CHEMICAL BIOLOGY
Fluorescence Techniques for Proteins
Carey K. Johnson, University of Kansas, Lawrence, Kansas
doi: 10.1002/9780470048672.wecb176
Biologists and chemists are increasingly turning to fluorescence to investigate proteins. Not only is fluorescence detection highly sensitive, but it can also report specific information about a range of properties of proteins. For example, fluorescence can be used to probe the environment of fluorescent molecules, interactions with other proteins, and the motions of proteins over time scales from picoseconds to seconds. This article surveys the properties of biologic fluorophores that can be used to study proteins and introduces the fluorescence techniques commonly applied to proteins, with an emphasis on the information generated. Techniques discussed include steady-state emission, time-resolved detection, fluorescence anisotropy or depolarization, energy transfer, fluorescence correlation spectroscopy, single-molecule spectroscopy, and fluorescence imaging.
Fluorescence is the basis for elegant and sensitive tools for studying proteins. Its applications range from detection of proteins at concentrations as low as single molecules to characterization of protein structure and dynamics. Since Gregorio Weber pioneered protein fluorescence studies in the 1950s (1), biologists and chemists have increasingly turned to fluorescence to probe the properties of proteins. Two fundamental reasons exist to use fluorescence. First, fluorescence is one of the most sensitive detection methods known. Second, fluorescence signals report a wealth of information on protein structure, dynamics, and interactions. This chapter contains a survey of fluorescence techniques with an emphasis on the information generated relevant to proteins. Additional surveys can be found listed in the Further Reading list. More focused review articles are referenced in the context of specific methods or techniques.
Biologic Fluorophores
Fluorescence measurements on proteins require both an appropriate fluorescence technique and the presence of a suitable fluorophore. The techniques used for the application of fluorescence to proteins are described later in this article. In this section, we briefly consider three classes of fluorophores that are used widely to study proteins: native fluorophores including fluorescent amino acids, extrinsic fluorescent labels, and autofluorescent proteins. Each has advantages for probing proteins and has distinct drawbacks: No perfect fluorophore exists for studying proteins.
Native fluorescent amino acids
The use of fluorophores intrinsic to the protein allows researchers to probe protein structure and dynamics without incorporating non-native fluorophores that could perturb the native structure of the protein. Proteins usually contain one or more fluorescent amino acids, which makes dynamic studies on the native protein feasible. Often, fluorescent amino acid residues can be introduced by site-directed mutation without altering protein structure significantly.
The three aromatic amino acids, tryptophan (Trp), tyrosine (Tyr), and phenylalanine (Phe), are the only native amino acids with useful fluorescence properties. Figure 1a shows their absorption and fluorescence spectra. Their fluorescence properties are summarized in Table 1. Note that the relative absorption coefficients increase in the order Phe < Tyr < Trp. The fluorescence quantum yields increase in the same order. The product of absorption coefficient and fluorescence quantum yield can be taken as a measure of the brightness of the fluorophore. By the standards of fluorescent dyes (see below), the brightness of all three amino acids is poor. Trp is the brightest of the three, and for proteins with a small number of Trp residues it may be possible to assign fluorescence decays to specific Trp residues. As a result, of the three fluorescent amino acids, Trp is by far the most widely exploited for its fluorescent properties. Fluorescence from Tyr is also detectable but may be masked by Trp fluorescence. Proteins often contain many Tyr residues, so it is often not possible to isolate the fluorescence from individual Tyr residues. Fluorescence from Phe is weak and not often used in fluorescence studies.

Figure 1. (a) Absorption and fluorescence spectra of fluorescent amino acids blocked with peptide bonds: N-acetyltryptophanamide (black), N-acetyltyrosinamide (green), and N-acetylphenylalaninamide (red) in aqueous solution. The absorption spectra (left) show the relative absorption strengths of the three amino acids. The fluorescence spectra (right) are normalized to the same peak intensity (see Table 1 for relative quantum yields). (b) Fluorescence emission spectra of the maleimide derivatives of four common fluorescent dyes: Alexa Fluor 488 (green), tetramethylrhodamine (yellow), Texas red (orange), and Cy 5 (red).
Table 1. Fluorescence properties of amino acids and other intrinsic fluorophores
|
Fluorophore |
Absorption λmax (nm) |
Absorption coefficient (M-1 cm-1) |
Fluorescence λmax (nm) |
Lifetime (ns) |
Quantum yield |
|
Tryptophan |
280 |
5,600 |
|
|
|
|
in water |
|
|
353 |
3.1 (mean) |
0.13 |
|
in proteins |
|
|
308-352 |
0.3-7 |
0.01-0.35 |
|
Tyrosine (in water) |
274 |
1,400 |
304 |
3.2 |
0.07 |
|
Phenylalanine (in water) |
257 |
200 |
260 |
6.8 |
0.02 |
|
NADH in water |
340 |
6,200 |
470 |
0.52 |
0.02 |
|
in protein |
330 |
|
440 |
0.6-10 |
0.08-0.2 |
|
FMN |
450 |
12,000 |
520 |
4.7 |
0.2 |
|
FAD |
|
|
|
0.5 |
0.02 |
Although Trp fluorescence is the strongest of the three fluorescent amino acids, its fluorescence is also the most complex. The Trp fluorescence spectrum is highly sensitive to its environment. Trp residues buried in the protein tend to fluoresce at shorter wavelengths (λmax as low as 308 nm), whereas solvent-exposed Trp residues emit at longer wavelengths (λmax = 353 nm in water). The environmental sensitivity of λmax results from the large increase of the Trp dipole moment in the fluorescing excited state (denoted the 1La state). The environment surrounding Trp, including H-bonds and charged groups, can therefore tune the emission wavelength over a large spectral range (see Reference 2). The fluorescence quantum yield of Trp is also highly sensitive to the environment, and depends on the distance and the orientation of the indole side chain of Trp relative to groups that quench Trp fluorescence, especially carbonyl groups of the peptide backbone. The fluorescence quantum yield of Trp is modulated further by the presence of two closely lying excited states, 1Lb and 1La. Several fine reviews of the fluorescence properties of Trp describe these properties in greater detail (see Further Reading).
Although the fluorescence from Tyr residues is often less intense than the fluorescence from Trp residues, Tyr fluorescence is not complicated by the presence of two close-lying excited states, and Tyr emission spectra are much less sensitive to the environment than those of Trp with peak emission typically around 303 nm to 305 nm. Fluorescence from Phe in proteins is extremely weak and is likely to be quenched by energy transfer to Tyr or Trp.
Some proteins contain other native fluorophores in addition to fluorescent amino acids. These include cofactors such as nicotinamide adenine dinucleotide (fluorescent in its reduced, NADH state) and flavin adenine dinucleotide (FAD). NADH is weakly fluorescent in water, but its fluorescence yield increases markedly on binding to a protein-binding site with an emission peak around 470 nm (3). FAD and flavin mononucleotide (FMN) are also fluorescent with an emission maximum around 520 nm, but fluorescence is quenched on binding to many flavoproteins (4).
Fluorescence labels
As described above, the intrinsic fluorophores that nature provides in proteins generally have rather low absorption coefficients and quantum yields. For many applications, brighter fluorescence probes are needed, and emission in the visible region is desirable. One way to meet these needs is by labeling with a fluorescent dye. A wide range of fluorescence probes is now available for this purpose (see Fig. 1b). A summary of the spectroscopic and photophysical properties of many fluorescence probes is available in References 5 and 6.
Extrinsic fluorophores have emerged as remarkable tools for studies of proteins in part because reactive derivatives are available to target labeling to specific functional groups in proteins. Derivatives (which include isothiocyanates, succinimidylesters, and sulfonyl chlorides) are available to label amino groups such as lysine residues or the N-terminus of the protein. However, proteins may contain many lysine residues, which makes it problematic to label a specific site selectively. Thiol-reactive fluorescent probes (iodoacetamides and maleimides) offer more selectivity because they can be targeted specifically to cysteine (Cys) residues in proteins. Additional details about the chemistry of labeling functional groups in proteins are available from Molecular Probes (Invitrogen Corp., Eugene, OR) (5).
Often, the greatest challenge in applying fluorescence probes to proteins is the labeling procedure itself. Although the chemical conditions for effective labeling of amino groups and thiols are well established, selectively targeted labeling may require thorough characterization of the reactivities of functional groups. Proteins often have multiple Cys or Lys residues with varying reactivities toward thiol-reactive or amine-reactive probes, respectively. Under the proper conditions, it may be possible to label the most reactive site. Other strategies include site-directed mutation to eliminate Cys residues from protein sites where labeling is undesired and to introduce a Cys residue at the desired location. More selective labeling motifs can also be introduced into proteins by site-directed mutation. For example, a tetra-cysteine motif can be introduced and labeled with a fluorophore derivatized with biarsenical ligands (7).
Over the past decade, semiconductor nanoparticles (e.g., CdSe) have emerged as alternative fluorescence labels for proteins. These particles, called quantum dots, are very bright, have a low susceptibility to photobleaching, and are available for a wide range of emission wavelengths. They are especially useful in imaging applications. Quantum dots derivatized for labeling proteins, for example by biotin or antibody conjugation, are now available commercially. Therefore, they are a very attractive option for some applications. However, quantum dots are large (>10 nm in diameter) and can be toxic in a cellular environment.
Autofluorescent proteins
A different approach to fluorescence labeling is to harness the protein-synthesizing machinery of cells to fuse an autofluorescent protein to the protein of interest. The original protein in this class was green fluorescent protein (GFP), isolated from the jellyfish Aequorea aequorea. GFP consists of a ^-barrel structure that encloses the fluorophore, which protects it from photo- bleaching reactions. Since the discovery of GFP, a menagerie of variants of GFP has been generated by site-directed mutations to alter their spectroscopic and photophysical properties. These variants include blue fluorescent protein (BFP), cyan fluorescent protein (CFP), and yellow fluorescent protein (YFP), as well as variants with enhanced brightness. A red-emitting fluorescent protein, dsRed, has been isolated from reef corals. Most variants of GFP, however, are susceptible to photochemical processes such as excited-state proton transfer that generate transient dark states (8). Tables of spectroscopic properties of fluorescent proteins are available in several sources (9, 10). Given the wide variety of absorption and emission wavelengths together with the improved brightness and photostability of several mutant fluorescent proteins, fluorescent proteins are now available for many applications. These fluorophores are particularly useful for fluorescence imaging in organisms because they are formed internally, which alleviates the need to introduce extrinsic fluorophores. Applications in live cells include photobleaching recovery and energy transfer. In the latter applications, pairs of fluorescent proteins (e.g., CFP and YFP) are used to detect protein interactions or conformational changes.
Fluorescence Primer
Excitation of a fluorophore by absorption of light generates an electronically excited state. This state can then relax by emitting a fluorescence photon. A schematic of the molecular states involved is shown in the Jablonski diagram (named after Polish physicist Aleksander Jablonski, 1898-1980) in Fig. 2a. Because fluorescence can be detected at right angles to the excitation beam (or any other direction), the emission can be detected with high sensitivity against a very low background. This section describes basic photophysics involved in fluorescence measurements, including methods of fluorescence excitation.

Figure 2. (a) Jablonski diagram of photophysical processes related to light absorption and emission. One-photon (green arrow) or two-photon (red arrows) exciting a vibronic level in the excited S1 electronic state is followed by rapid vibrational relaxation in the excited state. Nonradiative relaxation of the excited state can occur by internal conversion to the ground state (blue arrow) or intersystem crossing to the triplet state T1(gray arrow). Other processes that may deplete the excited state include photochemistry and energy transfer (light blue arrow). The excited state can also relax radiatively by emitting a photon (orange arrow). (b) Illustration of fluorescence generated by one-photon excitation (left) or two-photon excitation (right). The one-photon excitation probability is linear in light intensity, whereas the two-photon excitation probability is quadratic in light intensity and therefore occurs mostly in the focal region.
Fluorescence lifetime and quantum yield
Figure 2a illustrates the concepts of radiative and nonradiative decay, fluorescence quantum yield, and fluorescence decay. A molecule in an excited electronic state can relax by several channels. Molecules excited to a vibrational level in the excited state undergo vibrational relaxation (cooling, yellow arrows in Fig. 2a) to the lowest vibrational levels of the excited state in a few picoseconds or less. Other nonradiative relaxation channels include internal conversion with rate constant kic to generate another electronic state with the same electronic spin, or intersystem crossing with rate constant kisc to the triplet state T1. Other decay channels may include excited-state reactions such as electron transfer, proton transfer, or isomerization, with rate constants denoted kpc in Fig. 2a, or electronic energy transfer to another molecule, kfret. All processes compete kinetically with radiative decay from the excited state. Radiative decay has an intrinsic rate constant (denoted kr) that can be calculated from the integrated absorption coefficient (11).
The total decay rate constant kf of the excited state is simply the sum of the rate constants for all processes that deplete the excited state:
![]()
where... includes the rates of any other processes that deplete the excited state. The intensity of fluorescence is proportional to the population of excited states; therefore, after excitation the probability of fluorescence emission decays with the rate constant kf. The inverse of this value is called the fluorescence lifetime τf. The fluorescence decay is then described by:
![]()
The fraction of the decay rate that results in emission of a photon is the fluorescence quantum yield (or quantum efficiency ) фf:
![]()
Note that if the radiative rate kr can be calculated, then the fluorescence decay rate and fluorescence lifetime follow from the fluorescence quantum yield фf. Of course, the situation is often more complex. As will be described below, fluorescence decays for proteins often do not follow the single exponential decay model of Equation 2. The fluorescence quantum yield and Equation 3 then provide an average fluorescence lifetime.
Excitation sources
The simplest steady-state measurements of fluorescence properties such as the fluorescence emission spectrum or the steady-state anisotropy can be carried out in a standard fluorometer with excitation from a lamp source and a monochromator. Common lamp sources in commercial fluorometers include xenon lamps for excitation from the UV to the near-IR (250 nm to 1100 nm).
For many advanced fluorescence applications, the sample is excited with a laser. Lasers have several advantages over traditional lamp sources: 1) Laser beams often have low beam divergence and therefore are directed readily and focused onto a sample; 2) laser sources can generate narrow excitation band- widths, which allows excitation of the fluorophore of interest or photoselection of a subset of fluorophores; and 3) through a technique called mode-locking, some lasers can generate extremely short pulses of light, which allows time resolution of protein dynamics on time scales from less than one picosecond (10-12 s). The laser source selected for a given technique will depend on the wavelengths needed, the desired beam power, and, for time-resolved experiments, the required pulse width. The properties of some laser sources are summarized in Table 2.
Table 2. Common laser sources for fluorescence excitation
|
Laser |
Wavelength (nm) |
Typical average Power |
CW or pulse characteristics |
|
Ar ion |
457, 476, 488, 496, 502, 514, 529 (plus other weaker lines) |
10 mW to 10 W total for all lines |
CW Pulses can be generated for some lines (476, 496, 502, 514) by mode-locking (pulsewidth 100 s of picoseconds) |
|
Kr ion |
531, 568, 647, 676, 752 (plus other weaker lines) |
10 mW to 1 W total for all lines |
CW |
|
Diode pumped solid state |
460, 473, 488, 532, 561, 635, 660 (other wavelengths also available) |
1 mW-10 W |
CW or pulsed |
|
Diode |
400-1650 |
1-100 mW |
CW or pulsed |
|
Dye |
400-900 |
10mW-1 W |
CW, pulsed, or ultrafast |
|
He-Cd |
325 , 440 |
2-200 mW |
CW |
|
He-Ne |
543, 594, 633 |
1-10 mW |
CW |
|
Nd (Nd:YAG, Nd:YVO4) |
1064 and harmonics: 532, 355, 266 |
1-10 W |
CW or mode-locked (pulse width ~100 ps) |
|
Ti:sapphire |
700-1100 and harmonics: 350550, 233-366 |
100mW-1 W |
CW or modelocked (pulse widths ~10-200 fs) |
Two-photon excitation
With intense laser pulses, new nonlinear optical phenomena are possible. The prime example is two-photon excitation (TPE). The peak power in a laser pulse from a Ti:sapphire laser (pulse width ~100 fs) can readily reach 105 W or higher, with a focused intensity of 1014 W/cm2. Under these conditions, excitation can occur with two photons that have half of the energy (twice the wavelength) of the corresponding one-photon transition (see Fig. 2a). The rate of TPE is given by:
![]()
where N* is the population of the excited states, N the population of ground states, I is the peak intensity of the laser pulse in W/cm2, v is the optical frequency of the laser pulse, h is Planck’s constant, and δ(2) is the two-photon cross section. The two-photon cross section is a molecular property that depends on the nature of the ground state, the excited state, and the wavelength of light. It has typical magnitudes of 10-48 to 10-50 cm4 ∙ s at the peak excitation wavelength for strong two-photon absorbers. The two-photon excitation probability also depends on the polarizations of the photons, but S2-1 is usually given as an orientationally averaged value for linearly polarized light and an isotropic sample. The two-photon excitation spectra and cross sections have been described for fluorescence dyes (12), Trp (13), NADH and flavins (14), and GFP and other fluorescent proteins (15).
Why use TPE? The answer lies in Equation 4. First, because the excitation probability depends on the square of the laser intensity, molecules are excited predominantly at the focus of a lens. The result is highly localized excitation (see Fig. 2b). This property is particularly useful in two-photon fluorescence microscopy, which was pioneered by W. Denk et al. (16) and now widely used in imaging. A second advantage of TPE is the possibility of excitation with near-IR light, a region of the spectrum in which biologic samples are more transparent than in the visible or UV. This region also corresponds nicely to the output of Ti:sapphire lasers. Therefore, TPE with a near-IR laser source minimizes damage to cells or tissues and reduces significantly autofluorescent background in fluorescence microscopy. Third, the two-photon excitation spectra of many fluorophores are broad, which allows excitation of multiple fluorophores with the same excitation wavelength.
Fluorescence Techniques Applied to Proteins
Multiple parameters can be measured for fluorescence photons: count rate (or intensity), wavelength (λ), polarization (p), arrival time (ta), time delay after excitation (td), and location (x, y on an imaging detector). These parameters carry information about the fluorophore that includes the nature of its environment, its interactions with other molecules, and its motions. Fluorescence techniques that exploit each of these parameters are described below.
Steady-state fluorescence spectroscopy: intensity and wavelength
The most straightforward fluorescent technique is simply the measurement of the fluorescence intensity. Laser-induced fluorescence (LIF) is one of the most sensitive detection methods known. In LIF, the fluorescence intensity excited by a laser beam is used to quantify the amount of fluorophore present. When combined with a separation technique such as capillary electrophoresis, LIF is a powerful bioanalytic method. Detection limits can reach less than 100 femtomolar and potentially the single-molecule level (reviewed in Reference 17).
The fluorescence intensity resolved by wavelength constitutes the fluorescence spectrum. The wavelengths of fluorescence photons contain information about the environment of the fluorophore and the sample heterogeneity. For example, as described above, buried Trp residues tend to have blue-shifted emission bands (λmax < 330 nm), whereas Trp residues partially or fully exposed to water have red-shifted emission bands (λmax > 340 nm). Therefore, protein conformational changes or unfolding may be accompanied by shifts in the native fluorescence spectra. Fluorescence spectra can be measured on a standard fluorometer, which is available from many manufacturers.
Time-resolved fluorescence: time after excitation
In this section, we discuss methods that detect the time delay td between excitation of a fluorophore and arrival of a fluorescence photon. The distribution of td times constitutes the fluorescence decay profile of the fluorophore. The average time lag between the excitation event and the emission is the fluorescence lifetime τf of the fluorophore. The fluorescence decay contains information about dynamic processes that deplete the excited state (Fig. 2a). In time-resolved fluorescence experiments, the fluorescence decay is measured to gain information about these processes.
Often, experimentally measured fluorescence decays do not follow the simple single-exponential form predicted by Equation 2. In many cases, the fluorescence decays are better described by a multiexponential decay:
![]()
or by a distribution of decay times
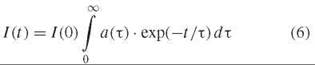
In Equation 5, TI is the decay time and ai is the relative amplitude for the i th decay component, and in Equation 6 a(τ) is the normalized distribution of decay times.
The observation of a nonsingle exponential fluorescence decay contains additional information about the nature of the system of fluorophores. Two general reasons exist for observation of nonsingle exponential decays. First, the sample may be heterogeneous, comprising different ground-state conformations or environments that have different intrinsic fluorescence lifetimes (Equation 5) or a distribution of lifetimes (Equation 6). Second, excited-state reactions may exist that are reversible (which allows return to the fluorescent state) or that generate a new fluorescent state that has a different decay rate.
A great deal of attention has been focused on the origin of nonsingle-exponential fluorescence decays in proteins. Some of the most intensely studied cases involve the fluorescence decays of the amino acids Trp and Tyr in proteins. Fluorescence decays for Trp in proteins are almost always multiexponential. The origins of the nonsingle-exponential fluorescence decays continue to be debated. In proteins with multiple Trp residues, multiexponential decay can be expected because of different environments and different decay times for each Trp. As with the emission wavelength, the fluorescence lifetime of a Trp residue is highly sensitive to its environment, notably the presence of quenching moieties (for example, histidine residues, disulfide bonds, or the carbonyl group of peptide bonds).
Even proteins that contain only a single Trp residue generally exhibit multiexponential decays. Several hypotheses have been proposed to explain why. First, multiple conformational states may exist for the single Trp such as different rotameric configurations (orientations about the Trp λ1 or λ2 C-C bond) (18). Even in the absence of multiple rotamers, the electron-transfer quenching rate is extremely sensitive to the local environment, so a distribution of local microconformational states may cause a nonexponential fluorescence decay. Other possible sources of nonexponential fluorescence decay include the response of the protein and surrounding solvent to the change in dipole moment of Trp on excitation (“solvation”) (19).
Time-resolved fluorescence measurement
Two common methods exist for measuring of fluorescence decays in proteins: time-domain and frequency-domain measurements. Signals are processed by time-correlated single-photon counting (TCSPC) for time-domain measurements, or by phase fluorimetry for frequency-domain measurements. For higher time resolution, both time-domain and frequency-domain methods generally employ the same light sources and the same detectors. Although one or the other technique may have some advantages in certain applications, a recent analysis showed that the two methods give essentially identical results for a series of fluorescence lifetime standards (20). More thorough treatments of TCSPC and frequency domain fluorometry are available in books cited in “Further Reading.”
TCSPC is illustrated in Fig. 3a. In addition to a mode-locked laser for pulsed excitation and a detector with high time resolution (usually a micro-channel plate photomultiplier tube capable of time-resolution of 20-30 ps), the required instrumentation includes constant-fraction discriminators to generate electrical pulses triggered by fluorescence photons and by the reference (the excitation pulse), a time-to-amplitude converter or other device to measure the time lag between reference and fluorescence counts, and a multichannel scaler to accumulate the histogram of lag times. Until recently, each device was a separate component. Now, it is possible to obtain the complete TCSPC electronic processing system on a single computer card. Manufacturers include Becker & Hickl (Berlin, Germany) and PicoQuant (Berlin, Germany).
In the frequency domain, fluorescence lifetime measurements are based on the phase shift and demodulation of a fluorescence signal with respect to a modulated excitation beam (21) (illustrated in Fig. 3b). For routine measurements with low time resolution, a fluorescence lamp or continuous laser source can be transmitted through an intensity modulator such as a Pockels’ cell. For high time-resolution, the modulation frequency achievable by this method is not high enough, and the method employs pulsed excitation, just as in TCSPC. The signal analysis consists in measurement of the fluorescence response at a range of frequencies (harmonics of the pulse repetition rate). The phase shift ф and modulation depth m are given as a function of the modulation frequency f by (ω = 2πf):
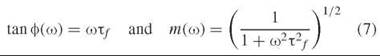
Accurate results require measurement of multiple modulation frequencies and fitting the resulting phase shifts and demodulations to characterize the fluorescence decay. At low frequencies, the fluorescence signal follows closely the modulation of the excitation pulse. At high frequencies, the time lag between excitation produces a phase shift in the fluorescence signal, and the distribution of time lags over the fluorescence decay manifests itself as a decrease in modulation. The instrumentation required, in addition to the mode-locked laser source and detector, includes mixing and tuning electronics to detect the fluorescence response as a function of frequency.
Both TCSPC and frequency-domain fluorimetry are limited in time resolution by the response of available detectors, typically >25 ps. For cases in which higher time resolution is needed, fluorescence up-conversion can be used (22). This technique uses short laser pulses (usually sub-picosecond) both to excite the sample and to resolve the fluorescence decay. Fluorescence collected from the sample is directed through a material with nonlinear optical properties. A portion of the laser pulse is used to “gate” the fluorescence by sum frequency generation. The fluorescence is up-converted to the sum frequency only when the gate pulse is present in the nonlinear material. The up-converted signal is detected. The resolution of the experiment therefore depends only on the laser pulse widths and not on the response time of the detectors. As a result, fluorescence can be resolved on the 100-fs time scale. For a recent application of fluorescence up-conversion to proteins, see Reference 23.

Figure 3. Time-resolved fluorescence techniques. (a) Time-correlated single-photon counting. The sample is excited with a short pulse of light. A fluorescence photon is detected by a microchannel-plate photomultiplier tube, which generates a voltage pulse. A portion of the excitation pulse is split off from the excitation beam and detected by a photodiode to generate a reference pulse. The time lag between excitation pulse and fluorescence photon is determined by a timer. After many counts are detected, the accumulated histogram of time lags gives the fluorescence decay convoluted with the instrument response function. (b) Illustration of the fluorescence response (solid red lines) to modulated excitation (dashed blue lines), simulated for a 1-ns fluorescence lifetime. At low modulation frequencies, the fluorescence response closely follows the modulation. High modulation frequencies generate a greater phase shift and demodulation of the fluorescence response. The phase shift and demodulation are analyzed as a function of modulation frequency to determine the fluorescence decay properties.
Fluorescence anisotropy: polarization
Here, we discuss the polarization of fluorescence emission. The spatial orientations of emitting fluorophores determine the polarization of photons emitted. This relationship is the basis of fluorescence depolarization experiments as illustrated in Fig. 4a. When a sample of randomly oriented molecules (e.g., proteins in solution) is excited by a polarized excitation beam, fluorophores whose transition dipole is parallel to the polarization are preferentially excited with a probability proportional to cos2 0, in which 0 is the angle between the polarization and the transition dipole for excitation (see Fig. 4b). If the fluorophore emits a fluorescence photon before reorienting, then the fluorescence emission will also be polarized preferentially in a direction parallel to the excitation polarization: The fluorescence is anisotropic. As the fluorophores reorient, the fluorescence emission becomes depolarized, and the fluorescence anisotropy decays. Thus, the rate of fluorescence depolarization measures the rate of reorientation of the molecule. Fluorescence depolarization or fluorescence anisotropy decay can therefore be applied to determine the rate of molecular reorientation, whether by rotational diffusion, domain motion, segmental reorientation, or local reorientational motion of the fluorophore.

Figure 4. (a) Fluorescence anisotropy is generated by excitation of the sample with vertically polarized light, which excites molecules preferentially whose transition dipole is aligned with the polarization (red arrows), generating an anisotropic distribution of excited molecular orientations. The fluorescence emission is therefore preferentially polarized vertically. (b) Definition of reorientation angles. The excitation probability is determined by the angle between the excitation polarization (z axis) and the absorption transition dipole μabs. If the emission dipole μem is not parallel to μabs (angle α), then the t = 0 anisotropy, r0, will be less than its maximum value of 0.4. Reorientation of μem (angle β) leads to decay of the anisotropy. (c) Schematic of anisotropy decay with two reorientational times: a fast reorientation caused by local segmental motion of the fluorophore or protein domain motion, and a slower component caused by tumbling of the entire protein by rotational diffusion.
Several parameters have been defined to describe fluorescence depolarization. One is the polarization, p, given by
![]()
Jablonski showed in 1960 that the fluorescence depolarization can be described naturally by another parameter, the fluorescence anisotropy r, defined by:

where ![]() denotes the steady-state (time-averaged) anisotropy, I|| is the fluorescence intensity with polarization parallel to the excitation polarization, and I⊥ is the fluorescence intensity with polarization perpendicular to the excitation polarization. Francis Perrin had derived an equation in 1926 that related the steady-state polarization p to the fluorescence lifetime. Cast in terms of the anisotropy, this equation takes on an especially simple form:
denotes the steady-state (time-averaged) anisotropy, I|| is the fluorescence intensity with polarization parallel to the excitation polarization, and I⊥ is the fluorescence intensity with polarization perpendicular to the excitation polarization. Francis Perrin had derived an equation in 1926 that related the steady-state polarization p to the fluorescence lifetime. Cast in terms of the anisotropy, this equation takes on an especially simple form:
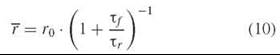
where τf is the fluorescence lifetime, τr is the rotational correlation time (sometimes denoted ф), and r0 is the short-time limiting anisotropy. Equation 10 can be used to estimate rotational correlation times from steady-state fluorescence measurements.
One application of fluorescence anisotropy is in fluorescence polarization assays, in which steady-state anisotropy measurements are used to detect binding of ligands to proteins (24). Fluorescence polarizations assays are effective for cases in which a fluorescence ligand binds to a much larger object such as a protein. On binding to the protein, the fluorescent ligand experiences a marked increase in its rotational correlation time, which results in an increased steady-state anisotropy. The technique lends itself to high-throughput assays of ligand binding, which permits rapid screening of binding interactions and sensitive determination of binding affinities.
For time-resolved measurements, Equation 9 can be expressed in time-dependent form:
![]()
where now the fluorescence intensity decays are measured as a function of time for parallel and perpendicular polarizations. The importance of Equation 11 lies in the relationship of the fluorescence anisotropy to the reorientation of the transition dipole moment:
![]()
where β(t) is the angle of reorientation of the emission transition dipole, and the brackets (∙∙∙)avg denote the average over all fluorophores. The short-time limiting anisotropy r0 is determined by the angle a between the absorption and emission transition dipoles at time t = 0 (see Fig. 4b). The maximum value for the anisotropy, obtained for parallel absorption and emission transition dipoles, is 0.4, which corresponds to a ratio I|| / I⊥ of 3 to 1.
The time dependence of the anisotropy r(t) depends on the underlying dynamics of reorientational motion. For rotational diffusion (tumbling) of a spherical object, the expected anisotropy decay is exponential with a rotational diffusion time given in the hydrodynamic limit by the Stokes-Einstein-Debye equation. For nonspherical molecules, more complex time dependence may be detected. (For more on these topics, see the book by Cantor and Schimmel in Further Reading.)
Interesting applications of anisotropy decays for proteins often develop not from tumbling of the protein as a whole, but from other reorientational degrees of freedom. These motions may include protein domain motions or segmental motions in proteins and peptides. The anisotropy decay in this case is non-single-exponential (see Fig. 4c) and takes the form:
![]()
where ai is the relative amplitude and τri the correlation time of the i th rotational component. The slowest rotational correlation time is usually the global tumbling time of the protein. Faster rotatational correlation times represent local motions. The amplitude ai contains information about the orientational freedom available for the fast motion of the fluorophore (25).
Time-resolved anisotropy decays can be recorded by time-correlated single-photon counting. The fluorescence signals I||(t) and I⊥(t) are both convolutions of the instrument response function (IRF) with the sample response and can be analyzed by nonlinear regression with iterative reconvolution of the fitting function with the IRF. In contrast, the anisotropy r(t), as calculated according to Equation 11 directly from the raw fluorescence signals, cannot be written as a convolution with an instrument function. For this reason, it is much more reliable to fit the signals I||(t) and I⊥(t) directly rather than to fit r(t) to determine the anisotropy decay properties.
Fluorescence anisotropy decay can also be measured by frequency-domain methods. In this approach, the polarized fluorescence intensities I||(ω) and I⊥(ω) are measured as a function of the modulation frequency of the polarized excitation beam. Even more information about frequency-domain anisotropy measurement and analysis can be found in the monograph by Lakowicz (see Further Reading).
Fluorescence correlation spectroscopy: photon arrival time
The arrival times of fluorescence photons contain information about correlations in fluorescence signals. Fluorescence correlation spectroscopy (FCS) (26) exploits these correlations to measure the magnitude and time scales of fluctuations in fluorescence. These fluctuations contain information about the dynamic time scales of the system and the concentration of fluorescing molecules. Correlations may span time ranges from nanoseconds to milliseconds, which extends the dynamic time window for fluorescence measurements far beyond what is achievable in fluorescence lifetime measurements. The autocorrelation function is calculated as:
![]()
where δF(t) is the fluctuation in the fluorescence intensity F(t) and (∙∙∙) here denotes the average over t. The recorded autocorrelation functions are typically fit to a function that describes diffusion through a three-dimensional Gaussian observation volume with adjustable parameters, which include the diffusion time τd, the average number of molecules (N) in the focal volume, and the focal volume dimensions r0/z.
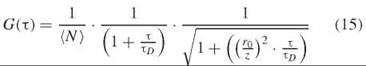
Because the magnitude of the correlation function in the short T limit is inversely proportional to the concentration of fluorophores, FCS can be used to follow changes in concentration. The diffusion time τd yields the translational diffusion coefficient. If the fluorophore undergoes intramolecular dynamics or photophysical processes, then Equation 15 must be modified accordingly (27). Analysis of the autocorrelation with a modified fitting function can then provide dynamic information about these processes.
Reviews listed in Further Reading provide excellent introductions to FCS. Related techniques have been developed to detect other molecular properties. These properties include fluorescence cross-correlation spectroscopy (FCCS) (28) to detect codiffusing fluorophores and photon-counting histograms (PCH) (29), or fluorescence intensity distribution analysis (FIDA) (29) to distinguish fluorescent species according to their brightness.
Resonance energy transfer: molecular calipers
Forster resonance energy transfer (FRET) is a remarkable tool for detecting changes in the distance between two fluorophores. FRET is the nonradiative transfer of excitation energy from the excited state of one fluorophore (the donor) to another (the acceptor). It is sensitive to the separation between fluorophores on the distance scale of tens of Angstroms, a distance range highly useful for proteins. The theory of dipole-dipole coupling between donor and acceptor (separated by distances that are large compared to the molecular sizes of the fluorophores) was developed by Theodor Forster. In this limit, the rate of energy transfer is given by:
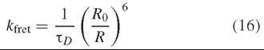
where TD is the lifetime of the donor when no acceptor is present, and Ro, known as the Forster radius, is the distance between fluorophores where the FRET rate is equal to the intrinsic donor decay rate (i.e., the efficiency of FRET is 50%). Additional information about the theory and application of FRET to proteins can be found elsewhere in this volume.
Single-molecule fluorescence
The intrinsic high sensitivity of fluorescence detection is illustrated dramatically by the detection of fluorescence from single fluorophores. Many fluorescence techniques described above have been applied with single-molecule sensitivity. In heterogeneous samples, where a distribution of molecular properties exists (structure, dynamics, and environment), single-molecule measurements can map out the distribution, which yields information that is not readily available from bulk or ensemble-averaged measurements. Single-molecule detection sensitivity can also be applied fruitfully in cases where the copy number of molecules is low, for example in detection of proteins in the contents of single cells. Single-molecule detection is often carried out with a fluorescence microscope equipped with a sensitive detector such as an avalanche photodiode or a CCD camera. Additional details about single-molecule measurements are available from several reviews (see Further Reading).
Imaging: location of emitted photons
Another area of rapid growth in application of the fluorescence techniques to proteins is in imaging of cells and tissues. Through use of an array detector such as a CCD camera with wide-field illumination or beam scanning with confocal detection, the spatial position of fluorophores can be recorded with a precision determined by the resolution of the microscope. (Single-particle tracking techniques can provide even greater precision by fitting the intensity profile of the emitted light (30).) A wide range of fluorescence techniques is now applied routinely to biologic imaging. These techniques include two-photon fluorescence microscopy, fluorescence recovery after photobleaching, fluorescence lifetime imaging, FRET imaging, and fluorescence anisotropy imaging. An introduction to topics within the broad area of fluorescence imaging techniques is available in many resources. See, for example the book by Pawley listed in Further Reading.
References
1. Teale FWJ, Weber G. Ultraviolet fluorescence of proteins. Biochem. J. 1959; 72:15.
2. Vivian JT, Callis PR. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 2001; 80:2093-2109.
3. Tolosa L, Nowaczyk K, Lakowicz J. Fluorescence probes for biochemical systems. In: Introduction to Laser Spectroscopy. Andrews DL, Demidov AA, eds. 2002. Kluwer Academic/Plenum Publishers, New York.
4. Visser AJWG, Ghisla S, Massey V, Muller F, Veeger C. Fluorescence properties of reduced flavins and flavoproteins. Eur. J. Biochem. 1979; 101:13-21.
5. Haugland RP. Handbook of fluorescent probes and research products. 2002. Molecular Probes, Inc., Eugene, OR.
6. Tsien RY, Ernst L, Waggoner A. Fluorophores for confocal microscopy: photophysics and photochemistry. In: Handbook of Biological Confocal Microscopy. Pawley JB, ed. 2006. Springer, New York.
7. Adams SR, Campbell RE, Gross LA, Martin Brent R, Walkup GK, Yao Y, Llopis J, Tsien RY. New biarsenical ligands and tetracys- teine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J. Am. Chem. Soc. 2002; 124:6063-6076.
8. Subramaniam V, Hanley QS, Clayton AHA, Jovin TM. Photophysics of green and red fluorescent proteins: Implications for quantitative microscopy. Methods Enzymol. 2003; 360:178-201.
9. Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat. Methods 2005; 2:905-909.
10. Ward TH, Lippincott-Schwartz J. The uses of green fluorescent protein in mammalian cells. In: Green Fluorescent Protein. Chalfie M, Kain SR, eds. 2006. Wiley, New York.
11. Birks JB, Dyson DJ. The relations between the absorption and fluorescence properties of organic molecules. Proc. R. Soc. London, Ser. A 1963; 275:135-148.
12. Xu C, Zipfel W, Shear JB, Williams RM, Webb WW. Multiphoton fluorescence excitation: new spectral windows for biological nonlinear microscopy. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:10763-10768.
13. Rehms AA, Callis PR. Two-photon fluorescence excitation spectra of aromatic amino acids. Chem. Phys. Lett. 1993; 208:276-282.
14. Kierdaszuk B, Malak H, Gryczynski I, Callis P, Lakowicz JR. Fluorescence of reduced nicotinamides using one- and two-photon excitation. Biophys. Chem. 1996; 62:1-13.
15. Blab GA, Lommerse PHM, Cognet L, Harms GS, Schmidt T. Two-photon excitation action cross-sections of the autofluorescent proteins. Chem. Phys. Lett. 2001; 350:71-77.
16. Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science 1990; 248:73-76.
17. Johnson ME, Landers JP. Fundamentals and practice for ultrasensitive laser-induced fluorescence detection in microanalytical systems. Electrophoresis 2004; 25:3513-3527.
18. Engelborghs Y. Correlating protein structure and protein fluorescence. J. Fluoresc. 2003; 13:9-16.
19. Lakowicz JR. On spectral relaxation in proteins. Photochem. Photobiol. 2000; 72:421-437.
20. Boens N, Qin W, Basaric N, Hofkens J, Ameloot M, Pouget J, Lefevre J-P, Valeur B, Gratton E, VandeVen M, Silva ND Jr., Engelborghs Y, Willaert K, Sillen A, Rumbles G, Phillips D, Visser AJWG, Van Hoek A, Lakowicz JR, Malak H, Gryczynski I, Szabo AG, Krajcarski DT, Tamai N, Miura A. Fluorescence lifetime standards for time and frequency domain fluorescence spectroscopy. Anal. Chem. 2007; 79:2137-2149.
21. Gratton E, Jameson DM, Hall RD. Multifrequency phase and modulation fluorometry. Ann. Rev. Biophys. Bioeng. 1984; 13:105-124.
22. Kahlow MA, Jarzeba W, DuBruil TP, Barbara PF. Ultrafast emission spectroscopy in the ultraviolet by time-gated upconversion. Rev. Sci. Instrum. 1988; 59:1098-1109.
23. Xu J, Toptygin D, Graver KJ, Albertini RA, Savtchenko RS, Meadow ND, Roseman S, Callis PR, Brand L, Knutson JR. Ultrafast fluorescence dynamics of tryptophan in the proteins monellin and iiaglc. J. Am. Chem. Soc. 2006; 128:1214-1221.
24. Jameson DM, Croney JC. Fluorescence polarization: Past, present and future. Comb. Chem. High Throughput Screening 2003; 6:167- 176.
25. Kinosita K, Jr., Kawato S, Ikegami A. A theory of fluorescence polarization decay in membranes. Biophys. J. 1977; 20:289-305.
26. Elson E, Magde D. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers 1974; 13:1-27.
27. Widengren J, Mets U, Rigler R. Fluorescence correlation spectroscopy of triplet states in solution: A theoretical and experimental study. J. Phys. Chem. 1995; 99:13368-13379.
28. Schwille P. Cross-correlation analysis in FCS. In: Fluorescence Correlation Spectroscopy. Rigler R, Elson ES, eds. 2001. Springer, Berlin.
29. Chen Y, Muller JD, So PTC, Gratton E. The photon counting histogram in fluorescence fluctuation spectroscopy. Biophys. J. 1999; 77:553-567.
30. Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002; 82:2775-2783.
Further Reading
Becker W. Advanced time-correlated single photon counting techniques. 2005. Springer-Verlag, Berlin.
Brand L, Johnson ML, eds. 1997. Flourescence spectroscopy. New York: Academic Press.
Callis PR. 1La and 1Lb transitions of tryptophan: applications of theory and experimental observations to fluorescence of proteins. Methods Enzymol. 1997; 278:113-150.
Cantor CR, Schimmel PR. Biophysical chemistry, pt. 2: techniques for the study of biological structure and function. 1980. W. H. Freeman, New York.
Chalfie M, Kain SR, eds. 2006. Green fluorescent protein: properties, applications, and protocols. 2nd ed. Wiley, New York.
Chen Y, Barkley MD. Toward understanding tryptophan fluorescence in proteins. Biochemistry 1998; 37:9976-9982.
Eftink MR. Intrinsic fluorescence of proteins. In: Topics in fluorescence spectroscopy. Lakowicz JR, ed. 2000. Kluwer Academic/Plenum Publishers, New York. pp. 1-15.
Engelborghs Y. The analysis of time resolved protein fluorescence in multi-tryptophan proteins. Spectrochim. Acta, Part A 2001; 57:2255- 2270.
Hotz CZ. Applications of quantum dots in biology: an overview. Methods Mol. Biol. 2005; 303:1-17.
Lakowicz JR. Principles of Fluorescence Spectroscopy. 2nd ed. 1999. Kluwer Academic/Plenum Publishers, New York.
Marriott G, Parker I. Biophotonics, PartA. Methods Enzymol. 2003; 360.
Marriott G, Parker I. Biophotonics, Part B. Methods Enzymol. 2003; 361
Michalet X, Kapanidis AN, Laurence T, Pinaud F, Doose S, Pflughoefft M, Weiss S. The power and prospects of fluorescence microscopies and spectroscopies. Annu. Rev. Biophys. Biomol. Struct. 2003; 32:161-182.
Moerner WE. A dozen years of single-molecule spectroscopy in physics, chemistry, and biophysics. J. Phys. Chem. B 2002; 106:910-927.
Nikon. Introduction to Fluorescent Proteins. http://www.Microscopyu.Com/articles/livecellimaging/fpintro.Html.
O’Conner DV, Phillips D. Time-Correlated Single Photon Counting. 1984. Academic, New York.
Pawley JB, ed. 2006. Handbook of biological confocal microscopy. In: Fluorescence Correlation Spectroscopy: Theory and Applications. 2001. Rigler R, Elson ES, eds. Springer-Verlag, Berlin.
Ross JBA, Laws WR, Rousslang KW, Wyssbrod HR. Tyrosine fluorescence and phosphorescence from proteins and polypeptides. In: Topics in Fluorescence Spectroscopy. Lakowicz JR, ed. 1992. Plenum Press, New York. pp. 1-63.
Schwille P, Haustein E. Fluorescence correlation spectroscopy. Biophysical Society. http://www.biophysics.org/education/schwille.pdf.
Valeur B, Editor. Molecular Fluorescence: Principles and Applications. 2000. Viley-VCH Verlag, Weinheim. 387p.
Xie XS, Lu HP. Single-molecule enzymology. In: Single Molecule Spectroscopy. Rigler R, Orrit M, Basche T, eds. 2001. Springer- Verlag, Berlin. pp. 227-240.
See Also
Fluorescence Resonance Energy Transfer (FRET) for Proteins
Fluorescence in Living Systems: Overview of Applications in Chemical Biology
Fluorescence Spectroscopy: Overview of Applications in Chemical Biology
Fluorescence Techniques: Lipids