CHEMICAL BIOLOGY
Forward Chemical Genetics
Young-Tae Chang, National University of Singapore and Singapore Bioimaging Consortium, A* STAR, Singapore
doi: 10.1002/9780470048672.wecb178
With the successful results of the Human Genome Project, we are now faced with the problem of handling numerous gene targets whose functions remain to be studied; this challenge is being undertaken by the field known as functional genomics. Chemical genetics is an emerging new paradigm to attack this challenging problem, of which there are two approaches: forward and reverse. Although reverse chemical genetics uses a ''cause-to-effect'' approach, forward chemical genetics uses an ''effect-to-cause'' approach. As opposed to conventional genetics where gene knock-outs or overexpression, forward chemical genetics uses a small-molecule library to produce a novel phenotype that eventually is employed in the elucidation of gene function. Compared with conventional genetics, chemical genetics holds several unique advantages. A successful forward genetic study provides not only knowledge about a novel gene's function, but also it provides a small molecule ON/OFFswitch that can regulate biologic processes. These small molecules will be extremely useful biologic probes as well as potential new drug candidates. Three main components make up forward chemical genetics: 1) chemical toolbox generation, 2) phenotypic screening, and 3) target identification. Although all three components require additional refinement, target identification poses the greatest challenge. Herein, the general concepts of chemical genetics, with a focus on the forward approach, and the technical overviews for each component are described.
Chemical genetics, in the simplest of terms, can be defined as a “genetics” study using “chemical” tools (1). Elucidating the function of every gene from the sequence data of tens of thousands of genes (so-called functional genomics) is the next major step for the human genome project. Geneticists conventionally have investigated the function of unknown genes by comparing the normal phenotype with that of knockouts, or through the overexpression of target genes. A novel approach using chemical library screening to find interesting phenotypic changes through the targeting of a specific gene product, a protein, has emerged as an alternative tactic—so-called chemical genetics. In chemical genetics, one chemical compound may specifically inhibit or activate one (or multiple) target proteins. Therefore, the compound is equivalent in conventional genetics to a gene knockout or to the overexpression of a gene. As in classic genetics, chemical genetics is divided into two approaches: forward and reverse.
Genetics and Chemical Genetics
Forward and reverse approach
Forward genetics (FG) operates “from effect to cause” or “phenotype (physically apparent characteristic) to genotype (genetic sequence)” and requires no specific gene target at the onset. It studies changes in phenotype(s) such as morphology, growth, or behavior resulting from random genomic DNA mutations or deletions induced from radioactive or chemical mutagenesis, and then it identifies the gene responsible through mutation mapping. Forward chemical genetics (FCG) mimics FG by substituting random mutagenesis with a collection of a library of typically unbiased (not targeted) compounds as protein function regulators in place of mutagens (2). The first step in both FG and FCG is to screen for changes induced by either the inhibition or the stimulation of a protein’s function, and both go on to identify the genetic cause but in different ways. FG goes after genetic mutation, which is a permanently retained marker, but FCG needs to identify the protein partner for the small molecule. This target identification is one of the greatest challenges in chemical genetics (3).
With the help of molecular biology techniques, reverse genetics (RG) was a later development in genetics and operates “from cause to effect” (genotype to phenotype) (4). Reverse genetics begins with selecting a gene of interest, manipulating it to produce an organism harboring the mutated gene, and characterizing the phenotypic differences between the mutant and the wild-type organisms (5). In the same context, reverse chemical genetics (RCG) begins with a known protein, which is analogous to a specific gene selection (6). This known protein is then screened with vast pools of library compounds to identify functional ligands that either stimulate or inhibit the target protein (7). Once a specific ligand is identified, it is introduced to a cell or organism, which is analogous to genetic mutation, and the resulting changes in phenotype are studied.
Advantages of forward chemical genetics
In RCG, the starting point is a selection of the protein of interest using previous information. The chemical libraries are mainly tested for only one selected protein target. This article is a more focused study of the known (most likely well-validated) target protein usually focused on improving on the chemical probes, which in a sense narrows down the scope of the work. In contrast, FCG studies whole cells or organisms, and thus, the compounds are screened against multiple potential targets simultaneously. A successful FCG will identify a novel gene product (target protein) and itsON/OFF switch, the small-molecule complement. Therefore, FCG promises an efficient “two-birds-with-one-stone” approach. However, the unpredictable pharmaceutically usable results have kept pharmaceutical companies from making a full commitment to FCG, leaving the field to be developed by the academic community thus far.
Compared with classic genetics, FCG offers several advantages and provides access to previously unstudied biologic space. Use of chemical tools offers greater ease and flexibility than does classic genetic modification. Classic genetic techniques are relatively difficult to employ especially in mammals because of their diploid genome, physical size, and slow reproduction rate. On the other hand, FCG studies may be conducted on any complex cellular or animal models without the need for any time-consuming genetic modifications that may prove lethal or in which the cell/animal can mask the phenotype through related gene functional compensation for the mutation. Especially important is FCG’s promise in operating in the relevant context of human cells at physiologic conditions that has strained traditional genetics techniques. Therefore, FCG fills a major gap in genetic studies where no, or suboptimal, model systems exist. Additionally, chemical genetics also allows for the possibility of “multiple knockouts” by adding multiple specific ligands, which is a situation often described as a “nightmare” for the geneticist (8).
Second, classic genetic knockouts, in principle, delete the protein entirely from the organism (9). Therefore, it is difficult to determine the effects that develop from the deletion separate from those that develop from merely a particular function of the protein (10). It is always possible that one protein may have multiple functions, and chemical genetics can potentially isolate and dissect particular functions of that protein while leaving others intact (11). Additionally, if a gene is essential for survival or development, a total knockout such as in classic genetics, may abolish the chance to study the later stage function of that gene because the deletion may be lethal (12). Chemical genetics allows the use of sublethal doses of the ligand and avoids full lethality, thus providing a partial knockout phenotype (13).
Another advantage of chemical genetics is real-time control. Chemical genetics allows for this control by the ability to introduce rapidly a cell-permeable ligand at any stage that may yield the desired phenotype as quickly as diffusion-limited kinetics will allow. The chemical perturber/ligand/probe is in effect a “switch” that can turn the event under study ON or OFF in real time and allows for kinetic in vivo analysis, which is something not usually possible in classic genetics. Although temporal control is available in classic genetic studies through conditional alleles, such as temperature-sensitive mutations, they often have unwanted broad side-effects that may interfere with the desired result (4). The antisense oligonucleotide and RNA interference (RNAi) are other popular alternatives for conditional knockouts (10) that work by inhibiting the synthesis of the target protein from mRNA. However, as their effects are delayed until all existing proteins are degraded, they are particularly ill-suited to time-sensitive studies, such as signal transduction, that occur on the milliseconds-to-hours time scale.
Chemical genetics and classic genetics are techniques that compliment each other well (14). One of the greatest advantages of classic genetics is the incredible specificity of a gene knockout. Although some chemical ligands can be specific switches with specificity approaching that of a gene knockout’s, the low specificity of many ligands often give “off-target” effects in which the probe may interact with proteins other than the protein(s) targeted. This low specificity makes defining specific protein functions very difficult because these off-target effects may lead to toxicity or false or unwanted positive/negative biologic results. In addition to this lack of specificity, chemical genetics cannot yet match the generality of genetics. Geneticists can, in theory, “knockout” any gene as long as the genomic sequencing is completed in the given species, which is an ability that at this point exists as nothing more than a dream for the chemical geneticist (8). These situations are the perfect place for the integration of chemical and classic genetics (10).
Importantly, unlike drug development where specificity is tantamount, in chemical genetics, ligands need not be completely specific, so long as they give an identifiable phenotype that allows for the deciphering of the target protein’s function and that its side effect are relatively small. Whereas one may desire compounds with affinity in the (sub)nanomolar range capable of producing the desired effect, in reality, compounds of low micromolar affinities are often accepted as good-to-reasonable candidates in chemical genetics (3), Despite this issue, and with an understanding of the necessary medicinal chemistry follow-up modifications and studies required, chemical genetics still has the advantage of immediately offering a potential drug lead, rather than simply a target gene or protein, as in classic genetics. Interestingly, a lead compound developed in drug discovery that may not possess pharmacokinetic properties suitable for therapeutic purposes may still be used as a probe in chemical genetics studies (11). In fact, the lower pharmacokinetic property requirements for chemical genetics probes compared with drugs allows for the use of a greater variety of functional groups and for a maximization of the chemical space in library constituents (15).
Components of Forward Chemical Genetics
With the advent of combinatorial library techniques that facilitate the synthesis of a large number of molecules (Fig. 1a), in combination with the success of HTS (high-throughput screening), a large number of compounds can be easily and rapidly screened to discover a novel small molecule (hit compound). Once the hit compound is selected in a cellular or organism system (Fig. 1b), the next step is to identify the target protein and the biochemical pathways involved. An on-bead affinity matrix or tagged molecule (photoaffinity, chemical affinity, biotin, or fluorescence), which is obtained through the modification of the lead compound (Fig. 1c), is commonly used in the identification of the target protein. The protein is identified by “fishing out” the target through the exploitation of the binding affinity of the proteins toward the immobilized molecule (Fig.ld), followed by gel separation (Fig. 1e) and mass-spectrometry. Some small molecules and their target proteins identified FCG thus far are summarized in Table 1 (16-46) In summary, the forward chemical genetics procedure is composed of 1) preparation of chemical toolboxes, 2) phenotypic screening, and 3) target identification and validation.
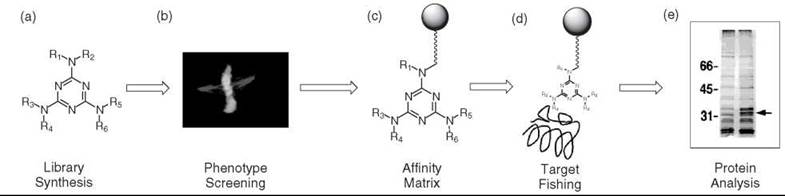
Figure 1. Forward chemical genetics scheme.
Chemical toolboxes
Collections of compounds, so-called libraries, are the absolute starting point for any chemical genetics study. These library compounds can be either natural products or synthetic compounds with various designs.
Natural products
Natural products have evolved alongside the various life forms on earth. Each surviving natural product may have its own reason for existence in the context of biologic function, so-called, biologically validated. That is why many natural products have been used for medicinal purposes for so long. For example, colchicine has been used as a drug for over 2000 years. Therefore, it is generally accepted that collections of natural products have a higher probability of delivering pharmacologically useful compounds than a typical synthetic combinatorial library. (9, 47)
Natural products are obtained from sources such as plants, soils, and marine sponges. The most difficult in natural product discovery is isolating any active components out of the whole mixture extract. The typical and time-consuming isolation route is known as bioassay-guided purification. The purification procedure involves iterative processes in which compounds undergo multiple rounds of extraction/chromatography guided by the screening results of the successive crude extracts. These studies are hampered by overlooking potentially highly active low-abundance compounds, the cytotoxicity of one component masking the desired effect of another component, and bioactivity resulting from complicated synergistic effects (2, 48). In addition, determining the structure of the compound is another laborious and challenging task. Over the past decade and a half, these drawbacks led to a retreat from natural products by most pharmaceutical companies. However, the disappointment in the number of drugs originating from combinatorial chemistry and the continued benefits of natural products are luring many companies back to them. Currently, major pharmaceutical companies are re-embracing natural products and natural product-like libraries. Natural products need not have their use restricted to therapeutics, but they can be of great value to chemical genetics studies. Extremely exciting is the integration of traditional natural products and their scaffolds with modern combinatorial and HTS tools. Perhaps success will lie in a balance of the old and the new.
Natural product-like compounds
Natural product-like libraries offer a highly desirable middle ground between those who seek powerfully bioactive compounds from natural libraries and those who seek the ease of synthesis found in libraries composed of small organic molecules. Natural products are typically chiral, are extremely complex, and contain many stereogenic centers. These structures are often highly potent and serve as attractive leads for drug development (49). Natural products can be considered privileged structures in a biologic context and an excellent starting point for library design with a high probability for biologic activity. These compounds often contain sets of related and homologous pharmacophoric groups throughout families of natural compounds (50). Natural product-like libraries are those collections of compounds whose structures are based on or share high structural homology with natural products. These libraries may be designed to generate derivatives of a natural product scaffold. In addition, some have sought to generate natural product like-libraries not to improve on known activity but to expand a molecule’s functionality into a previously unknown area of biologic space (51). The synthesis of natural product-like libraries, however, encounters many of the difficulties of synthesizing natural products, along with the challenge of synthesizing these complex structures on the solid phase.
Table 1. Small molecules and their target proteins identified in forward chemical genetic screening
|
Small molecules |
Compound source |
Library size |
Activity |
Target protein |
References |
|
|
DOS of Triazine |
1,536 |
Inhibition of brain/eye development in zebrahsh |
Ribosomal subunit proteins (S5, SI3, S18, L28) |
(16) |
|
|
Commercial |
16,320 |
Inhibition of rapamycin activity |
Teplp (PTEN) & Ybr077cp |
(17) |
|
|
Commercial |
6,000 |
Yeast growth inhibition |
Sir2p (HDAC) |
(18) |
|
|
Commercial and in-house small-molecule library |
100,000 |
Suppression of FK506 activity |
Porlp (VDAC) |
(19) |
|
|
Commercial and in-house small-molecule library |
100,000 |
Suppression of FK506 activity |
Ald6p (aldehyde dehydrogenase) |
(20) |
|
|
DOS |
1,600 |
Auxin signal increase |
Sirtuin |
(21, 22) |
|
|
DOS |
Single compound |
BR biosynthesis inhibition in plant |
DWF4 (cytochrome P450) |
(23) |
|
|
Known inhibitor (vascular endothelial growth factor receptor inhibitor) |
Single compound |
Block blood vessel formation |
VEGFR inhibitor |
(24) |
|
|
DOS of Triazine |
> 100 |
Slow epiboly development in zebrahsh |
tubulin |
(25) |
|
|
Commercial |
50,240 |
SARS-CoV inhibition |
SARS-CoV protease |
(26) |
|
|
Commercial |
50,240 |
SARS-CoV inhibition |
SARS-CoV protease |
(26) |
|
|
DOS of purine |
Single compound |
Myotube disassembly |
tubulin |
(27, 28) |
|
|
DOS of Triazine |
1,536 |
Pigmenting albino melanocyte |
Mitochondrial FIFO-ATPase |
(29) |
|
|
DOS of Triazine |
1,170 |
Pigmenting melanocyte |
Prohibitin |
(30) |
|
|
Commercial |
16,320 |
Mitosis perturbation |
tubulin |
(31) |
|
|
DOS of 1,3-dioxane library |
7,392 |
Inhhibition of β-tubulin acetylation |
HDAC6 |
(32) |
|
|
Commercial |
16,320 |
Block mitosis ordering |
Mitotic kinesin Eg5 |
(33) |
|
|
DOS |
5,000 |
Downregulation of β-catenin |
cAMP response element-binding protein |
(34) |
|
|
DOS |
> 100,000 |
Neuronal differentiation |
GSK-3β |
(35) |
|
|
DOS |
12 |
AP-1 inhibition |
REF-1 |
(36) |
|
|
DOS |
140,000 |
Inhibition of Smo |
Smo |
(37) |
|
|
DOS of Triazine |
1,120 |
Daf-2 insulin signaling in C. elegans |
GAPDH |
(44) |
|
|
DOS |
Not specified |
NF-KB inhibition |
thioredoxin |
(45, 46) |
|
|
Commercial |
16,320 |
Inhibition of actin assembly |
N-WASP |
(38, 39) |
|
|
DOS of purine library 150 |
|
Cell cycle arrest at G2/M |
CDK1 (cyclin dependent kinase 1) |
(40) |
|
Small molecules
|
Compound source DOS of purine library 1,561 |
Library size |
Activity Inhibition of mitotic spindle assembly |
Target protein NQO1 (quinine oxidoreductase) |
References (41) |
|
|
Commercial |
109113 |
Inhibition of mitotic entry |
Ubiquitin |
(42) |
|
|
Not specified |
20,000 |
Reprogramming of cellular life span |
Ataxia telangiectasia-mutated (ATM)protein |
(43) |
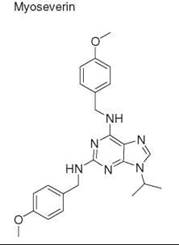
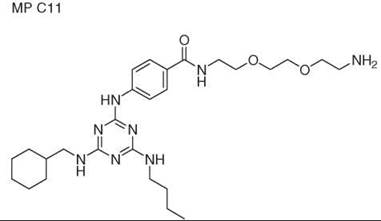
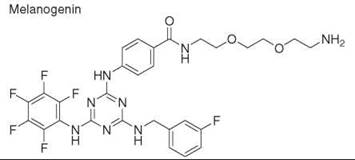
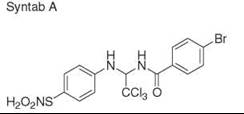
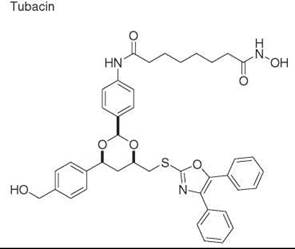
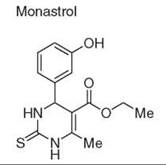
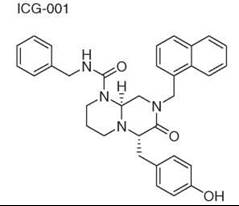
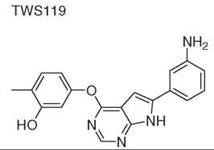
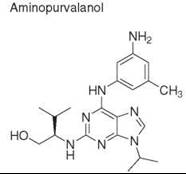
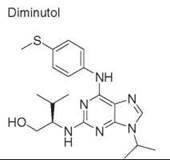
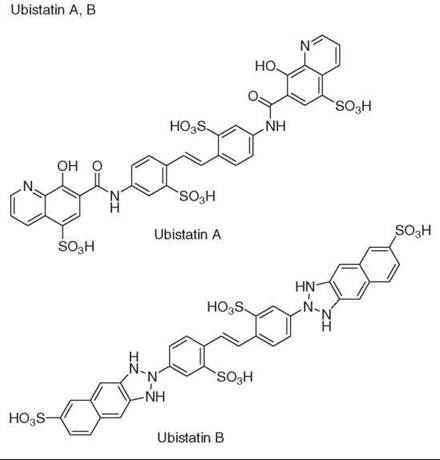
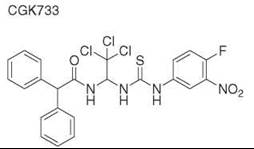
Diversity-oriented synthesis
After the combinatorial boom of the 1990s, a well-documented disappointment in the number of quality leads has set in (52). The compounds coming out of early combinatorial libraries simply did not perform up to the designer’s expectations. Guidelines such as Lipinski’s rules are helpful for generating more drug-like small molecules, but it has been argued that compounds from solid-phase organic synthesis may not be chemically diverse enough to generate the desired selectivity and potency (53).
Diversity-oriented synthesis (DOS) is a new term for a method of library construction in chemical genetics. Advocates of the DOS approach point to the archetypal case of a flat aromatic or heterocyclic core dotted with various diverse appendages that have largely failed to deliver the promised drugs. The discrepancy between the success of natural products and the traditional combinatorial libraries’ as yet disappointing rate of lead generation has forced the question of whether natural products occupy regions of chemical space evolutionarily fine-tuned to be the most potent and active compounds (54). Thus, one of the stated goals of DOS is to create extremely diverse libraries populating the maximum amount of chemical space in order to explore the greatest amount of biologic space.
DOS draws its name from its contrast to the traditionally used approach of target-oriented synthesis (TOS). Beginning with a known target (often natural product), TOS uses retrosynthetic analysis to work backward from a complicated product to simple and available starting materials. The goal of TOS is to get to a precise region of chemical space—a single target or a few closely related derivatives. Retrosynthetic analysis is not applicable to DOS because no target structure is available, and thus, the library cannot be targeted. Therefore, new thinking and planning strategies are required, and the idea of forward synthetic analysis has been proposed, which moves in the direction of simple to complex, or reactants to products, in contrast to TOS. DOS strategies focus on maximizing diversity by using branched and divergent pathways where the products of one reaction are common substrates for the next and where any follow-up chemistry must be efficient and systematic (55, 56). Molecules that occupy a greater degree of 3-D space, or those that are more globular/spherical, are a popular goal of DOS, as opposed to the relatively flat or circular molecules traditionally used in combinatorial chemistry.(54)
Tagged libraries
Tagged libraries represent unique opportunities in library design (57). In this approach, libraries may be synthesized by any means and designed around any type of scaffold, but they must contain some functional tags integrated into the library. These tags are incorporated into the library compounds from the beginning, and they bestow some additional function into the molecules. The most common example of a tag is a fluorophore, but many more variations exist that allow the compounds to have functions extending beyond their biologic activity that may aid in areas such as target identification or ligand assembly.
Peptide nucleic acids (PNAs) are oligonucleotide-like molecules that have their DNA backbone removed and replaced with an achiral polyamide backbone that can hybridize with DNA through strand displacement (58). PNA tags have been used in the development of tagged libraries that allow for the spatially addressable localization and identification of probes on a DNA microarray.
Click chemistry offers unique tags that can be used to generate library members through strategies such as target-guided ligand assembly. Sharpless developed click chemistry as a rapid reaction that proceeds in a short amount of time through “spring-loaded” highly exothermic, irreversible reactions to form carbon-hetero bonds (59). One of the most common bond is a (2+3) Huisgen 1,3-dipolar cycloaddition between an azide and an alkyne moiety, which is described as an energetically “near-perfect” reaction. Click chemistry’s main advantage is the swift and clean nature of these reactions that can greatly assist library synthesis and lead discovery. Click chemistry-based tags have also been incorporated into studies of activity-based protein profiling (ABPP) (60). ABPP is a chemical genetic method that uses probes to monitor and visualize changes in protein functions/levels in the cell, especially changes that occur on a posttranslational level. In a traditional ABPP study, the probes carry a tag/reporter, such as biotin or fluorescence, whose size and physical properties may adversely affect probe-target interaction, cellular uptake, and probe distribution. In the click chemistry-based approach, the probe is functionalized with a tag bearing a simple azide moiety, which presents much less of a disturbance in the system. After screening and binding to the protein target, an acetylene bearing reporter is introduced where it is covalently attached to the probe-target complex for visualization.
An intrinsic linker tag approach has been developed for facilitated target identification. All library compounds carry a linker tag built in, and the compounds were tested in phenotypic screening in the presence of the tag. These linker tags contain a functional group at the end of the linker, and this facilitates the conversion of a hit compound to affinity matrix without the need for a time-consuming structure-activity relationships (SAR) study (44). The tagged strategy is applicable to any type of library scaffold and allows for rapid transfer from biologic screening to target identification.
Instead of tags being external to the active portion of the molecule, internally tagged fluorescent compound libraries were also demonstrated in a chemical genetic study. In many cases, the addition of an exogenous reporter or tag can alter the effect or cellular distribution of the original molecule. Tagging the molecule intrinsically avoids this problem. In this strategy, the probes being studied are all intrinsically fluorescent through their known fluorescent scaffold, and no additional tagging is required to visualize cellular localization (61) or to visualize a bound protein (62).
Dynamic combinatorial libraries
Traditional combinatorial libraries are synthesized primarily by parallel or split-pool techniques as static pools of discrete molecules. Although using compound mixtures fell out of favor in combinatorial chemistry, they found renewed use in supramolecular chemistry in the form of dynamic combinatorial chemistry (DCL). The dynamism of DCLs results from the reversible interchangeability possible with their components. In these systems, every member of the library and the targets themselves affect all other members of the library, particularly in terms of library composition (63). Any stabilization of one member will result in an equilibrium shift and thermodynamic redistribution, by LeChatelier’s principle, of the library mixture favoring the best binder. Advantageously, library construction and screening can be combined in one step, because amplification of the best binder can be analytically detected. Therefore, the use of DCLs is as much an application of HTS as is library development. Whereas traditional combinatorial libraries rely on their sheer numbers, DCL libraries offer an alternative approach through self-replication and amplification.
The use of dynamic combinatorial libraries requires three steps: 1) the collection/design of a library capable of undergoing reversible constituent interchange, 2) conditions whereby the library may undergo interconversion, and 3) a step that selects the “fittest” binder and possibly involves its amplification. When a template or ligand is used to amplify the concentration of a member of the library, called “survival of the fittest,” the Darwinian implications of this often lead to it being referred to as “molecular evolution.” In a molecular evolution system, the fittest binder is amplified with each successive round of screening, whereas the poor binders’ concentration will either be unaffected or decrease. Although widely applied for many other purposes, DCLs have also been used to identify protein ligands, which make them a promising tool in chemical genetics library development.
Annotated chemical libraries
Generally, libraries require careful design followed by extensive screening. Truly daunting is the follow-up work to identify a hit compound’s mechanistic mode of action or target protein. An alternative approach to designing and building large collections of compounds of which no known biologic activity information exists is the use of an annotated chemical library (ACL). An ACL is a collection of compounds of diverse structure from various sources possessing experimentally bona fide biologic activities and mechanisms. An ACL contains compounds with diverse sets of biologic activity, whereas a typical library would even in a best-case scenario be composed of just a fraction of a percent of active compounds. One need not synthesize even a single compound to generate an ACL; one merely needs to identify, collect, and annotate. ACLs operate by assigning previously reported biologic activity to each compound without any required regard for the pathway or phenotype under study.
Phenotypic screening
Once libraries of compounds are assembled, the next step in chemical genetics is evaluating them for their biologic activity (i.e., assay or screening). Although virtually any kind of organisms can be used for phenotypic screenings in FCG, the most popular model systems have been Saccharomyces cerevisiae (budding yeast), Arabidopsis (plant), zebrafish embryo, drosophila (fruit fly), Caenorhabditis elegans (worm), and various mammalian cell cultures.
Assays must be designed with maximum sensitivity, selectivity, reproducibility, and cost effectiveness in mind (64). Given the size of today’s libraries, the option of doing anything other than HTS is something less than desirable. Screening technologies have progressed in miniaturization from high-throughput (384 well plates) to ultra-high-throughput (3456 well plates) screening (u-HTS), and their limits have not yet been reached. However, although screening methods have matched the enormity of many libraries, the screens must be reliable and reproducible, and the data must also be manageable. Not only have the number of assays increased, but assays have increased in the dimensionality of the data produced, so-called “high-content screening.” Therefore, a significant challenge in HTS, and especially in u-HTS, is the development of tools that allow for data management and analysis in chemical genetics.
Yeast
Budding yeast (S. cerevisiae) is a robust and powerful tool in chemical genetics. Three significant advantages of yeast as a model organism in understanding cellular responses to chemical perturbation include its ease of growth, high genetic conservation with humans, and the size of the collection, up to 6000 genes, which makes their use a very high-throughput screen.
Plants
Plants offer an attractive platform for phenotypic screening in chemical genetics: 1) All known plant growth regulators are small molecules, the experimental protocols for analyzing plant growth regulators are well defined and can be easily adapted to unbiased chemical genetic screens; 2) the genomes of the most common systems are already sequenced and that significantly aids in target identification; and 3) plant roots readily uptake small molecules avoiding many of the permeability and transport issues in traditional chemical genetic systems.
Zebrafish
Zebrafish (Danio rerio) have become a promising whole- organism screening method in chemical genetics for many of the same reasons they are popularly used in developmental biology and genetics. Whole-organism screening is preferred in some cases over target-based screening because it allows for a more unbiased discovery in a relevant physiologic context. However, although mammals provide an excellent relevant screening context, their use is expensive, requires a great deal of space, large quantities of compounds, strict regulations, is laborious, and often raises ethical questions. Because of these limitations in mammals, systems such as zebrafish have been popularized as a result of the many advantages they present. First, zebrafish, unlike other systems such as yeast or round worms, are vertebrates with discrete organs such as the brain, sensory organs, heart, muscles, and bones. These organ systems are very close to their human counterparts, in terms of their high level of structure, and this aids in their suitability for chemical genetics and drug discovery. Additionally, zebrafish are small enough in their early embryonic stages to live in a well of a microtiter plate. They are also prolific reproducers, which allows for the screening of large libraries. Lastly, zebrafish embryos are a desirable model because of their complete transparency that allows for the multiple observations of dynamic processes in every organ and structure without the need for dissection or for sacrificing the animal.
Drosophila
Drasophila has been used by geneticists in many studies. Their short life cycle and low cost make them a desirable animal model. Although they bear less genes than humans, cases exist of one Drasophila gene representing several human genes, and this kind of feature makes it a popular model system for studying human disease pathways. Additionally, Drosophila is an excellent compliment to C. elegans RNAi studies in which some knockouts are unavailable, such as genes expressed in the nervous system.
C. elegans
C. elegans has had little use by researchers in chemical genetics thus far, but it is of growing use in drug discovery and is poised to make a large contribution. It is merely a matter of time until it becomes a widespread tool in chemical genetics. C. elegans was the first multicellular organism to have its genome completely sequenced. The knowledge and the experience that comes with it will prove invaluable in future C. elegans applications in chemical genetics. The worm is small and transparent, which allows for full visualization of its developmental processes and contains complex structures such as a digestive tract, nervous system, and muscles. Additionally, it has a short life cycle and produces many progeny, which makes it more compatible with high-throughput screening. Advantageously, RNAi screens were first used and developed in C. elegans and the large amount of experience in this field will greatly aid multipronged chemical genetic approaches, particularly target identification.
Target identification and validation
Target identification is an integral part of chemical genetics studies and is its most rate-limiting and challenging step (57). Much of the difficulty originates from the weakly binding compounds often identified in FCG screening. The possibility of success in target identification greatly increases with increasing binding affinity. Whereas nM or pM binding constants will make target identification much easier, mid-μM results are much more common. Some simple approaches exist such as “the guess and test” where one hypothesizes a target and performs in vitro tests to validate it, or “guilt by association” identification where targets are implied or hinted at through studies such as mRNA transcription profiling (12). However, as in most FCG, without a clue toward the identity of the target protein, an ab intio target identification technique is required.
Affinity matrix
The most commonly used tool in target identification is the pull-down experiment, which is also known as the classic “fishing experiment” using an affinity matrix. This procedure typically involves the attachment of the “hit” molecule to a solid-phase resin such as agarose gels. The solid-linked material is then exposed to a cell extract. This process is commonly performed by passing the extract over a column of the immobilized material.
Although commonly used, affinity experiments are beset with drawbacks. First, the compounds need to be derivatized to include a handle for attachment to the resin, unless they intrinsically bear some functional tag that allows for it. The so-called tether effect can alter the activity of the compound, and tedious structure-activity relationships (SAR) studies are required to optimize the attachment point. As a solution, this SAR work could be avoided by introducing the intrinsic linker tag strategy (57). Two other requirements are needed for any hope of success in affinity matrix experiments: 1) high-affinity ligands and 2) high abundance of target. The end result of the affinity matrix relies on the multiplication of affinity and on the abundance. Also, during the cell extract preparation, a serious dilution occurs, at least 100 times. It is totally possible that the target protein exists in a complex with other protein(s) in intact cells, but it may be segregated into a monomer in the cell extract, and thus, it no longer binds to the small molecule. Therefore, a new technique, which can be performed in vivo without breaking the cellular integrity, is highly desirable.
Photoaffinity
Another interesting alternative is photoaffintity labeling, which involves attaching a photoaffintity moiety and a reporter tag. Although this method does require significant SAR knowledge, it does not require immobilization of the compound on solid support (3). Photoaffinity simply uses a photo-activated cross-linking group that forms a covalent bond during irradiation and a reporter, such as a radioactive isotope or biotin, that allows for isolation or ease of identification. Libraries have been designed bearing photoaffinity groups to exploit this approach.
Tag for mass spectrometry
Labeling proteins with “heavy” and “light” tags and screening the “hit” compound versus an inactive control, followed by mass spectrometric comparison of the two samples, is another approach that avoids many of the common pitfalls in affinity methods (3). Techniques such as stable isotope labeling with amino acids in cell culture and isotope-coded affinity tagging (ICAT) exemplify these techniques.
Yeast three-hybrid system
Genetic approaches also exist to tackle this problem. A promising approach is the yeast three-hybrid system (Y3H). This work evolved from yeast two-hybrid screens and has grown in use (65). Three components are required for these studies: 1) a protein containing a DNA binding domain fused to a small molecule with a ligand binding domain, 2) a protein with a transcriptional activation domain fused to another ligand binding domain, and 3) a bivalent small molecule. The bivalent small molecule is composed of a known ligand with an affinity for the protein containing the DNA binding domain, a probe portion of the molecule that is being tested for novel protein binding, and a linker connecting the two faces of the bivalent small molecule. Should the probe portion of the small molecule bind to the protein bearing the transcriptional activation domain, that protein is brought into a proximal relationship with the DNA and allows for the activation of the downstream reporter gene, which thus indicates successful target identification (66). The advantages of the Y3H system are that the identification of ligand binding proteins is linked to the selection of the cDNAs that encode the proteins, that phenotype and genotype are closely linked, and that these systems are explored in vivo. However, one drawback of this approach is that it is limited to simple, unicellular organisms, but work is underway to overcome this hurdle (3).
Drug western
In this approach, tagged small molecules are used to probe electrophoretically resolved cell extracts or cDNA expression libraries. Drug westerns involve bacteriophages infecting bacteria grown in a Petri dish with a cDNA library. Lysis caused by a viral infection leads to a clearing, called a plaque, containing a single member of the library. The proteins in the plaque are transferred to nitrocellulose where they are screened with tagged small molecules. Any hit plaques are isolated, a single virus is purified, and the target protein is identified by DNA sequencing. An example is the screening of two million plaques in which a sulfonamide drug was identified as inhibiting the transcription factor NF-YB (67).
Protein microarray
Protein microarrays are derived from and are complimentary to DNA microarrays. Here, collections of proteins are immobilized on a microarray surface and are probed for specific binding with tagged small molecules or by comparing the profiles of healthy versus diseased tissues (68). Although a very direct approach, protein microarrays suffer from the lack of large numbers of purified and stable proteins available for immobilization on the microarray surface. Although they have found great utility in proteomics, their development has been slow because of several technical challenges.
Magnetism-based interaction capture (MAGIC)
A unique in vivo target identification method in mammalian cells has been reported recently. The hit compound was attached covalently to magnetic nanoparticles and a green fluorescent protein-(GFP)-fused protein library was expressed in mammalian cells. When mixed together, the target proteins bind to the nanoparticle. An exogenously applied magnetic field assembles the GFP fused proteins whereby the signal can be visualized (69).Although a chemical modification of the hit compounds is still required, this is the only technique reported so far to visualize a small-molecule-target-protein interaction in mammalian cells.
Display cloning
A phage display-based direct cloning of cellular protein has been demonstrated as a possible technique for target identification (70). The surface protein library was constructed using human brain cDNA, and a biotinylated FK506 probe molecule was used as a chemical bait. During the affinity selection, the FKBP12 (FK506-binding protein) gene emerged as the dominant library member and was the only sequence identified after the second round of selection. Although the approach has been demonstrated successfully by isolating a full-length gene clone of FKBP12, its broad application potential to novel target identification remains to be explored.
References
1. Schreiber SL. Chemical genetics resulting from a passion for synthetic organic chemistry. Bioorg. Med. Chem. 1998; 6:1127-1152.
2. Lokey RS. Forward chemical genetics: progress and obstacles on the path to a new pharmacopoeia. Curr. Opin. Chem. Biol. 2003; 7:91-96.
3. Burdine L, Kodadek T. Target identification in chemical genetics: The (often) missing link. Chem. Biol. 2004; 11:593-597.
4. Zheng XF, Chan TF. Chemical genomics in the global study of protein functions. Drug Discov. Today 2002; 7:197-205.
5. Flaumenhaft R, Sim DS. The platelet as a model for chemical genetics. Chem. Biol. 2003; 10:481-486.
6. Blackwell HE, Zhao YD. Chemical genetic approaches to plant biology. Plant Physiol. 2003; 133:448-455.
7. Mayer TU. Chemical genetics: tailoring tools for cell biology. Trends Cell Biol. 2003; 13:270-277.
8. MacBeath G. Chemical genomics: what will it take and who gets to play? Genome Biol. 2001; 2(6):COMMENT2005.
9. Darvas F, Dorman G, Krajcsi P, Puskas LG, Kovari Z, Lorincz Z, Urge L. Recent advances in chemical genomics. Curr. Med. Chem. 2004; 11:3119-3145.
10. Shokat K, Velleca M. Novel chemical genetic approaches to the discovery of signal transduction inhibitors. (see comment). Drug Discov. Today 2002; 7:872-879.
11. Alaimo PJ, Shogren-Knaak MA, Shokat KM. Chemical genetic approaches for the elucidation of signaling pathways. Curr. Opin. Chem. Biol. 2001; 5:360-367.
12. Ward GE, Carey KL, Westwood NJ. Using small molecules to study big questions in cellular microbiology. Cell. Microbiol. 2002; 4:471-482.
13. Zheng XS, Chan TF, Zhou HH. Genetic and genomic approaches to identify and study the targets of bioactive small molecules. Chem. Biol. 2004; 11:609-618.
14. Strausberg RL, Schreiber SL. From knowing to controlling: A path from Genomics to drugs using small molecule probes. Science 2003; 300:294-295.
15. Zanders ED, Bailey DS, Dean PM. Probes for chemical genomics by design. Drug Discov. Today 2002; 7:711-718.
16. Khersonsky SM, Jung DW, Kang TW, Walsh DP, Moon HS, Jo H, Jacobson EM, Shetty V, Neubert TA, Chang YT. Facilitated forward chemical genetics using a tagged triazine library and zebrafish embryo screening. J. Am. Chem. Soc. 2003; 125:11804-11805.
17. Huang J, Zhu H, Haggarty SJ, Spring DR, Hwang H, Jin F, Snyder M, Schreiber SL. Finding new components of the target of rapamycin (TOR) signaling network through chemical genetics and proteome chips. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:16594-16599.
18. Bedalov A, Gatbonton T, Irvine WP, Gottschling DE, Simon JA. Identification of a small molecule inhibitor of Sir2p. Proc. Natl. Acad. Sci. 2001; 98:15113-15118.
19. Butcher RA, Schreiber SL. A small molecule suppressor of FK506 that targets the mitochondria and modulates ionic balance in Saccharomyces cerevisiae. Chem. Biol. 2003; 10:521-531.
20. Butcher RA, Schreiber SL. Identification of Ald6p as the target of a class of small-molecule suppressors of FK506 and their use in network dissection. Proc. Natl. Acad. Sci. 2004; 101:7868-7873.
21. Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 2001; 276:38837-38843.
22. Zhao YD, Dai XH, Blackwell HE, Schreiber SL, Chory J. SIR1, an upstream component in auxin signaling identified by chemical genetics. Science 2003; 301:1107-1110.
23. Asami T, Mizutani M, Fujioka S, Goda H, Min YK, Shimada Y, Nakano T, Takatsuto S, Matsuyama T, Nagata N, Sakata K, Yoshida S. Selective interaction of triazole derivatives with DWF4, a cytochrome P450 monooxygenase of the brassinosteroid biosynthetic pathway, correlates with brassinosteroid deficiency in Planta. J. Biol. Chem. 2001; 276:25687-25691.
24. Chan J, Bayliss PE, Wood JM, Roberts TM. Dissection of angiogenic signaling in zebrafish using a chemical genetic approach. Cancer Cell 2002; 1:257-267.
25. Moon HS, Jacobson EM, Khersonsky SM, Luzung MR, Walsh DP, Xiong WN, Lee JW, Parikh PB, Lam JC, Kang TW, Rosania GR, Schier AF, Chang YT. A novel microtubule destabilizing entity from orthogonal synthesis of triazine library and zebrafish embryo screening. J. Am. Chem. Soc. 2002; 124:11608-11609.
26. Kao RY, Tsui WHW, Lee TSW, Tanner JA, Watt RM, Huang JD, Hu LH, Chen GH, Chen ZW, Zhang LQ, He T, Chan KH, Tse H, To APC, Ng LWY, Wong BCW, Tsoi HW, Yang D, Ho DD, Yuen KY. Identification of novel small-molecule inhibitors of severe acute respiratory syndrome-associated coronavirus by chemical genetics. Chem. Biol. 2004; 11:1293-1299.
27. Rosania GR, Chang YT, Perez O, Sutherlin D, Dong H, Lockhart DJ, Schultz PG. Myoseverin, a microtubule-binding molecule with novel cellular effects.[see comment). Nat. Biotechnol. 2000; 18:304-308.
28. Perez OD, Chang YT, Rosania G, Sutherlin D, Schultz PG. Inhibition and reversal of myogenic differentiation by purine-based microtubule assembly inhibitors. Chem. Biol. 2002; 9:475-483.
29. Williams D, Jung DW, Khersonsky SM, Heidary N, Chang YT, Orlow SJ. Identification of compounds that bind mitochondrial F1F0 ATPase by screening a triazine library for correction of albinism. Chem. Biol. 2004; 11:1251-1259.
30. Snyder JR, Hall A, Ni-Komatsu L, Khersonsky SM, Chang YT, Orlow SJ. Chem. Biol. 2005; 12:477-484.
31. Haggarty SJ, Mayer TU, Miyamoto DT, Fathi R, King RW, Mitchison TJ, Schreiber SL. Dissecting cellular processes using small molecules: identification of colchicine-like, taxol-like and other small molecules that perturb mitosis.[erratum appears in Chem. Biol. 2001;8: 1265). Chem. Biol. 2000;7:275-286.
32. Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. 2003; 100:4389-4394.
33. Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999; 286:971-974.
34. Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Oh SW, Kim HY, Moon SH, Ha JR, Kahn M. A small molecule inhibitor of beta-catenin/cyclic AMP response element-binding protein transcription. Proc. Natl. Acad. Sci. 2004; 101:12682-12687.
35. Ding S, Wu TYH, Brinker A, Peters EC, Hur W, Gray NS, Schultz PG. Synthetic small molecules that control stem cell fate. Proc. Natl. Acad. Sci. 2003; 100:7632-7637.
36. Chong T, McMillan M, Teo JL, Henderson WR Jr., Kahn M. Chemogenomic investigation of AP-1 transcriptional regulation of LTC4 synthase expression. Lett. Drug Design Discov. 2004; 1:211-216.
37. Weitzman JB. Agonizing Hedgehog. J. Biol. 2002; 1:10.
38. Peterson JR, Lokey RS, Mitchison TJ, Kirschner MW. A chemical inhibitor of N-WASP reveals a new mechanism for targeting protein interactions. Proc. Natl. Acad. Sci. 2001; 98:10624-10629.
39. Peterson JR, Bickford LC, Morgan D, Kim AS, Ouerfelli O, Kirschner MW, Rosen MK. Chemical inhibition of N-WASP by stabilization of a native autoinhibited conformation. Nat. Struct. Mol. Biol. 2004; 11:747-755.
40. Rosania GR, Merlie J Jr., Gray N, Chang YT, Schultz PG, Heald R. A cyclin-dependent kinase inhibitor inducing cancer cell differentiation: biochemical identification using Xenopus egg extracts. Proc. Natl. Acad. Sci. 1999; 96:4797-4802.
41. Wignall SM, Gray NS, Chang YT, Juarez L, Jacob R, Burlingame A, Schultz PG, Heald R. Identification of a novel protein regulating microtubule stability through a chemical approach.[see comment). Chem. Biol. 2004; 11:135-146.
42. Verma R, Peters NR, D’Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang MS, Coffino P, Fushman D, Deshaies RJ, King RW. Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science 2004; 306:117-120.
43. Won J, Kim M, Kim N, Ahn JH, Lee WG, Kim SS, Chang KY, Yi YW, Kim TK. Small molecule-based reversible reprogramming of cellular lifespan. Nat. Chem. Biol. 2006; 2:369-374.
44. Min J, Kim YK, Cipriani PG, Kang M, Khersonsky SM, Walsh DP, Lee JY, Niessen S, Yates JR 3rd, Gunsalus K, Piano F, Chang YT. Forward chemical genetic approach identifies new role for GAPDH in insulin signaling. Nat. Chem. Biol. 2007; 3:55-59.
45. Misra-Press A, McMillan M, Cudaback E, Qabar M, Ruan F, Nguyen M, Vaisar T, Nakanishi H, Kahn M. Identification of a novel inhibitor of the NF-kappaB pathway. Curr. Med. Chem. 2002; 1:29.
46. Henderson WR Jr, Chi EY, Teo JL, Nguyen C, Kahn M. A Small Molecule Inhibitor of Redox-Regulated NF-{kappa}B and Activator Protein-1 Transcription Blocks Allergic Airway Inflammation in a Mouse Asthma Model. J. Immunol. 2002; 169:5294-5299.
47. Breinbauer R, Vetter IR, Waldmann H. From protein domains to drug candidates-natural products as guiding principles in the design and synthesis of compound libraries. Angew. Chem., Int. Ed. Eng. 2002; 41:2879-2890.
48. Abel U, Koch C, Speitling M, Hansske FG. Modern methods to produce natural-product libraries. Curr. Opin. Chem. Biol. 2002; 6:453-458.
49. Arya P, Joseph R, Chou DTH. Toward high-throughput synthesis of complex natural product-like compounds in the genomics and proteomics age. Chem. Biol. 2002; 9:145-156.
50. Lee ML, Schneider G. Scaffold architecture and pharmacophoric properties of natural products and trade drugs: Application in the design of natural product-based combinatorial libraries. J. Comb. Chem. 2001; 3:284-289.
51. Pelish HE, Westwood NJ, Feng Y, Kirchhausen T, Shair MD. Use of biomimetic diversity-oriented synthesis to discover galanthamine-like molecules with biological properties beyond those of the natural product. J. Am. Chem. Soc. 2001; 123:6740- 6741.
52. Ortholand JY, Ganesan A. Natural products and combinatorial chemistry: back to the future. Curr. Opin. Chem. Biol. 2004; 8:271-280.
53. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev. 1997; 23:3-25.
54. Burke MD, Schreiber SL. A planning strategy for diversity- oriented synthesis. Angew. Chem., Int. Ed. Eng. 2004; 43:46-58.
55. Lo MM-C, Neumann S, Nagayama S, Perlstein EO, Schreiber SL. A library of spirooxindoles based on a stereoselective three-component coupling reaction. J. Am. Chem. Soc. 2004; 126: 16077-16086.
56. Burke MD, Berger EM, Schreiber SL. A synthesis strategy yielding skeletally diverse small molecules combinatorially. J. Am. Chem. Soc. 2004; 126(43):14095-14104.
57. Mitsopoulos G, Walsh DP, Chang YT. Tagged library approach to chemical genomics and proteomics. Curr. Opin. Chem. Biol. 2004; 8:26-32.
58. Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-Selective Recognition of DNA by Strand Displacement with a Thymine- Substituted Polyamide. Science 1991; 254:1497-1500.
59. Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003; 8:1128-1137.
60. Speers AE, Cravatt BF. Chemical strategies for activity-based proteomics. Chembiochem 2004; 5:41-47.
61. Rosania GR, Lee JW, Ding L, Yoon HS, Chang YT. Combinatorial approach to organelle-targeted fluorescent library based on the styryl scaffold. J. Am. Chem. Soc. 2003; 125:1130-1131.
62. Li QA, Lee JS, Ha C, Park CB, Yang G, Gan WB, Chang YT. Solid-phase synthesis of styryl dyes and their application as amyloid sensors. Angew. Chem., Int. Ed. Eng. 2004; 43:6331-6335.
63. Ramstrom O, Bunyapaiboonsri T, Lohmann S, Lehn JM. Chemical biology of dynamic combinatorial libraries. Biochim. Biophys. Acta 2002; 1572:178-186.
64. Verkman AS. Drug discovery in academia. Am. J. Physiol. Cell Physiol. 2004; 286:465-474.
65. Licitra EJ, Liu JO. A three-hybrid system for detecting small ligand-protein receptor interactions. Proc. Natl. Acad. Sci. 1996; 93:12817-12821.
66. Kley N. Chemical dimerizers and three-hybrid systems: scanning the proteome for targets of organic small molecules. Chem. Biol. 2004; 11:599-608.
67. Tanaka H, Ohshima N, Hidaka H. Isolation of cDNAs encoding cellular drug-binding proteins using a novel expression cloning procedure: Drug-western. Mol. Pharmacol. 1999; 55:356-363.
68. MacBeath G, Schreiber SL. Printing proteins as microarrays for high-throughput function determination. Science 2000; 289:1760- 1763.
69. Won J, Kim M, Yi YW, Kim YH, Jung N, Kim TK. A magnetic nanoprobe technology for detecting molecular interactions in live cells. Science 2005; 309:121-125.
70. Sche PP, McKenzie KM, White JD, Austin DJ. Display cloning: functional identification of natural product receptors using cDNA-phage display. Chem. Biol. 1999; 6:707-716.
Further Reading
Crews CM, Splittgerber U. Chemical genetics: exploring and controlling cellular processes with chemical probes. Trends Biochem. Sci. 1999; 24:317-320.
Khersonsky SM, Chang YT. Strategies for facilitated forward chemical genetics. Chembiochem. 2004 ;5:903-908.
Smukste I, Stockwell BR. Advances in chemical genetics. Annu. Rev. Genom. Hum. Genet. 2005; 6:261-286.
Specht KM, Shokat KM. The emerging power of chemical genetics. Curr. Opin. Cell Biol. 2002; 14:155-159.
Stockwell BR. Frontiers in chemical genetics. Trends Biotechnol. 2000; 18:449-455.
Thorpe DS. Forward and reverse chemical genetics using SPOS-based combinatorial chemistry. Comb. Chem. High T. Scr. 2003; 6:623-647.
Walsh DP, Chang YT. Chemical genetics. Chem. Rev. 2006; 106:2476-2530.
See Also
Chemical Libraries: Screening for Biologically Active Small Molecules
Diversity-Oriented Synthesis of Small Molecules
Reverse Chemical Genetics
Small Molecule-Biological Target Interactions, Tool to Study
Small Molecules to Elucidate Biological Function