CHEMICAL BIOLOGY
Chemistry of Fatty Acid Desaturases
Peter H. Buist, Carleton University, Ottawa, Ontario, Canada
doi: 10.1002/9780470048672.wecb167
The mid-chain dehydrogenation of saturated fatty acyl derivatives is carried out by a large family of 02-dependent, nonheme diiron-containing enzymes known as desaturases. Both soluble and membrane-bound desaturases have been characterized. The mechanism of desaturation is thought to involve the stepwise syn removal of vicinal hydrogen atoms via a short-lived carbon-centered radical intermediate. The most common desaturase inserts a (Z)-double bond between the C-9,10 carbons of a stearoyl thioester;however, many variations of this prototypical reaction have been discovered. Accounting for this diversity in terms of subtle alterations in active-site architecture constitutes a new frontier for research in this area.
The regioselective and stereoselective introduction of an olefinic link into fatty acyl side chains is catalyzed by a unique set of enzymes known as desaturases (1). The overall biochemical equation for this transformation can be depicted as follows:
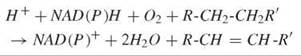
Unlike other dehydrogenases, desaturases attack unactivated C-H bonds with the concomitant reduction of molecular oxygen; two reducing equivalents are derived from NADH, and two are derived from the substrate. Most fatty acid desaturases are integral, membrane-bound proteins found in the endoplasmic reticulum and accept substrates bearing phospholipid or Coenzyme A headgroups. In plants, a soluble, plastidial desaturase operates exclusively on acyl carrier protein ACP substrates. Common to all of these enzymes is a nonheme diiron-containing catalytic core (vide infra) that is used to generate the active oxidant in situ. Considerable insight into the detailed chemical mechanism of desaturation has been gained in the last 15 years (2). This knowledge has lead to a more sophisticated understanding of lipid biochemistry in various contexts, including chemical ecology, plant biotechnology, and medical lipidology.
Biologic Context
Fatty acid desaturation is a ubiquitous lipid modification that is critically important to aerobic life forms in two major ways: 1) adjustment of lipidic biophysical properties and 2) biosynthesis of chemical messengers. Saturated fatty acids, produced by the fatty acid synthase assembly line, typically are dehydrogenated first at C-9,10 to give an oleate derivative that is additionally desaturated to generate the w-6 and w-3 group of the so-called “essential fatty acids” (Fig. 1a). Various highly bioactive signaling molecules (Fig. 1b) containing unsaturated sites are biosynthesized from this collection of primary fatty acids. In addition, desaturase-derived natural products with antifeedant properties have been discovered (Fig. 1c). It should be noted that the introduction of double bonds into biomolecules also dramatically enhances their susceptibility to free radical autoxidation with all attendant deleterious effects (3).
Unsaturated lipids and membrane fluidity
To function properly, cell membranes must exist primarily in the liquid-crystalline state. Model studies using synthetic lipids have demonstrated that the presence of a (9Z)-olefinic fatty acyl side chain in a diacylphospholipid lowers its gel-liquid phase temperature (Tc) by some 50° C relative to its fully saturated analog (4). The presence of the ∆9,12 fatty acyl side chain lowers the Tc even more and constitutes an important adaptation to chilling for plants. The regulation of the response to chilling temperatures has been studied in some detail in various organisms (4).
Chemical signaling and the carbon-carbon double bond
Nowhere is the “chemical biology” of the carbon-carbon double bond more exquisitely displayed than in the area of insect pheromone biosynthesis (5). Species-specific signals are generated by using a chemical language dictated by the number, position, and stereochemistry of internal double bonds located in a hydrocarbon backbone of varying lengths. Increased differentiation is achieved through additional functionalization and the use of multiple components in specific ratios. The blend of (Z)- and (E)-11-tetradecenoates shown in Fig. 1b is a case in point. In the area of sphingolipid biochemistry, it has been discovered that the position and stereochemistry of the double bond in ceramide is critically important to biologic function (6).

Figure 1. Role of fatty acid desaturases in the biosynthesis of unsaturated fatty acids: Typical fatty acids found as components of cell membranes and storage lipids (a); signaling agents (b); and plant-derived antifeedants (c).
Plant defense
The involvement of desaturase-type enzymes in the biosynthesis of some important lipidic antifeedants now has been firmly established (Fig. 1c) (7). An instructive example of this phenomenon is the production of ricinoleate (a purgative) by a desaturase homolog found in the castor plant. Here, a subtle variation in mechanistic pathway is responsible for the introduction of a C-12 hydroxyl group rather than a 12,13-double bond (7). Another interesting case features the putative dehydrogenation of a cyclopropyl fatty acid to produce sterculate, a potent inhibitor of the mammalian but not the plant ∆9 desaturase (8). Finally, the biosynthesis of polyacetylenes with antifungal properties such as falcarindiol is thought to involve a sequence of unique desaturase-mediated oxidations (9).
Chemistry of Fatty Acid Desaturation
The soluble castor stearoyl-ACP A9 desaturase has served as an important enzyme for detailed structural and biochemical studies (10). A crystal structure of this protein confirmed the presence of a nonheme, carboxylate-bridged diiron cluster in close proximity to a narrow, curved hydrophobic channel (11). The latter can accommodate a stearoyl-ACP substrate that adopts a gauche conformation around the 9,10-carbon-carbon single bond. Delivery of electrons from nicotinamide adenine dinucleotide phosphate (NADPH) via ferredoxin as required by Equation 1 is gated by substrate binding—an event that alters the redox properties of the catalytic center and ultimately triggers oxygen binding. Generation of a potent oxidant then can proceed (12). It is assumed that a similar diiron-based cluster exists in membrane-bound desaturases; Detailed comparison of amino acid sequences for this class has revealed the presence of a highly conserved, catalytically essential 8-histidine motif that is capable of binding two iron atoms. In addition, Mossbauer studies of a closely related alkane ω-hydroxylase revealed the presence of two catalytically active iron atoms (13). However, reconstitution of purified membranous desaturases to permit more detailed structural study has been extremely difficult. Initial design and testing of mechanistic probes was carried out using convenient in vivo microbial systems with subsequent application to in vitro desaturase preparations.
Consensus mechanistic model for C-H activation
The generally accepted mechanism for fatty acid desaturation (Fig. 2) was developed by analogy with similar models advanced for the closely related iron-containing hydroxylase enzymes (2). It is assumed that the active oxidant in desaturases is similar to that postulated for soluble methane monoxygenase—a well-characterized nonheme diiron-containing enzyme that also oxidizes unactivated C-H bonds. Dehydrogenation is thought to proceed by initial hydrogen atom abstraction, followed by a rapid collapse of the short-lived, carbon-centered radical to produce the olefinic product. The intermediacy of a carbocationic intermediate derived by an additional one-electron oxidation of the corresponding radical cannot be ruled out. Attempts to show that a discrete hydroxyl intermediate might be on the pathway to olefin have failed. Interestingly, in the case of membranous desaturases, regioselective formation of a minor hydroxyl by-product frequently is observed (14). Both soluble and membrane-bound desaturases are thought to follow a stepwise mechanism, although only in the latter set of enzymes is the initial C-H cleavage step kinetically important relative to other enzymic events.

Figure 2. Consensus mechanism for fatty acid desaturation. The related minor hydroxylation pathway also is shown.
Stereochemistry of hydrogen removal
All desaturases studied to date feature the syn removal of vicinal hydrogens as depicted in Fig. 2. For example, in the case of both soluble castor and membranous ∆9 desaturases, it could be shown via the use of stereospecifically labeled substrates that dehydrogenation proceeds with removal of proR hydrogens at both C-9 and C-10 (15, 16). This stereopreference seems highly conserved. A striking example of this phenomenon has been provided through extensive studies in the area of insect pheromone biosynthesis. Here, a mixture of (Z)- and (E)-isomers (Fig. 1b) are formed by removal of a topologically equivalent set of vicinal hydrogens (17). Similar stereochemical rules are obeyed in the biosynthesis of conjugated dienes by 1,4-dehydrogenation of mono-alkenes (18).
Cryptoregiochemistry of fatty acid desaturation
The term “cryptoregiochemistry” was coined to describe which substrate hydrogen is removed first in fatty acid desaturation (19). In the case of membrane-bound desaturases, the site of initial oxidation could be determined by examining the magnitude of the primary deuterium kinetic isotope effect (KIE) on C-H cleavage as a function of isotope location. According to the mechanistic model (Fig. 2), abstraction of the first C-H bond should be energetically more difficult, and hence more sensitive to isotopic substitution, than the subsequent C-H bond cleavage step. Thus, a large KIE was observed at C-9 but not at C-10 in the desaturation of regiospecifically deuterated substrates by a yeast stearoyl CoA A9 desaturase (Fig. 3a) (19). Corroborating evidence for initial attack at C-9 by this desaturase was obtained by observing preferential sulfoxidation of an S-9 substrate analog (Fig. 3b) (20). Additional proof for a C-9-initiated ∆9 desaturation was the observation of low level (1%) regioselective 9-hydroxylation along with the production of the major 9,10-olefinic product (14). The former pathway presumably occurs by hydroxyl trapping of the putative C-9 radical intermediate (Fig. 2).
The cryptoregiochemistry of a large number of membrane-bound desaturating systems with varying positional specificities has been determined (2), which include desaturases found in species of bacteria, blue-green and green algae, fungi, nematode, plants, insects, and mammals. The somewhat surprising trend that has emerged is that the carbon closest to the C-1 (acyl) terminus always is attacked first during these dehydrogenation reactions. This result points to a highly conserved active-site architecture that is used by desaturases from a wide range of life forms. The corresponding hydroxyl by-product (Fig. 2) with the predicted regiochemistry is observed routinely (14).
The determination of the cryptoregiochemistry for the soluble castor ∆9 desaturase has been more difficult because the C-H bond cleavage is masked kinetically by other events in the catalytic cycle, which nullifies the use of an approach that relies on the measurement of an intermolecular KIE. Nevertheless, the results of experiments that use sulfur, oxygen, and fluorine-labeled substrates suggest that, in this case, the initial site of oxidation is at C-10 rather than C-9 (10, 16).

Figure 3. Evidence for a stepwise removal of hydrogen by membranous stearoyl A9 desaturase (SCD) initiated at C-9: Primary deuterium isotope effect observed at C-9 but not at C-10 (a); Sulfoxidation of S-9 preferred over S-10 thiasubstrates (b).
Variations on an oxidative theme: the oxygenation/dehydrogenation connection
Perhaps the most important mechanistic question that remains unanswered with respect to desaturases relates to the switch that controls the choice of dehydrogenation/hydroxylation pathways (Fig. 2). The study of the FAD2 subgroup of plant desaturases has been particularly instructive in this context. Pairs of enzyme homologs that are tuned to catalyze either dehydrogenation or oxygenation as the major pathway have been identified (7). Thus, oleate ∆12 desaturase introduces a double bond at C-12,13 by initial hydrogen abstraction at the C-12 position, as determined by a KIE study (21). However, an enzyme homolog, oleate 12-hydroxylase, forms 12-hydroxyoleate (ricinoleate), a major component of castor oil (Fig. 4a). The results of site-directed mutagenesis experiments that use a bifunctional ∆12 oleate desaturase/12-hydroxylase demonstrated that relatively conservative changes in a few amino acids have a major impact on the hydroxylation/desaturation ratio (7, 14). An even more dramatic example of this mechanistic dichotomy is the discovery of FAD2-type enzyme homologs in the Crepis species that can execute either the dehydrogenation or epoxidation of the C-12,13-olefinic bond (Fig. 4b) (22). Interestingly, the former reaction is initiated also at C-12, as indicated by a large primary deuterium kinetic isotope effect at this position (23). A similar trend in kinetic isotope effects is exhibited by an insect acetylenase (24). It is tempting to account for these observations in terms of “fine control” of substrate position relative to oxidant in the active site. Alternatively, subtle changes in the coordination chemistry at the catalytic centers, similar to that postulated for clavaminate synthase, could be responsible for the observed divergence in reaction outcome (25).

Figure 4. Variations on an oxidative theme: the dehydrogenation/oxygenation connection. C-12-initiated oxidation catalyzed by oleate ∆12 desaturase and its homolog oleate 12-hydroxylase (a) and ∆12 acetylenase and its homolog ∆12 epoxidase (b).
Regiochemistry of desaturation
Regioselective remote functionalization of unactivated C-H bonds has long been a goal of synthetic chemists. Indeed, much early work in this area (26) was inspired by the ability of fatty acid desaturases to introduce double bonds in a regioselective manner. Three modes of regiocontrol have been identified (27, 28). The positional specificity of the ∆n class of desaturases is determined by the location (carbons n, n + 1) of the incipient double bond relative to the acyl head group (C-1) independent of the chain length of the substrate. For example, both yeast and rat liver membrane-bound ∆9desaturases dehydrogenate a range of fatty acyl CoA thioesters (C-15 to C-19) at the 9,10-position with no measurable regiochemical “error” and at comparable rates. Similar regioselectivity is observed for the corresponding soluble castor enzyme; but in this case, the C-18 substrate is a highly preferred substrate (10). Both the regioselectivity and the chain-length specificity of the latter enzyme can be altered through protein engineering experiments (1). The ω-n group desaturases inserts a double bond “n” carbons from the methyl terminus of substrate. Yet a third group of desaturases, the v + n class uses an adjacent double bond as the primary reference point and introduces unsaturation “n” carbons from a preexisting double bond.
Chemical Tools and Techniques
The design of probes for the mechanistic study of fatty acid desaturases had to take into account that this class of enzymes cannot tolerate large alterations in substrate structure. Consequently, use of isotopic labeling and the isomorphic replacement of the -CH2- unit by the sulfur atom or CHF moiety have been the most versatile approaches. Additional challenges were the high endogenous lipid content of membrane-bound desaturases and the need to enzymatically prepare substrate ACP thioesters in the study of soluble desaturases (16). To understand the bioinorganic chemistry of desaturases, a new methodology for probing nonheme iron-containing enzymes also was required (29).
Stereospecific and regiospecific isotopic labeling
To determine the stereochemistry of desaturase-mediated hydrogen removal, the synthesis of stereospecifically monodeuterated fatty acid substrates and a suitable gas chromatography-mass spectrometry (GC-MS) or liquid chromatography-mass spectrometry (LC-MS) method of product analysis is needed (2). The most common method of introducing deuterium stereospecifically is through deuteride displacement of suitable activated chiral alcohols. Occasionally, the latter compounds are available from natural sources; however, when they are not available from natural sources, total synthesis of substrate is necessary. In addition, the frequent problem of mass spectral interference because of high endogenous do-product content must be overcome through additional remote mass labeling of substrate using deuterium, sulfur, or fluorine substitution.
An examination of the primary deuterium kinetic isotope effect on each C-H bond cleavage involved in desaturation can reveal the site of initial oxidation, provided other kinetically more important enzymic steps do not mask these effects. This methodology relies on monitoring the loss of deuterium from regiospecifically deuterated fatty acids via GC-MS. General strategies for the synthesis of the required labeled compounds were devised during early work on assignments of 13C nuclear magnetic resonances (NMRs) for various fatty acids (30).
Use of fluorine as a probe and as a tag
Monofluorinated fatty acids function as fatty acyl substrates for desaturases and can be synthesized in enantiomerically enriched form via diethylaminosulfur trifluoride (D.A.S.T.) treatment of the suitable chiral alcohols. An example of the use of fluorine-substituted fatty acids as mechanistic probes was demonstrated by the use of chiral 9-fluorostearoyl substrates to induce latent stereoselective 10-hydroxylation by the castor stearoyl ACP ∆9 desaturase (16). In addition, the advantages of 1H-decoupled 19F-NMR, such as wide chemical shift range, high sensitivity, and lack of interferences, allow one to monitor desaturase-mediated transformations at |xMolar fluoro-substrate concentrations. In this context, M-fluorine-tagged fatty acid analogs have proven extraordinarily useful in tracking remote functional group transformations (2).
Use of sulfur as a methylene isostere
Thia fatty acid analogs are synthesized easily by thiolation of the corresponding bromoacids. When the sulfur atom in thia substrates is remote from the site of desaturation, normal olefinic thia products are produced, with the observed chemoselectivity being a consequence of a strict, desaturase-imposed, regiochemical imperative. Thia compounds also can be used to determine the site of initial oxidation in desaturase-mediated reactions because oxo transfer occurs most efficiently when the sulfur atom is located at the site of initial oxidation (20). As a bonus, the enantioselectivity of the oxidant can be ascertained by determining the absolute configuration of the resultant dialkyl sulfoxides (20). It was found that the stereochemistry of sulfoxidation matched that of hydrogen removal for the parent substrate. These analyses can be carried out on a microscale by using M-fluorine-tagged thia derivatives and suitable Pirkle-type NMR shift reagents (31).
Future Directions
Currently, efforts are underway to obtain more structural information on desaturases to address the mechanistic issues that have been raised through substrate-based studies. The need for more detailed 3-D active-site information is acute, particularly in the case of membrane-bound desaturases for which only hypothetical models currently are available. Design of mechanism-based inhibitors of medically relevant desaturases such as stearoyl CoA ∆9 desaturase (SCD) (metabolic syndrome) (32), DesA3 (tuberculosis) (33), and dihydroceramide ∆4 desaturase (apoptosis) (34, 35) also will be aided by new structural data.
The vision of plants as chemical factories that produce high value seed oils through protein engineering remains an active area of research (1). Identification of the first plant acetylenase enzyme has renewed interest in the large polyacetylenic group of natural products (9). The testing of plausible biosynthetic pathways for these intriguing phytochemicals and isolation of new genes can be anticipated.
References
1. Shanklin J, Cahoon EB. Desaturation and related modifications of fatty acids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998; 149:611-641.
2. Buist PH. Fatty acid desaturases: selecting the dehydrogenation channel. Nat. Prod. Rep. 2004; 21:249-262.
3. Gurr MI, Harwood JL, Frayn KN. Lipid Biochemistry: An Introduction. 5th edition. 2002. Blackwell Science, Oxford, UK. pp. 70.
4. Gurr MI, Harwood JL, Frayn KN. Lipid Biochemistry: An Introduction. 5th edition. 2002. Blackwell Science, Oxford, UK. pp. 242-244.
5. Howard RW, Blomquist GJ. Ecological, behavioral, and biochemical aspects of insect hydrocarbons. Ann. Rev. Entomol. 2005; 50:371-393.
6. He L, Byun H-S, Smit J, Wilschut J, Bittman RJ. Enantioselective synthesis of a novel trans double bond ceramide analogue via catalytic asymmetric dihydroxylation of an enyne. The role of the trans double bond of ceramide in the fusion of Semliki Forest virus with target membranes. J. Am. Chem. Soc. 1999; 121:3897-3903.
7. Broun P, Shanklin J, Whittle E, Somerville C. Catalyic plasticity of fatty acid modification enzymes underlying chemical diversity of plant lipids. Science 1998; 282:1315-1317.
8. Bao XM, Katz S, Pollard M, Ohlrogge J. Carbocyclic fatty acids in plants: biochemical and molecular genetic characterization of cyclopropane fatty acid synthesis of Sterculia foetida. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:7171-7177.
9. Cahoon EB, Schnurr JA, Huffmann EA, Minto RE. Fungal responsive fatty acid acetylenases occur widely in evolutionarily distant plant families. Plant J. 2003; 34:671-683.
10. Fox BG, Lyle KS, Rogge CE. Reactions of the diiron enzyme stearoyl-acyl carrier protein desaturase. Acc. Chem. Res. 2004; 37:421-429.
11. Lindqvist Y, Huang W, Schneider G, Shanklin J. Crystal Structure of ∆9 stearoyl-acyl carrier protein desaturase from castor seed and its relationship to other di-iron proteins. EMBO J. 199; 15:4081-4092.
12. Reipa V, Shanklin J, Vilker V. Substrate binding and the presence of ferredoxin affect the redox properties of the soluble plant Delta9-18:0-acyl carrier protein desaturase. Chem. Commun. (Camb). 2004; 21:2406-2407.
13. Shanklin J, Achim C, Schmidt H, Fox BG, Munck E. Mossbauer studies of alkane omega-hydroxylase: evidence for a diiron cluster in an integral-membrane enzyme. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:2981-2986.
14. Broadwater JA, Whittle E, Shanklin J. Desaturation and hydroxylation. Residues 148 and 324 of Arabidopsis FAD2, in addition to substrate chain length, exert a major influence in partitioning of catalytic specificity. J. Biol. Chem. 2002; 277:15613-15620.
15. Schroepfer GJ, Bloch K. The stereospecific conversion of stearic acid to oleic acid. J. Biol. Chem. 1965; 240:54-63.
16. Behrouzian B, Savile CK, Dawson B, Buist PH, Shanklin J. Exploring the hydroxylation-dehydrogenation connection: novel catalytic activity of castor stearoyl-ACP ∆9 desaturase. J. Am. Chem. Soc. 2002; 124:3277-3283.
17. Navarro I, Font I, Fabrias G, Camps F. Stereospecificity of the (E) and (Z)-11 Myristoyl CoA desaturase in the biosynthesis of Spodoptera littoralis sex pheromone. J. Am. Chem. Soc. 1997; 119:11335-11336.
18. Rodriguez S, Clapes P, Camps F, Fabrias G. Stereospecificity of an enzymatic monoene 1,4-dehydrogenation reaction: conversion of (Z)-11-tetradecenoic acid into (E,E)-10,12-tetradecadienoic acid. J. Org. Chem. 2002; 67:2228-2233.
19. Buist PH, Behrouzian B. Use of deuterium kinetic isotope effects to probe the cryptoregiochemistry of ∆9 desaturase. J. Am. Chem. Soc. 1996; 118:6295-6296.
20. Buist PH, Marecak DM Stereochemical analysis of sulfoxides obtained by diverted desaturation. J. Am. Chem. Soc. 1992; 114:5073-5080.
21. Buist PH, Behrouzian B. Deciphering the cryptoregiochemistry of oleate Oleate ∆12 Desaturase: a kinetic isotope effect study. J. Am. Chem. Soc. 1998; 120:871-876.
22. Lee M, Lenman M, Banas A, Bafor M, Singh S, Schweizer M, Nilsson R, Liljenberg C, Dahlqvist A, Gummeson P-O, Sjodahl S, Green A, Stymne S. Identification of nonheme diiron proteins that cata yze triple bond and epoxy group formation. Science 1998; 280:915-918.
23. Reed DW, Polichuk DR, Buist PH, Ambrose SJ, Sasata RJ, Savile CK, Ross AR, Covello PS. Mechanistic study of an improbable reaction: alkene dehydrogenation by the ∆12 acetylenase of Crepis alpina. J. Am. Chem. Soc. 2003; 125:10635-10640.
24. Abad JL, Rodriguez S, Camps F, Fabrias G. Synthesis and use of probes to investigate the cryptoregiochemistry of the first animal acetylenase. J. Org. Chem. 2006; 71:7558-7564.
25. Zhou J, Kelly WL, Bachmmann BO, Gunsior M, Townsend CA, Solomon EI. Spectroscopic studies of substrate interactions with clavaminate synthase 2, a multifunctional alpha-KG-dependent non-heme iron enzyme: correlation with mechanisms and reactivities. J. Am. Chem. Soc. 2001; 123:7388-7398.
26. Breslow R. Biomimetic chemistry. Chem. Soc. Rev. 1972; 1:553-580
27. Howling D, Morris LJ, Gurr MI, James AT. The specificity of fatty acid desaturases and hydroxylases: the dehydrogenation and hydroxylation of monoenoic acids. Biochim. Biophys. Acta 1972; 260:10-19.
28. Schwartzbeck JL, Jung S, Abbott AG, Mosley E, Lewis S, Pries GL, Powell GL. Endoplasmic oleoyl-PC desaturase references the second double bond. Phytochemistry 2001; 57:643-652.
29. Yang Y-S, Broadwater J, Pulver SC, Fox BG, Solomon EI. Circular dichroism and magnetic circular dichroism studies of the reduced binuclear non-heme iron site of stearoyl-ACP. ∆9-desaturase: substrate binding and comparison to ribonucleotide reductase. J. Am. Chem. Soc. 1999; 121:2770-2783.
30. Tulloch AP. Synthesis, analysis and application of specifically deuterated lipids. Prog. Lipid Res. 1983; 22:235-256.
31. Lao KY, Hodgson DJ, Dawson B, Buist PH. A micromethod for the stereochemical analysis of fatty acid desaturase-mediated sulfoxidation reactions. Bioorg. Med. Chem. Lett. 2005;15: 2799-2802.
32. Dobrzyn A, Ntambi JM. The role of stearoyl-CoA desaturase in the control of metabolism. Prostaglandins Leukot. Essent. Fatty Acids 2005; 73:35-41.
33. Phetsuksiri B, Jackson M, Scherman H, McNeil M, Besra GS, Baulard AR, Slayden RA, DeBarber AE, Barry CE, Baird MS, Crick DC, Brennan PJ. Unique mechanism of action of the thiourea drug isoxyl on Mycobacterium tuberculosis. J. Biol. Chem. 2003; 278:53123-53130.
34. Delgado A, Casas J, Llebaria A, Abad JL, Fabrias G. Inhibitors of sphingolipid metabolism enzymes. Biochim. Biophys. Acta 2006; 1758:1957-1977.
35. Zheng W, Kollmeyer J, Symolon H, Momin A, Munter E, Wang E, Kelly S, Allegood JC, Liu Y, Peng Q, Ramaraju H, Sullards MC, Cabot M, Merrill AH Jr. Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim Biophys Acta. 2006; 1758:1864-1884.
Further Reading
Heinz E. Biosynthesis of polyunsaturated fatty acids. In: Lipid Metabolism in Plants. Moore TSJr, ed. 1993. CRC Press, Boca Raton, FL.
Cook HW, McMaster CR. Fatty acid desaturation and chain elongation in eukaryotes. In: Biochemistry of Lipids, Lipoproteins and Membranes, 4th edition. Vance DE, Vance JE, eds. 2002. Elsevier, Amsterdam, The Netherlands.
Somerville CR, Bonetta D. Plants as factories for technical materials. Plant Physiol. 2001; 125:168-171.
Wallis JG, Browse J. Mutants of Arabidopsis reveal many roles for membrane lipids. Prog. Lipid. Res. 2002; 41:254-278.
Martin CE, Oh CS, Jang Y. Regulation of long chain unsaturated fatty acid synthesis in yeast. Biochim. Biophys. Acta 2007; 1771:271-285.
Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Muller J, Lippard SJ. Dioxygen activation and methane hydroxylation by soluble methane monooxygenase: a tale of two irons and three proteins. Angew. Chem. Int. Ed. 2001; 40:2782-2807.
See Also
Oxygen-Activating Enzymes, Chemistry of
GC-MS of Lipids
Membranes, Fluidity of
Natural Products: An Overview
Organic Chemistry in Biology