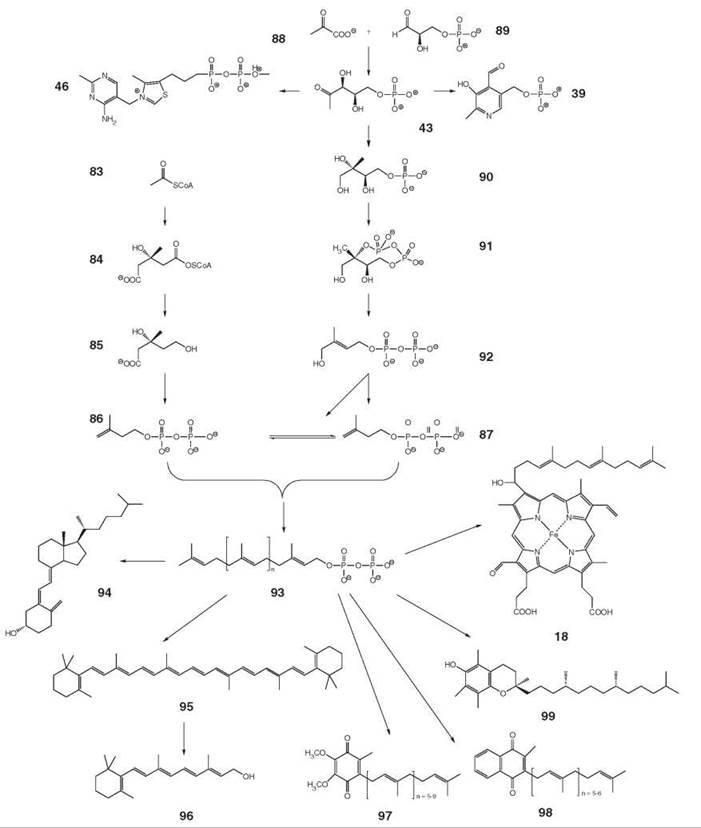
Figure 11. Biosynthesis of isoprenoid type cofactors. 18, Heme a; 39, pyridoxal 5’-phosphate; 43, 1-deoxy-D-xylulose 5-phosphate; 46, thiamine pyrophosphate; 83, acetyl-CoA; 84, (S)-3-hydroxy-3-methylglutaryl-CoA; 85, mevalonate; 86, isopentenyl diphosphate (IPP); 87, dimethylallyl diphosphate (DMAPP); 88, pyruvate; 89, D-glyceraldehyde 3-phosphate; 90, 2C-methyl-D-erythritol 4-phosphate; 91, 2C-methyl-erythritol 2,4-cyclodiphosphate; 92, 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate; 93, polyprenyl diphosphate; 94, cholecalciferol; 95, β-carotene; 96, retinol; 97, ubiquinone; 98, menaquinone; 99, α-tocopherol.
Biosynthesis of archaeal coenzymes
Archaebacteria that were discovered only during recent decades are now recognized as a third kingdom of life besides eubacteria and eukaryotes. An important subgroup of archaea can generate energy by conversion of carbon dioxide or low molecular weight organic acids into methane. The pathway of methanogenesis has been shown to implicate several unique coenzymes (55). Specifically, 5-deaza-8-hydroxy-10-ribitylisoalloxazine (factor F0, 35), which is an analog of riboflavin, serves as the business end of coenzyme F420 (36, Fig. 3), whose designation is based on its characteristic absorption maximum at 420 nm. Factor F0 is biosynthesized from the pyrimidine type intermediate 23 of the riboflavin biosynthetic pathway, which affords the pyrimidine ring and the ribityl side chain, whereas the carbocyclic moiety is derived from the shikimate pathway via 4-hydroxyphenylpyruvate (56, 57). In contrast to the coenzymes described below, deazaflavin-type coenzymes are not strictly limited to methanogenic bacteria and are also found in streptomycetes and mycobacteria.
The tetrapyrrole-type coenzyme F430 (19) was named on basis of its absorption maximum at 430 nm. The nickel-chelating factor is biosynthesized via the porphyrin biosynthetic pathway (Fig. 2) (19). For the handling of one-carbon fragments that play a central role in their metabolism, methanogenic bacteria use methanopterin (34, Fig. 3). The tetrahydropterine system that serves as the business end of the methanopterin coenzyme family is structurally similar to tetrahydrofolate, and the biosynthetic pathway starting from GTP is similar to that of tetrahydrofolate (Fig. 3). The ribitylaniline moiety is derived from ribose and from the shikimate pathway via 4-aminobenzoate (55).
Coenzymes as biosynthetic precursors
Some coenzymes serve as biosynthetic precursors that afford structural parts of other coenzymes. Thus, the benzenoid moiety of the flavocoenzyme FMN serves as a precursor for the lower ligand 26 of the central cobalt ion in vitamin B12 (20) (Fig. 3) (5). Pyridoxal and NAD are used as precursors for the biosynthesis of thiamine in yeast (Fig. 4) (23, 24).
Branched coenzyme biosynthesis pathways
Branching of pathways is relevant in several cases. Thus, intermediates of the porphyrin biosynthetic pathway serve as precursors for chlorophyll (17, Fig. 2) and for the corrinoid ring systems of vitamin B12 (20, Fig. 2) (17). 1-Deoxy-D-xylulose 5-phosphate (43) serves as an intermediate for the biosynthesis of pyridoxal 5'-phosphate (39, Fig. 5), for the terpenoid precursor IPP (86) via the nonmevalonate pathway (Fig. 11), and for the thiazole moiety of thiamine pyrophosphate (46, Fig. 4). 7,8-Dihydroneopterin triphosphate (29, Fig. 3) serves as intermediate in the biosynthetic pathways of tetrahydrofolate (33) and tetrahydrobiopterin (31). The closely related compound 7,8-dihydroneopterin 2',3'-cyclic phosphate is the precursor of the archaeal cofactor, tetrahydromethanopterin (34) (58). A common pyrimidine-type intermediate (23) serves as precursor for flavin and deazaflavin coenzymes. Various sulfur-containing coenzymes (thiamine (9), lipoic acid (7), biotin (6), Fig. 1) use a pyrosulfide protein precursor that is also used for the biosynthesis of inorganic sulfide as a precursor for iron/sulfur clusters (12).
Requirement of coenzymes for their own biosynthesis
Several coenzymes are involved in the biosynthesis of their own precursors. Thus, thiamine is the cofactor of the enzyme that converts 1-deoxy-D-xylulose 5-phosphate (43) (the precursor of thiamine pyrophosphate, pyridoxal 5'-phosphate and of isoprenoids via the nonmevalonate pathway) into 2 C-methyl-D-erythritol 4-phosphate (90, Fig. 11). Similarly, two enzymes required for the biosynthesis of GTP, which is the precursor of tetrahydrofolate, require tetrahydrofolate derivatives as cofactors (Fig. 3). When a given coenzyme is involved in its own biosynthesis, we are faced with a “hen and egg” problem, namely how the biosynthesis could have evolved in the absence of the crucially required final product. The answers to that question must remain speculative. The final product may have been formed via an alternative biosynthetic pathway that has been abandoned in later phases of evolution or that may persist in certain organisms but remains to be discovered. Alternatively, the coenzyme under study may have been accessible by a prebiotic sequence of spontaneous reactions. An interesting example in this respect is the biosynthesis of flavin coenzymes, in which several reaction steps can proceed without enzyme catalysis despite their mechanistic complexity.
In terms of coenzyme evolution, it is also noteworthy that the biosynthesis of a given coenzyme frequently requires the cooperation of other coenzymes. For example, the biosynthesis of riboflavin (24) requires tetrahydrofolate (33) for the biosynthesis of GTP serving as precursor (Fig. 3). Pyridoxal 5'-phosphate is required for the biosynthesis of the activated pyrosulfide type protein (2) that serves as the common precursor for iron/sulfur clusters and various sulfur-containing organic coenzymes (Fig. 1).
Covalent coenzymes
Whereas many coenzymes form noncovalent complexes with their respective apoenzymes, various flavoenzymes are characterized by covalently bound FMN (25) or FAD (Fig. 3). Covalent linkage involves the position 8a methyl group or the benzenoid carbon atom 6 of the flavin and a cysteine or histidine residue of the protein. The covalent CN or CS bond can be formed by autoxidation of the noncovalent apoenzyme/coenzyme precursor complex as shown in detail for nicotine oxidase (59).
Biotin (6, Fig. 10) and lipoic acid (7, Fig. 1) are attached enzymatically to apoenzymes via carboxamide linkage to specific lysine residues (60, 61). The pantothenyl moiety (64, Fig. 7) can also be linked covalently to proteins via amide linkage (62). Covalently bound heme is involved in heme M (63) and heme L-catalyzed reactions (64, 65).
Several covalently bound coenzymes, including pyruvoyl, methylidine imidazolone, topaquinone, and tryptophan tryptophyl quinine-type prosthetic groups are generated by posttranslational modification (66).
Cellular topology of coenzyme biosynthesis in eukaryotes
In bacteria, coenzyme biosynthesis is located in the cytoplasm. In eukaryotic cells, organelles play important roles in the biosynthesis of certain cofactors. For example, certain steps of iron/sulfur cluster biosynthesis proceed in mitochondria (10, 13). In plants, some steps of the biosynthesis of tetrahydrofolate, biotin, and lipoate proceed in mitochondria (7, 15), whereas the biosynthesis of vitamin B2 is operative in plastids (67). In apicomplexan protozoa, enzymes in mitochondria, in the apicoplast (an organelle that is believed to have a common evolutionary origin with chloroplasts) and in the cytoplasmic compartment must cooperate for the biosynthesis of tetrapyr- role cofactors, thiamine, and isoprenoid cofactors (68, 69). The biosynthesis of carotenoids proceeds via the nonmevalonate pathway in the chloroplasts of plants (52). The same holds true for the biosynthesis of thiamine, pyridoxal, and chlorophyll (70).
Three-dimensional structures of coenzyme biosynthesis enzymes
The rapid technological progress in X-ray crystallography has enabled the structural analysis of numerous enzymes involved in coenzyme biosynthesis. Complete sets of structures that cover all enzymes of a given pathway are available in certain cases such as riboflavin, tetrahydrobiopterin, and folic acid biosynthesis. Structures of orthologs from different taxonomic groups have been reported in certain cases. X-ray structures of enzymes in complex with substrates, products, and analogs of substrates, products, or intermediates have been essential for the elucidation of the reaction mechanisms. Structures of some coenzyme biosynthesis enzymes have been obtained by NMR-structure analysis.
Enzymes that are addressed by major drugs have been studied in particular detail. Thus, well above one hundred structures have been reported for dihydrofolate reductases from a variety of organisms, including major pathogens such as Mycobacterium tuberculosis, which is the causative agent of tuberculosis, and of Plasmodium falciparum, which is the most important of the Plasmodium spp. that causes malaria. The interaction of mammalian dihydrofolate reductases with inhibitors that are used as cytostatic agents and/or immunosuppressants is also documented extensively by X-ray structures.
The rapidly growing number of three-dimensional coenzyme biosynthesis enzyme structures in the public domain and the cognate publications are best addressed via the internet server of Brookhaven Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). Queries can be targeted to individual enzymes or to entire pathways.
Medical Aspects
Genetic deficiency of coenzyme biosynthesis
Genetic defects have been reported for the biosynthesis of several coenzymes in humans. Typically, these rare anomalies cause severe neurological deficits that become apparent at birth or in early childhood.
Specifically, the deficiency of certain enzymes of tetrahydrobiopterin biosynthesis (GTP cyclohydrolase I, pyruvoyltetrahydrobiopterin synthase, Fig. 3) result in severe neurological and developmental deficits designated as atypical phenylketonuria caused by the ensuing deficiency in catecholamine type neurotransmitter biosynthesis. The condition can be treated with some success by the oral application of synthetic tetrahydrobiopterin in large amounts. More recently, tetrahydrobiopterin therapy has also been advocated for certain patients with classic phenylketonuria that results from mutations of phenylalanine hydroxylase (71). This therapeutic approach is based on the concept that the function of certain defective phenylalanine hydroxylases can be bolstered by increased amounts of the cognate coenzyme tetrahydrobiopterin. In fact, the relatively large number of patients with classic phenylketonuria may provide an economic incentive for the development of a biotechnological process for the bulk production of the coenzyme.
Deficiencies of enzymes involved in the transformation of the vitamin pantothenic acid (64) into the cognate coenzyme forms (66, Fig. 7) result in severe developmental and neurological deficits that affect few human patients (34, 72). Therapy with megadoses of pantothenic acid has been advocated, but their efficiency has yet to be demonstrated by stringent clinical studies (73).
Genetic defects of molybdopterin biosynthesis (Fig. 3) also result in severe neurologic and developmental deficits (74). Genetic defects in the biosynthesis of the quinine-type coenzyme Q10 (97, Fig. 11) can result in encephalopathy, myopathy, and renal disease (53).
Inherited defects or porphyrine biosynthetic enzymes can cause the accumulation of pathway intermediates that cannot be converted anymore with sufficient velocity. Various genetic forms of porphyria have been reported and result in liver toxicity, neurological damage and photosensitivity (75). Acquired forms of porphyria can be caused by a variety of toxic and pharmacologic agents.
The absorption of vitamin B12 (20) requires a glycoprotein-designated intrinsic factor that is secreted by the gastric mucosa (7, 76). The factor binds the vitamin and enables its subsequent transport across the ileal mucosa. Acquired failure to produce the intrinsic factor results in a complex disease that can present with hematological (macrocytotic anemia and pernicious anemia), neurological, or psychiatric symptoms or a combination thereof. Prior to the discovery of vitamin B12, pernicious anemia was lethal. Initial treatment was based on the consumption of large amounts of uncooked liver. If diagnosed timely, the disease can now be cured easily by the parenteral administration of vitamin B12. Notably, the liver can store large amounts of the vitamin.
Coenzyme biosynthesis enzymes as anti-infective drug targets
Enzymes involved in coenzyme biosynthesis represent targets for anti-infective agents (77). The sulfonamides that were discovered in the 1930s were the first group of synthetic agents with a broad spectrum of activity against pathogenic bacteria and protozoa. Their mode of action, via inhibition of dihy- dropteroate synthetase in the biosynthetic pathway of tetrahydrofolate biosynthesis (Fig. 3), was elucidated only much later. Subsequent studies on compounds with antifolate activity afforded inhibitors of dihydrofolate reductase, which is the enzyme in that pathway that catalyzes the formation of tetrahydrofolate from dihydrofolate in organisms that synthesize the coenzyme de novo and from folate in organisms that rely on dietary sources. Trimethoprim, which is an inhibitor of dihydrofolate reductase (that is required for the use of nutritional folate and dihydrofolate as well as for the metabolic recycling of tetrahydrofolate coenzymes) became a widely used antimicrobial agent that is typically applied in combination with a sulfonamide.
Fosmidomycin, initially discovered as a product of Streptomyces lavendulae with antibacterial and herbicide activity, was recently shown to act via the inhibition of IspC protein that catalyzes the first committed step in the nonmevalonate pathway of isoprenoid biosynthesis that is absent in humans (43 → 90, Fig. 11). Based on these findings, the compound is now under clinical evaluation as an antimalarial drug (78).
In principle, other coenzyme biosynthetic pathways that occur in pathogenic bacteria but not in humans should qualify as anti-infective drug targets with a favorable toxicity profile. Novel anti-infective principles would be highly desirable in light of the rapid spread of resistant pathogens.
Coenzyme biosynthesis as a target for cytostatic agents
The development of the sulfonamides as antibacterial and antiprotozoan agents had preceded the discovery of its metabolic target in the biosynthesis of tetrahydrofolate (that was per se unknown in the 1930s) (Fig. 3). The discovery of the vitamin in the 1940s triggered a wave of research directed at additional inhibitors of its biosynthesis. This work resulted in the discovery of methotrexate that is widely used as a cytostatic agent predominantly for hematological malignancy, and also as an immunosuppressive agent used in the therapy of autoimmune disease such as Crohn’s disease (79).
Drug interaction with coenzyme biosynthesis pathways
Mevastatin is an inhibitor of 3-hydroxy-3-methylglutaryl-CoA reductase (Fig. 11, 84 → 85) that was isolated from Penicillium citrinum in 1971. A group of structurally related compounds designated as statins is now widely used for the prevention and treatment of cardiovascular disease (80). Statins exert their desired effects via the lowering of low-density lipoprotein and probably also via reduced prenylation of small G-proteins of the Ras protein family that are involved in proinflammatory signaling. The highly pleiotrophic mevalonate pathway is the source of numerous other highly important metabolites including coenzyme Q10 (97). However, a recent review concludes that the suppression of coenzyme Q10 biosynthesis, which can be expected as a side result of statin therapy, is not a significant cause of rhabdomyolysis, a dreaded side effect of statin therapy (81).
Modulation of vitamin D biosynthesis by environmental factors
Fair-skinned humans face a dilemma because ultraviolet light exposure carries the risks of carcinogenesis and skin aging, whereas insufficient ultraviolet light carries the risk of vitamin D3 (94, Fig. 11) insufficiency. The case is unique in so far as an endogenous biosynthetic pathway is subject to regulation by external lifestyle factors such as ultraviolet exposure and the use of chemical and physical sunscreens. Recent studies indicate that vitamin D deficiency is wide spread in the human population and is a risk factor for a wide variety of conditions, including cancer and autoimmune disease (82). The dilemma of ultraviolet protection and vitamin D sufficiency can be addressed easily by vitamin D supplements.
Biotechnology
Harnessing biosynthetic pathways for vitamin production
Certain vitamins that serve as coenzymes (vitamin C) or as precursors of coenzymes (all B group vitamins and certain carotenoids) are commercially produced in bulk amounts, which are used for human nutrition and animal husbandry, as antioxidants (vitamins C and E) and food colorants (vitamin B2, carotenoids). Only a fraction of technically manufactured vitamins is used for inclusion in drugs.
Vitamin B12 is produced exclusively by bacterial fermentation technology. The chemical synthesis of vitamin B2 has been superseded during the past two decades by fermentation processes using bacteria and yeasts. Carotene, various other carotenoids and vitamin A are produced by chemical synthesis, but a variety of biotechnological processes have been also been explored for their production. Notably, vitamin A deficiency continues to be a major cause for acquired blindness in developmental countries, although vitamin A and P-carotene, which serves a provitamin, can be produced at modest cost and in virtually unlimited quantity by existing technology. At least certain steps of the various technical vitamin C processes are also conducted by fermentation. For various other vitamins, biotechnological production may become competitive in the future.
References
1. Leeper FJ, Smith AG. Editorial: vitamins and cofactors - chemistry, biochemistry and biology. Nat. Prod. Rep. 2007; 24:923-926.
2. Thony B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 2000; 347:1-16.
3. Scott IA. Discovering nature’s diverse pathways to vitamin B12: a 35-year odyssey. J. Org. Chem. 2003; 68:2529-2539.
4. Roth JR, Lawrence JG, Bobik TA. Cobalamin (coenzyme B12): synthesis and biological significance. Annu. Rev. Microbiol. 1996; 50:137-181.
5. Taga ME, Larsen NA, Howard-Jones AR, Walsh CT, Walker GC. BluB cannibalizes flavin to form the lower ligand of vitamin B12. Nature 2007; 446:449-453.
6. Brown KL. Chemistry and enzymology of vitamin B12. Chem. Rev. 2005; 105:2075-2149.
7. Rebeille F, Ravanel S, Marquet A, Mendel RR, Smith AG, Warrene MJ. Roles of vitamins B5, B8, B9, B12 and molybdenum cofactor at cellular and organismal levels. Nat. Prod. Rep. 2007; 24:949-962.
8. Mendel RR. Biology of the molybdenum cofactor. J. Exp. Bot. 2007; 58:2289-2296
9. Raschle T, Arigoni D, Brunisholz R, Rechsteiner H, Amrhein N, Fitzpatrick TB. Reaction mechanism of pyridoxal 5'-phosphate synthase. Detection of an enzyme-bound chromophoric intermediate. J. Biol. Chem. 2007; 282:6098-6105.
10. Johnson DC, Dean DR, Smith AD, Johnson MK. Structure, function and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005; 74:247-281.
11. Imlay JA. Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 2006; 59:1073-1082
12. Kessler D. Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol. Rev. 2006; 30:825-840.
13. Lill R, Muhlenhoff U. Iron-sulfur protein biogenesis in eukaryotes: components and mechanisms. Annu. Rev. Cell Dev. Biol. 2006; 22:457-486.
14. Leeper FJ. The biosynthesis of porphyrins, chlorophylls, and vitamin B12. Nat. Prod. Rep. 1989; 6:171-203.
15. Holliday GL, Thornton JM, Marquet A, Smith AG, Rebeille F, Mendel RR, Schubert HL, Lawrence AD, Warren MJ. Evolution of enzymes and pathways for the biosynthesis of cofactors. Nat. Prod. Rep. 2007; 24:972-987.
16. Panek H, O’Brian MR. A whole genome view of prokaryotic haem biosynthesis. Microbiology 2002; 148(Pt 8):2273-2282.
17. Tanaka R, Tanaka A. Tetrapyrrole biosynthesis in higher plants. Annu. Rev. Plant Biol. 2007; 58:321-346.
18. Mendel RR, Smith AG, Marquet A, Warren MJ. Metal and cofactor insertion. Nat. Prod. Rep. 2007; 24:963-971.
19. Warren MJ, Scott AI. Tetrapyrrole assembly and modification into the ligands of biologically functional cofactors. Trends Biochem. Sci. 1990; 15:486-491.
20. Webb ME, Marquet A, Mendel RR, Rebeille F, Smith AG. Elucidating biosynthetic pathways for vitamins and cofactors. Nat. Prod. Rep. 2007; 24:988-1008
21. Roje S. Vitamin B biosynthesis in plants. Phytochem 2007; 68:1904-1921.
22. Bacher A, Fischer M. Biosynthesis of flavocoenzymes. Nat. Prod. Rep. 2005; 22:324-350.
23. Begley TP. Cofactor biosynthesis: an organic chemist’s treasure trove. Nat. Prod. Rev. 2006; 23:15-25.
24. Chatterjee A, Jurgenson CT, Schroeder FC, Ealick SE, Begley TP. Biosynthesis of thiamin thiazole in eukaryotes: conversion of NAD to an advanced intermediate. J. Am. Chem. Soc. 2007; 129:2914-2922.
25. Raschke M, Burkle L, Muller N, Nunes-Nesi A, Fernie AR, Arigoni D, Amrhein N, Fitzpatrick TB. Vitamin B1 biosynthesis in plants requires the essential iron sulfur cluster protein, THIC. Proc. Natl. Acad. Sci. U. S. A. 2007; 104:19637-19642.
26. Fitzpatrick TB, Amrhein N, Kappes B, Macheroux P, Tews I. Two independent routes of de novo vitamin B6 biosynthesis: not that different after all. Biochem. J. 2007; 407:1-13.
27. Scott DE, Ciulli, Abell C. Coenzyme biosynthesis: enzyme mechanism, structure and inhibition. Nat. Prod. Rep. 2007; 24:1009-1026.
28. Ehrenshaft M, Bilski P, Li MY, Chignell CF, Daub ME. A highly conserved sequence is a novel gene involved in de novo vitamin B6 biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 1999; 96:9374-9378.
29. Osmani AH, May GS, Osmani SA. The extremely conserved pyroA gene of Aspergillus nidulans is required for pyridoxine synthesis and is required indirectly for resistance to photosensitizers. J. Biol. Chem. 1999; 274:23565-23569.
30. Tambasco-Studart M, Titiz O, Raschle T, Forster G, Amrhein N, Fitzpatrick TB. Vitamin B6 biosynthesis in higher plants. Proc. Natl. Acad. Sci. U. S. A. 2005; 102:13687-13692.
31. Denu JM. Vitamins and aging: pathways to NAD+ synthesis. Cell. 2007; 129:453-454.
32. Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell. 2007; 129:473-484.
33. Begley TP, Kinsland C, Mehl RA, Osterman A, Dorrestein P. The biosynthesis of nicotinamide adenine dinucleotides in bacteria. Vitam. Horm. 2001; 61:103-119.
34. Webb ME, Smith AG, Abell C. Biosynthesis of pantothenate. Nat. Prod. Rep. 2004; 21:695-721.
35. Chakauya E, Coxona KM, Whitney HM, Ashurst JL, Abell C, Smith AG. Pantothenate biosynthesis in higher plants: advances and challenges. Physiol. Plant. 2006; 126:319-329.
36. Linster CL, Van Schaftingen E. Vitamin C: Biosynthesis, recycling and degradation in mammals. FEBS J. 2007; 274:1-22.
37. Smirnoff N. L-ascorbic acid biosynthesis. Vitam. Horm. 2001; 61:241-266.
38. Wheeler GL, Jones MA, Smirnoff, N. The biosynthetic pathway of vitamin C in higher plants. Nature 1998; 393:365-369.
39. Linster CL, Gomez TA, Christensen KC, Young BD, Brenner C. Arabidopsis VTC2 encodes a GDP-L-galactose phosphorylase, the last unknown enzyme in the Smirnoff-Wheeler pathway to ascorbic acid in plants. J. Biol. Chem. 2007; 282:18879-18885.
40. Davidson VL. Electron transfer in quinoproteins. Arch. Biochem. Biophys. 2004; 428:32-40.
41. Felton LM, Anthony C. Role of PQQ as a mammalian enzyme cofactor? Nature 2005; 433:E10.
42. Kasahara T, Kato T. A new redox-cofactor vitamin for mammals. Nature 2003; 422:832.
43. Rucker, R, Storms, D, Sheets, A, Tchaparian, E and Fascetti, A. Is pyrroloquinoline quinone a vitamin? Nature. 2005; 433:E10-E11.
44. Magnusson OT, Toyama H, Saeki M, Schwarzenbacher R, Klinman JP. The structure of a biosynthetic intermediate of pyrroloquinoline quinone (PQQ) and elucidation of the final step of PQQ biosynthesis. J. Am. Chem. Soc. 2004; 126:5342-5343.
45. Magnusson OT, Toyama H, Saeki M, Rojas A, Reed JC, Liddington RC, Klinman JP, Schwarzenbacher R. Quinone biogenesis: Structure and mechanism of PqqC, the final catalyst in the production of pyrroloquinoline quinone. Proc. Natl. Acad. Sci. U. S. A. 2004; 101:7913-7918
46. Marquet A, Bui BT. Biosynthesis of biotin and lipoic acid. Vitam. Horm. 2001; 61:51-101.
47. Marquet A, Tse Sum Bui, B, Smith AG, Warren MJ. Iron-sulfur proteins as initiators of radical chemistry. Nat. Prod. Rep. 2007; 24:1027-1040.
48. Sacchettini JC, Poulter CD. Creating isoprenoid diversity. Science 1997; 277:1788-1789.
49. Slater, EE and MacDonald, JS. Mechanism of action and biological profile of HMG CoA reductase inhibitors. A new therapeutic alternative. Drugs 1988; 36 Suppl 3:72-82.
50. Stancu C, Sima A. Statins: mechanism of action and effects. J. Cell. Mol. Med. 2001; 5:378-387.
51. Qureshi N, Porter JW. Conversion of acetyl-coenzyme A to isopentenyl pyrophosphate. In: Biosynthesis of isoprenoid Compounds. J. W. Porter and S. L. Spurgeon, eds. 1981. John Wiley, New York. pp. 47-94.
52. Eisenreich W, Bacher A, Arigoni D, Rohdich F. Biosynthesis of isoprenoids via the non-mevalonate pathway. Cell. Mol. Life Sci. 2004; 61:1401-1426.
53. Siemieniuk E, Skrzydlewska E. Coenzyme Q10: its biosynthesis and biological significance in animal organisms and in humans. Postepy. Hig. Med. Dosw. 2005; 59:150-159.
54. Szkopinska A, Ubiquinone. Biosynthesis of quinone ring and its isoprenoid side chain. Intracellular localization. Acta Biochim. Polonica. 2000; 47:469-480.
55. Graham DE, White RH. Elucidation of methanogenic coenzyme 76. biosyntheses: from spectroscopy to genomics. Nat. Prod. Rep. 2002; 19:133-147.77.
56. Eisenreich W, Schwarzkopf B, Bacher A. Biosynthesis of nucleotides, flavins, and deazaflavins in Methanobacterium ther-moautotrophicum. J. Biol. Chem. 1991; 266:9622-9631.78.
57. Reuke B, Korn S, Eisenreich W, Bacher A. Biosynthetic precursors of deazaflavins. J. Bacteriol. 1992; 174:4042-4049.79.
58. Grochowski LL, Xu H, Leung K, White RH. Characterization of an Fe2+-dependent archaeal-specific GTP cyclohydrolase, MptA, from Methanocaldococcus jannaschii. Biochemistry 2007; 46:6658-6667.
59. Koetter JW, Schulz GE. Crystal structure of 6-hydroxy-D-nicotine 81. oxidase from Arthrobacter nicotinovorans. J. Mol. Biol. 2005; 352:418-428.
60. Jitrapakdee S, Wallace JC. The biotin enzyme family: conserved 82. structural motifs and domain rearrangements. Curr. Protein Pept. Sci. 2003; 4:217-229.
61. Rucker RB, Wold F. Cofactors in and as posttranslational protein modifications. FASEB J. 1988; 2:2252-2261.
62. Vagelos PR, Majerus PW, Alberts AW, Larrabee AR, Ailhaud GP. Structure and function of the acyl carrier protein. Fed. Proc. 1966; 25:1485-1494.
63. Blair-Johnson M, Fiedler T, Fenna R. Human myeloperoxidase: structure of a cyanide complex and its interaction with bromide and thiocyanate substrates at 1.9 A resolution. Biochemistry 2001; 40:13990-13997.
64. Singh AK, Singh N, Sharma S, Singh SB, Kaur P, Bhushan A, Srinivasan A, Singh TP. Crystal structure of Lactoperoxidase at 2.4 A resolution. J. Mol. Biol. 2007; 376:1060-1075.
65. Furtmuller PG, Zederbauer M, Jantschko W, Helm J, Bogner M, Jakopitsch C, Obinger C. Active site structure and catalytic mechanisms of human peroxidases. Arch. Biochem. Biophys. 2006; 445:199-213.
66. Davidson VL. Protein-derived cofactors. Expanding the scope of post-translational modifications. Biochemistry 2007; 46:5283-5292.
67. Fischer M, Bacher A. Biosynthesis of vitamin B2 in plants. Physiol. Plant. 2006; 126:304-318.
68. Ralph SA, van Dooren GG, Waller RF, Crawford MJ, Fraunholz, MJ, Foth, BJ, Tonkin, CJ, Roos, DS and McFadden, GI. Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat. Rev. Microbiol. 2004; 2:203-216.
69. van Dooren GG, Stimmler LM, McFadden GI. Metabolic maps and functions of the Plasmodium mitochondrion. FEMS Microbiol. Rev. 2006; 30:596-630.
70. Smith AG, Croft MT, Moulin M, Webb ME. Plants need their vitamins too. Curr. Op. Plant Biol. 2007; 10:266-275.
71. Muntau AC, Roschinger W, Habich M, Demmelmair H, Hoffmann B, Sommerhoff CP, Roscher AA. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N. Engl. J. Med. 2002; 347:2122-3212.
72. Tahiliani AG, Beinlich CJ. Pantothenic acid in health and disease. Vitam. Horm. 1991; 46:165-228.
73. Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KH, Gitschier J. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N. Engl. J. Med. 2003; 348:33-40.
74. Schwarz G. Molybdenum cofactor biosynthesis and deficiency. Cell Mol. Life Sci. 2005; 62:2792-2810.
75. Sarkany RP. Porphyria. From Sir Walter Raleigh to molecular biology. Adv. Exp. Med. Biol. 1999; 455:235-241.
76. Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N. Engl. J. Med. 1997; 337:1441-1448.
77. Lange RP, Locher HH, Wyss PC, Then RL. The targets of currently used antibacterial agents: lessons for drug discovery. Curr. Pharm. Des. 2007; 13:3140-3154.
78. Wiesner J, Ortmann R, Jomaa H, Schlitzer M. New antimalarial drugs. Angew. Chem. Int. Ed. Engl. 2003; 42:5274-5293.
79. Gangjee A, Jain HD. Antifolates - past, present and future. Curr. Med. Chem. Anticancer Agents. 2004; 4:405-410.
80. Mazighi M, Lavallee PC, Labreuche J, Amarenco P. Statin therapy and stroke prevention: what was known, what is new and what is next? Curr. Opin. Lipidol. 2007; 18:622-625.
81. Harper CR, Jacobson TA. The broad spectrum of statin myopathy: from myalgia to rhabdomyolysis. Curr. Opin. Lipidol. 2007; 18:401-408.
82. Holick MF. Vitamin D deficiency. N. Engl. J. Med. 2007; 357:266-281.